

Abstract
Background and aims:
Progranulin is a circulating protein that modulates inflammation and is found in atherosclerotic lesions. Here we determined whether inflammatory cell–derived progranulin impacts atherosclerosis development.
Methods:
Ldlr−/− mice were transplanted with bone marrow from wild-type (WT) or Grn−/− (progranulin KO) mice (referred to as Tx-WT and Tx-KO, respectively).
Results:
After 10 weeks of high-fat diet feeding, both groups displayed similarly elevated plasma levels of cholesterol and triglycerides. Despite abundant circulating levels of progranulin, the size of atherosclerotic lesions in Tx-KO mice was increased by 47% in aortic roots and by 62% in whole aortas. Aortic root lesions in Tx-KO mice had increased macrophage content and larger necrotic cores, consistent with more advanced lesions. Progranulin staining was markedly reduced in the lesions of Tx-KO mice, indicating little or no uptake of circulating progranulin. Mechanistically, cultured progranulin-deficient macrophages exhibited increased lysosome-mediated exophagy of aggregated low-density lipoproteins resulting in increased cholesterol uptake and foam cell formation.
Conclusions:
We conclude that hematopoietic progranulin deficiency promotes diet-induced atherosclerosis in Ldlr−/− mice, possibly due to increased exophagy-mediated cholesterol uptake. Circulating progranulin was unable to prevent the increased lesion development, consistent with the importance of progranulin acting via cell-autonomous or local effects.
Keywords: progranulin, atherosclerosis, macrophage, exophagy, lysosome, aggregated LDL
Graphical Abstract
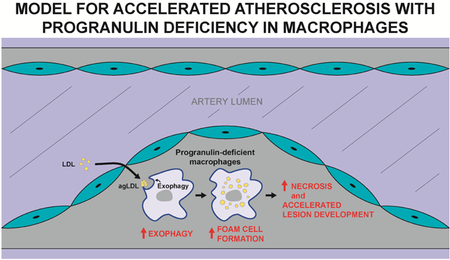
Introduction
Heterozygous mutations in the human progranulin gene (GRN) cause frontotemporal dementia with high penetrance (1–3). The encoded progranulin is a lysosomal (4–6) and secreted (7,8) protein of unclear function. It is required for normal lysosome homeostasis, and Grn−/− cells and tissues have increased lysosomal content, based on staining of the lysosomal proteins LAMP1 (4,6,9,10) and CD68 (11). In mice, nervous system phenotypes associated with progranulin deficiency include the accumulation of lipofuscin (10,12–14), behavioral changes (13,15–17), neuroinflammation (9,11–13,15,16,18), increased complement activation (4,10,11), and increased synaptic pruning (11). Grn−/− mice also exhibit decreased bone mass (19). Complete progranulin deficiency in humans causes neuronal ceroid lipofuscinosis, a form of lysosome storage disease (20,21). The biological function of progranulin and how its absence leads to disease remain largely unknown.
Progranulin has been detected in human and murine atherosclerotic plaques (22,23). It is expressed by multiple cell types of atherosclerotic lesions, including macrophages (22–24), smooth muscle cells (22,23), and endothelial cells (25). Global deficiency of progranulin increases atherosclerosis in Apoe−/− mice (23), although the mechanism(s) responsible for this phenotype are unclear. In atherosclerotic lesions, progranulin could be derived from the uptake of circulating progranulin or from the local synthesis and secretion of progranulin by cells, such as immune cells, residing within and around lesions. The relative contributions of these sources are unknown. Because increasing evidence suggests that the primary function of progranulin is in lysosomes, we hypothesized that the effects of progranulin are largely cell-autonomous. In agreement with this, reconstituting progranulin expression in Grn−/− immune cells is sufficient to attenuate their hyper-inflammatory phenotype (26), whereas these effects are lacking or diminished by treatment with exogenously provided progranulin (27) (also unpublished observations by T.N., R.F.).
In this study, we sought to test the hypothesis that immune cell–derived progranulin modulates the development of murine atherosclerosis. We utilized bone marrow-transplantation studies to reconstitute immune cells of mice prone to atherosclerosis (low-density lipoprotein receptor–deficient [Ldlr−/−] mice) (28), generating mice in which the immune cells, such as lesion macrophages, either expressed or lacked endogenous progranulin. After feeding these mice a high-fat diet for 10 weeks, we analyzed their plasma parameters and lesion formation. We also examined the effects of cholesterol loading in cultured WT and Grn−/− macrophages and show that Grn−/− cells exhibit increased exophagy, a mechanism that may contribute to their increased atherosclerosis.
Materials and methods
Reagents
AlexaFluor546 (Alexa546), LipidTOX red, Alexa488-phalloidin and biotin-fluorescein-dextran (10,000 MW) were purchased from Invitrogen. All other chemicals were purchased from Sigma Aldrich.
Mice and facilities
Animal work was approved by the Institutional Animal Care and Use Committees at the University of California, San Francisco, and Harvard University and followed NIH guidelines. Mice were housed in a pathogen-free barrier facility with a 12-h light/12-h dark cycle and allowed food and water ad libitum. Female Ldlr−/− mice (28) on a C57BL/6J background were obtained from the Jackson Laboratory at 5 weeks of age and initially fed a chow diet (Harlan Teklad, #5053). Grn−/− mice (26) and GrnR493X knock-in mice, which lack detectable progranulin protein (29), were on a C57BL/6J background (backcrossed more than eight generations).
Bone marrow transplantation
At 9 weeks of age, Ldlr−/− recipient mice were irradiated with two doses of 600 Rad (6 Gy) per mouse spaced 4 h apart. After irradiation, mice were placed in their home cage and given water that contained antibiotics [neomycin and polymyxin B sulfate (Gibco, #21850–029, 8024 U/mg)] diluted in acidic water for 2 weeks; subsequently, the mice were given acidic water without antibiotics. The morning after irradiation (within 24 h), mice were transplanted with donor bone marrow and placed in their home cages. Mice were monitored daily, and body weight was measured weekly during the recovery period. At 7 weeks post-transplantation (16 weeks of age), mice were placed on a high-fat, western purified diet consisting of 21% total fat, 34% sucrose, and 0.2% cholesterol by weight (Harlan Teklad, #88137) for 10 weeks.
Assessment of atherosclerosis
Mice were fasted overnight, anesthetized by intraperitoneal injection of tribromoethanol (Avertin, 250 mg/kg of body weight) and sacrificed by heart blood puncture, followed by perfusion with 10 ml of ice-cold PBS. For histological analysis, the entire aortic root was fixed with 4% paraformaldehyde, cryopreserved in sucrose, and embedded in OCT. Aortic roots were sectioned serially at 9-μm intervals from the base of the aortic sinus and mounted on slides (Leica X-tra). For each mouse, on average of six sections were quantified. Aortic root lesion area was quantified in Oil Red O (ORO)–stained sections, and nuclei were counterstained with Mayer’s Haematoxylin (American Mastertech, HXMMH100). Images were acquired using a Nikon E600 microscope with a 40x objective, a QImaging Retiga CCD color camera, and Q-Capture imaging software. Lesion area was quantified from images with Image J software. For analysis of necrotic cores, acellular lipid-rich areas were visually identified and outlined, and the area was determined with Image J software.
For en face analysis of aorta area containing atherosclerotic plaques, aortas were perfused and post-fixed in formalin (4% paraformaldehyde). Aortas were dissected and opened longitudinally from the aortic arch to the iliac bifurcation, and then pinned flat on a black surface. Aortas were photographed at a fixed magnification and lesions were quantified with ImageJ software as described (30).
For fluorescence immunohistochemistry of LAMP1, aortic root sections were washed with PBS, followed by blocking in a TBS solution with 10% normal goat serum, 3% BSA, 1% glycine, and 0.4% Triton X-100 to prevent nonspecific binding. Rabbit anti-LAMP1 antibody (Abcam, ab24170) was diluted in TBS blocking solution at 1:300. After overnight incubation in humidified chambers at RT, slides were washed three times for 5 min each in TBS. Alexa568 donkey anti-rabbit IgG (Invitrogen, A10042) was diluted in TBS blocking solution at 1:300 to visualize LAMP1. After 1 h incubation in the dark at RT, slides were washed 5 times for 2 min each in TBS, counterstained with DAPI (diluted 1:5000 in TBS) for 2–5 min, and cover slips were applied with aqueous mounting medium (Fluoromount-G, Southern Biotech). Images were acquired using a Nikon C2 confocal microscope with 10x and 60x objectives. LAMP1 fluorescence intensities were quantified using NIH ImageJ by measuring the pixel intensity of the LAMP1 channel in DAPI-positive cell using a 60x objective. On average, the fluorescent signal intensity of LAMP1 was quantified in 3–6 cells per field, and 7–10 fields were captured per animal. Each data point represents the average LAMP1 fluorescent signal intensity from one animal.
To quantify CD68-positive macrophages, aortic root sections were stained as above. Primary antibodies were used at the following concentrations: sheep anti-mouse progranulin (R&D, AF2557) at 1:100 and rat anti-CD68 antibody (Serotec, MCA1957) at 1:600. Secondary antibodies were used at the following concentrations: Alexa488 chicken anti-rat IgG (Invitrogen, A21470) at 1:300 to visualize CD68; Alexa568 donkey anti-sheep IgG (Invitrogen, A21099) at 1:300 to visualize progranulin. Quantification of CD68-positive cells was performed by counting the number of DAPI-positive nuclei co-localized with CD68-flourescence within a 60x field. Seven Tx-WT mice and 7 Tx-KO mice were used in this quantification, and 7–10 fields were captured per animal.
Statistical analysis
Data are presented as mean ± SD. Data were analyzed with GraphPad Prism or Excel using student’s t tests, Mann-Whitney U tests, or two-way ANOVA, followed by Tukey post hoc tests. Differences were considered statistically significant when p<0.05.
Additional methods are available in the Supplementary Materials.
Results
Macrophage progranulin deficiency increases atherosclerotic lesion size
To determine whether macrophage progranulin contributes to the development of atherosclerosis, we performed bone marrow transplantation to establish progranulin deficiency in the hematopoietic compartment, including myeloid cells, in a mouse model of atherosclerosis. We transplanted bone marrow from either WT or Grn−/− mice into irradiated female Ldlr−/− recipient mice (referred to as Tx-WT mice and Tx-KO mice, respectively) (Fig. 1A). We assessed the extent of bone marrow reconstitution 17 weeks after transplantation by using real-time quantitative PCR (qPCR) to measure Grn and Ldlr mRNA levels in blood cells and livers of recipient mice. We confirmed that Grn mRNA was not expressed in blood cells (undetectable–0.02 relative units for Tx-KO mice versus 1.2 ± 0.6 for Tx-WT mice) of mice transplanted with Grn−/− bone marrow (Fig. 1B). In contrast, Grn mRNA expression was detected in the livers of Tx-KO mice, although it trended lower (0.6 ± 0.2 relative units versus 1.0 ± 0.3 for Tx-WT mice). As expected, Ldlr mRNA was markedly reduced in the livers of recipient mice (0.1 ± 0.1 for Tx-WT mice and 0.1 ± 0.1 relative units for Tx-KO mice, versus 1.0 ± 0.2 for WT control mice) (Fig. 1B). This detectable level of Ldlr mRNA was likely from Kupffer cells derived from transplanted marrow cells that express Ldlr. In contrast, Ldlr mRNA was readily detected in blood cells of recipients of both WT and Grn−/− bone marrow (0.9 ± 0.3 relative units for Tx-WT mice and 0.9 ± 0.6 for Tx-KO mice), indicating that hematopoietic cells in the Ldlr−/− recipient mice were successfully reconstituted.
Fig. 1. Bone marrow transplantation and successful reconstitution of immune cells in Ldlr−/− recipient mice.

(A) Experimental paradigm. (B) mRNA levels of progranulin (Grn) and LDL receptor (Ldlr) in blood cells and livers of Ldlr−/− recipient mice 17 weeks after transplantation with wild-type (Tx-WT) or Grn−/− bone marrow (Tx-KO). (C) Plasma progranulin levels are reduced in Tx-KO mice, as measured by ELISA at 17 weeks after bone marrow transplantation (including 10 weeks of western diet feeding). (D) Grn−/− bone marrow donor mice (chow-fed) lack plasma progranulin, as measured by ELISA. (E) Tx-KO mice have markedly reduced progranulin staining (red) in lesion macrophages (green). Scale bar, 200 μm. (F) Quantification of progranulin-positive macrophages. Values are mean ± SD (n=12 per group in C, n=3–4 in D, n=7 in F); *p<0.05, as determined by Mann-Whitney U test. BM, bone marrow.
After 7 weeks of recovery from transplantation, we fed both Tx-WT and Tx-KO mice a western diet (containing higher levels of fat, cholesterol, and sucrose) for 10 weeks and then examined the metabolic parameters and development of atherosclerosis. In Tx-KO mice, plasma progranulin levels were ~15% less than in Tx-WT mice (10.0 ± 1.9 μg/ml versus 13.0 ± 1.8 μg/ml) (Fig. 1C), indicating that hematopoietic cells contribute to circulating levels of progranulin under these conditions. Both transplanted groups had higher progranulin levels than chow-fed WT donor mice and chow-fed non-transplanted Ldlr−/− mice (Fig. 1D). This may be a result of the high-fat, western diet that increases progranulin expression in the liver and adipose tissue (24). To confirm progranulin deficiency in lesion macrophages, we co-stained aortic root sections with the macrophage marker CD68 (Fig. 1E and F). Progranulin was readily detected in the atherosclerotic lesions of Tx-WT mice, where it showed extensive co-localization with nearly all CD68-positive macrophages (99.2 ± 1.3%). In contrast, progranulin staining was markedly reduced in lesion macrophages of Tx-KO mice (4.1 ± 2.0%). These results indicate that Grn−/− macrophages in atherosclerotic lesions take up very little circulating progranulin.
After 10 weeks on the western diet, Tx-WT and Tx-KO mice developed similar degrees of hypercholesterolemia (818 ± 134 mg/dl for Tx-WT mice versus 881 ± 163 mg/dl for Tx-KO mice) and hypertriglyceridemia (155 ± 40 mg/dl for Tx-WT mice versus 138 ± 53 mg/dl for Tx-KO mice) (Fig. S1A and B). Additionally, the distribution of cholesterol across the different lipoprotein classes was similar for both groups (Fig. S2C). Metabolic parameters, such as body weight and fasting blood glucose, were also similar between the groups (Table S1). Body weights after 8 weeks on the western diet were similar to what was reported for similar transplantation studies of Ldlr−/− mice (31), and our study mice appeared to be healthy and eating normally. Plasma cytokines and blood leukocytes were largely similar between the groups, except TNFα was decreased in Tx-KO mice (Table S1). These findings indicate that loss of progranulin in hematopoietic cells did not significantly affect systemic energy, glucose, or lipid metabolism, or impact systemic total leukocyte counts.
We quantified atherosclerotic lesions by two methods. First, we assessed the size of atherosclerotic lesions in aortic roots by Oil Red O staining and found that Tx-KO mice had ~47% more mean lesion area (5.5 ± 1.2 × 105 μm2 versus 3.7 ± 0.6 × 105 μm2, P<0.001) (Fig. 2A and C). Second, we assessed lesions in whole aortas by en face analysis and similarly found a ~62% increase in lesions in Tx-KO mice (2.7 ± 0.9% versus 1.7 ± 1.2%, P<0.01) (Fig. 2B and D). The area of necrosis within the core of lesions, an indicator of advanced lesions, was greater and more variable in Tx-KO mice (2.0 ± 1.3 × 105 μm2 versus 1.0 ± 0.5 × 105 μm2) (Fig. 2E). We observed no differences in the number of apoptotic cells in lesions as determined by TUNEL staining (Fig. 2F). Additionally, the total number of CD68-positive cells was unchanged (311 ± 92 cells per field versus 272 ± 119 cells per field) within lesions (Fig. 2G), suggesting that macrophage recruitment to lesions was not affected by macrophage progranulin deficiency.
Fig. 2. Increased atherosclerosis in Ldlr−/− mice transplanted with progranulin-deficient bone marrow.

(A) Increased lesion area in Tx-KO mice, as measured by Oil Red O staining of lesions in aortic roots. Scale bars, 500 μm. (B) Increased lesion area in Tx-KO mice, as measured by en face analysis of aortic lesions. Arrows indicate lesions. Scale bars, 5 mm. (C–G) Quantification of lesion area in aortic roots (C) and aortas (D) and necrotic core area (E), TUNEL-positive cells (F), and CD68-positive cells (G) in aortic roots. Representative images are shown for each transplanted group (n=12 mice per group in C-E, n=6–8 in F–G). Values are mean ± SD; *p<0.01, **p<0.001, as determined by Mann-Whitney U test.
Lysosomal abnormalities in lesions of Tx-KO mice and in Grn−/− macrophages
Recent evidence linking complete progranulin deficiency to neuronal ceroid lipofuscinosis (20), a form of lysosomal storage disease, suggests that progranulin functions in the lysosome. Moreover, staining for the lysosomal markers LAMP1 and CD68 is increased in cultured Grn−/− microglia (6) and in the brains of Grn−/− mice (4,9–11). Therefore, we stained for the lysosomal markers LAMP1 in aortic root macrophages. In Tx-KO mice, we found a trend toward increased LAMP1 fluorescence although this did not reach significance (1.5 ± 0.8 relative units versus 1.0 ± 0.4, P=0.1246) (Fig. 3A and B).
Fig. 3. Lysosomal abnormalities in lesions of Tx-KO mice and in Grn−/− macrophages.

(A–B) Trend toward increased lysosome staining in lesions of Ldlr−/− mice transplanted with progranulin-deficient bone marrow. (A) Staining of LAMP1 in aortic root lesions. Scale bar, 10 μm. (B) Quantification of cellular LAMP1 fluorescence. Values are normalized to Tx-WT levels and presented as mean ± SD (n=9 mice per group); p=0.1246, as determined by Mann-Whitney U test. (C–E) Lysosomal abnormalities in Grn−/− macrophages. (C) Increased lysosomal content in Grn−/− macrophages, as measured by LAMP1 staining. Staining of LAMP1 (red), progranulin (green), and nuclei with Hoechst (blue) in WT and Grn−/− BMDMs. Scale bar, 50 μm. (D) Quantification of cellular LAMP1 fluorescence. (E) Increased expression of TFEB target genes in Grn−/− BMDMs. Values are mean ± SD; *p<0.05, **p<0.01, ****p<0.0001, as determined by student’s t test. n.s., not significant.
We also examined the effects of progranulin deficiency on lysosomes in cultured macrophages by staining bone marrow–derived macrophages (BMDMs) from WT and Grn−/− mice with antibodies against LAMP1 and found increased LAMP1 fluorescence in Grn−/− macrophages (Fig. 3D and E). Because LAMP1 expression is regulated by the transcription factor TFEB (32), a master regulator of lysosome biology, we measured expression of additional TFEB target genes. In Grn−/− BMDMs, we observed increased expression of several additional TFEB target genes (Fig. 3F), including cathepsins (CtsD, CtsZ), hexosaminidase A (HexA), and glucocerebrosidase (Gba), which encode enzymes that degrade proteins, GM2 gangliosides, and glycolipid intermediates, respectively. These findings are consistent with previous reports of increased levels of these and other TFEB target genes in the brains of Grn−/− mice (4,9,10) and in progranulin-deficient fibroblasts (29), and suggest that progranulin-deficient macrophages increase their LAMP1 expression in lysosomes, perhaps to compensate for impairment of a lysosomal function.
Progranulin deficiency increases foam cell formation in response to aggregated LDL in cultured macrophages
We next explored the mechanism underlying the accelerated atherosclerosis in Tx-KO mice by investigating atherosclerosis-relevant pathways in progranulin-deficient macrophages. Since altered lipid accumulation could promote atherosclerosis in the Tx-KO mice, we first assessed lipid handling by Grn−/− macrophages. We treated BMDMs with acetylated LDL (acLDL), oxidized LDL (oxLDL), or oleic acid and observed no differences in the amount of lipid stored in WT and progranulin-deficient macrophages, either by staining with BODIPY or by analyzing cellular lipid content with thin-layer chromatography (Fig. 4A and Fig. S2). To determine if the acLDL was trapped in lysosomes of progranulin-deficient macrophages due to compromised lysosomal function, we stained cells with both LysoTracker and LipidTox, a fluorescent dye for neutral lipids. This staining revealed no co-localization, indicating that neutral lipids did not accumulate in lysosomes of acLDL-treated Grn−/− macrophages (Fig. S2B).
Fig. 4. Progranulin deficiency enhances agLDL-induced foam cell formation in cultured macrophages.

(A) Similar neutral lipid levels in WT and Grn−/− BMDMs after treatment with oxLDL (25 μg/ml), acLDL (50 μg/ml), or oleic acid (500 μM) for 20 h. Lipids were analyzed by thin layer chromatography. (B) Increased neutral lipid staining (LipidTOX) in Grn−/− BMDMs treated with agLDL (250 μg/ml) for 12 h. Scale bar, 20 μm. (C) Quantification of LipidTOX fluorescence per field. Values are mean ± SD (n≥30 fields per condition). ***p<0.001, as determined by student’s t test. Sal, saline; oxLDL, oxidized LDL; acLDL, acetylated LDL; OA, oleic acid; CE, cholesterol ester; TG, triglyceride; CHOL, free cholesterol; n.s., not significant.
Recent evidence suggests that cholesterol uptake by exophagy, a lysosome-related cellular uptake pathway, may be an important trigger of macrophage foam cell formation (33,34). Exophagy refers to a process in which large macromolecular substrates (such as aggregated LDL (agLDL) and dead/dying adipocytes) are degraded in a plasma membrane-bound extracellular compartment by extruded lysosomal contents (35–38). The degraded proteins and lipids are subsequently taken up into cells via endocytosis. In vitro studies show that uptake and storage of agLDL via exophagy leads to the formation of macrophage foam cells (33), a key event in development of atherosclerosis. We assessed the exophagy pathway in WT and Grn−/− BMDMs treated with agLDL (250 μg/ml). As a control, we treated cells with acLDL (50 μg/ml), which macrophages take up through scavenger receptors (39). After 12 h, we stained cells with LipidTOX to assess lipid loading. With agLDL treatment, Grn−/− BMDMs had ~2.7-fold greater LipidTOX fluorescence than WT BMDMs (Fig. 4B and C), consistent with increased catabolism, uptake, and storage of the cholesterol derived from agLDL in lipid droplets. In contrast, no difference in LipidTOX fluorescence was observed in WT and Grn−/− BMDMs treated with acLDL. The exophagy-mediated uptake of agLDL did not trigger an increased inflammatory response in either WT or Grn−/− BMDMs (Fig. S3), after a 7-h treatment with 250 μg/ml agLDL. In contrast, treatment of BMDMs with acLDL resulted in a robust pro-inflammatory response, as previously reported (40,41), and this response was increased in some measurements (MCP-1) in Grn−/− BMDMs. These data demonstrate that Grn−/− macrophages have increased foam cell formation when treated with agLDL, but not when treated with other modified LDL species (namely, acLDL or oxLDL).
Progranulin-deficient macrophages exhibit increased exophagy of agLDL
To determine whether progranulin-deficient macrophages exhibit increased exophagy, we measured lysosome-associated exocytosis after agLDL treatment. We utilized an established assay (34,36) that specifically detects labeled lysosomal content that is exocytosed from cells and contacts extracellular labeled agLDL. In this assay, lysosomes were first labeled by incubating BMDMs overnight with biotin-fluorescein-dextran. The extent of dextran loading was marginally decreased (by 5.5%) in Grn−/− BMDMs (Fig. S4). To assess exophagy, we then measured the amount of dextran that bound exogenously provided AlexaFluor (Alexa)546-labeled agLDL outside the cell during a 90-min incubation. Specifically, we determined the dextran-fluorescein intensity co-localizing in the extracellular compartment formed around Alexa546-labeled agLDL (conjugated with streptavidin to capture secreted biotin-fluorescein-dextran). Using this assay, we found that exophagy was markedly increased (~2.5-fold) in Grn−/− BMDMs compared with WT BMDMs (Fig. 5A). We also assessed the amount of actin polymerization associated with sites of exophagy via staining with Alexa488-phalloidin (35) and found this was similar in WT and Grn−/− BMDMs (Fig. 5B), suggesting that this process is not impacted by progranulin deficiency. Together these results suggest a model in which macrophage progranulin deficiency enhances exophagy of agLDL, resulting in increased foam cell formation and accelerated atherosclerotic lesion development.
Fig. 5. Progranulin deficiency enhances lysosome exocytosis during exophagy of agLDL.

(A) Increased lysosome exocytosis during exophagy of agLDL in Grn−/− BMDMs. Arrows indicate regions of macrophage lysosome exocytosis to agLDL during exophagy. Quantification of biotin-fluorescein-dextran fluorescence co-localized with streptavidin-Alexa546-agLDL per field (right). (B) Actin polymerization during exophagy of agLDL was similar in WT and Grn−/− macrophages. Arrows indicate regions of macrophage actin polymerization during exophagy. Quantification of Alexa488-phalloidin fluorescence co-localized with Alexa546-agLDL per field (right). Values are mean ± SD. ***p<0.001 student’s t test. n.s., not significant. Scale bar, 20 μm.
Discussion
In the current study, we show that progranulin deficiency in the hematopoietic compartment significantly accelerates atherosclerosis in a murine model. This worsening occurs despite high circulating levels of progranulin and is not caused by changes in circulating cholesterol or triglyceride levels. Our in vitro studies revealed that progranulin-deficient macrophages exhibit increased foam cell formation via exophagy-mediated catabolism, uptake, and storage of agLDL-derived cholesterol. Together, our results indicate that immune cell–derived progranulin plays a key role in modulating the progression of atherosclerotic lesions, apparently via a cell-autonomous or local manner.
A previous study in Apoe−/− mice showed that global progranulin deficiency worsens atherosclerosis (23). Our study corroborates this study in a different model (Ldlr−/−) and, additionally, shows that deficiency of progranulin in hematopoietic cells alone is sufficient to cause this disease phenotype. Importantly, the levels of circulating progranulin were still relatively high in the Tx-KO mice, but this source of progranulin was not found in lesions and was unable to suppress the accelerated atherosclerosis. These findings are consistent with an emerging model for progranulin in which the major effects of the protein are cell-autonomous effects in the lysosome. Supporting this, we also found increases in exophagy, a potential atherosclerosis-modulating process in cells lacking progranulin. Taken together, our findings argue strongly that progranulin deficiency promotes atherosclerosis by altering macrophage biology. This model could be tested further in future studies by examining whether overexpressing progranulin in macrophages protects against lysosomal dysfunction, foam cell formation, and atherosclerosis, although it is not clear whether normal levels of progranulin are limiting.
Our findings are consistent with an emerging model in which progranulin functions primarily in lysosomes. Although progranulin’s molecular function is unclear, it localizes to lysosomes (4–6), and its trafficking to this organelle involves sortilin (42) and prosaposin (5). Homozygous deficiency of progranulin in humans causes neural ceroid lipofuscinosis, a severe neurodegenerative disease of multiple etiologies that is linked to lysosomal dysfunction (20). Previous studies have shown that progranulin-deficient cells and tissues have increased lysosomal content, based on staining of the lysosomal proteins LAMP1 (4,6,9,10) and CD68 (11). We similarly found increased lysosomal content in atherosclerotic lesions of mice lacking macrophage progranulin (Fig. 3), as well as in cultured Grn−/− macrophages (Fig. 4). Expanding on these observations, we also show that Grn−/− macrophages exhibit increased exophagy, a lysosome-related cellular uptake pathway.
To our knowledge, progranulin deficiency is the first known condition that results in increased exophagy, demonstrated here both in vitro and, in correlation, by enhanced disease pathogenesis in vivo. How progranulin deficiency increases exophagy in macrophages is unclear. One possibility is that exophagy is enhanced as a result of lysosomal alterations in Grn–/– macrophages. While lysosomal content as measured by dextran loading was not increased, LAMP1 staining was increased in Grn−/− macrophages, possibly consistent with lysosomal activation since LAMP1 is a TFEB target gene (32). Supporting this concept, Grn−/− BMDMs had increased expression of multiple TFEB target genes. Alternatively, progranulin deficiency could alter signaling pathways activating Rho family GTPases (Rac1 and Cdc42) to promote actin assembly in a way that primes macrophages for enhanced exophagy (35,43). However, since we did not observe changes in actin assembly, it is less likely that signaling to the exophagy machinery is altered. Given that hydrolysis of the cholesteryl esters in agLDL by lysosomal acid lipase is required for growth of the lysosomal synapse (35,44), increased cholesterol ester hydrolysis may promote growth of lysosomal synapses in Grn−/− BMDMs. Moreover, our studies suggest that progranulin deficiency specifically increases cholesterol uptake via the exophagy pathway, as progranulin deficiency did not affect uptake of acLDL through receptor-mediated endocytosis (Fig. 4 and Fig. S2).
Several studies support the model that progranulin deficiency causes increased exophagy. In bone homeostasis, osteoclasts play a key role by resorbing bone through a process very similar to exophagy (45–47). Grn−/− mice exhibit decreased bone mass (19) and exaggerated bone resorption by osteoclasts (48), effects that may be the result of increased exophagy of bone by myeloid–derived Grn−/− osteoclasts. In the adipose tissue, macrophages take up dead and/or dying adipocytes through exophagy (38). It is not known if progranulin deficiency affects this process, but Grn−/− macrophages may increase clearance of apoptotic thymocytes in vitro (49), suggesting a possible broader role for exophagy in clearance of dead and/or dying cells. In the brain, synaptic pruning is carried out by microglia (50), which are myeloid–derived resident macrophages. Because synapses are relatively large, roughly on the order of several microns in diameter, we hypothesize that microglia use the exophagy pathway for synaptic pruning and that this process is enhanced in Grn−/− mice. In support of this idea, Grn−/− mice were recently reported to have increased synaptic pruning in the ventral thalamus (11). Thus, our findings of increased exophagy might be a central mechanism uniting a number of pathological phenotypes found in progranulin deficiency.
Supplementary Material
Fig. S1. Similar levels of plasma lipids in Ldlr−/− mice transplanted with WT or Grn−/− bone marrow. (A–B) Plasma cholesterol (A) and triglyceride (B) levels in Tx-WT and Tx-KO mice after 10 weeks of western diet feeding (n=12–13 mice per group). (C) Cholesterol distribution in plasma lipoproteins separated by fast protein liquid chromatography (n=3 pools of 4 mice each). LDL, low-density lipoprotein; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; VLDL, very low-density lipoprotein. Values are mean ± SD.
Fig. S2. Grn−/− macrophages take up and store acLDL and oxLDL normally in vitro. (A) Similar neutral lipid levels in WT and Grn−/− BMDMs after treatment with 25 μg/ml oxLDL, 50 μg/ml acLDL, or 500 μM oleic acid for 14 h. Lipids were analyzed by imaging with the neutral lipid dye BODIPY. (B) Double-staining with LysoTracker (green) and LipidTOX (red, neutral lipids) indicates no accumulation of neutral lipids in lysosomes of Grn−/− macrophages. Scale bar, 5 μm. acLDL, acetylated LDL.
Fig. S3. agLDL does not trigger a pro-inflammatory response in cultured macrophages. WT and Grn−/− BMDMs were treated with agLDL (250 μg/ml) or acLDL (100 μg/ml) for 7 h, and gene expression was determined by qPCR. Values are mean ± SD. ****p<0.0001, as determined by two-way ANOVA with Tukey post hoc test. n.s., not significant.
Fig. S4. Lysosome loading of biotin-fluorescein-dextran is marginally decreased in Grn−/− macrophages. WT and Grn−/− BMDMs were incubated in media containing biotin-fluorescein-dextran (1 mg/ml) overnight, followed by a 2-h chase. Lysosome loading of biotin-fluorescein-dextran was assessed by confocal microscopy, and the fluorescence per field was quantified (right). Values are mean ± SD (n≥30 fields per genotype). ***p<0.001, as determined by student’s t test. Scale bar, 20 μm.
HIGHLIGHTS.
Progranulin deficiency in the hematopoietic compartment results in accelerated atherosclerosis in Ldlr−/− mice
Progranulin deficiency in macrophages results in increased exophagy-mediated uptake of aggregated low-density lipoprotein (LDL) and foam cell formation
Progranulin is a modulator of exophagy, a lysosome-related cellular uptake pathway
Acknowledgments
We thank Robert Mahley and Laura Mitic for helpful discussions, Marina Vayner and Grisell Diaz-Ramirez for mouse husbandry, Caroline Miller and Kathryn Bummer for histology, and Gary Howard for editorial assistance.
Financial support
This work was supported by grants from the NIH (P50 AG023501 to RVF and R01 HL093324 to FRM), the Consortium for Frontotemporal Dementia Research (to RVF, TCW, EJH), and a VA Merit Award (BX002978 to EJH), and by postdoctoral fellowships from the NIH (F32 HL116197, supporting ADN), the American Diabetes Association (supporting TAN), and the American Heart Association (supporting RKS). TCW is an investigator of the Howard Hughes Medical Institute. Initial studies were carried out at the Gladstone Institutes, which received support for animal care from the National Center for Research Resources (RR18928).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
References
- 1.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, and Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 [DOI] [PubMed] [Google Scholar]
- 2.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, and Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 [DOI] [PubMed] [Google Scholar]
- 3.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, and Rademakers R (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet 15, 2988–3001 [DOI] [PubMed] [Google Scholar]
- 4.Tanaka Y, Matsuwaki T, Yamanouchi K, and Nishihara M (2013) Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 250, 8–19 [DOI] [PubMed] [Google Scholar]
- 5.Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown W, Smolka M, Sun Y, and Hu F (2015) Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol 210, 991–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka Y, Suzuki G, Matsuwaki T, Hosokawa M, Serrano G, Beach TG, Yamanouchi K, Hasegawa M, and Nishihara M (2017) Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum. Mol. Genet 26, 969–988 [DOI] [PubMed] [Google Scholar]
- 7.Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, and Rademakers R (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Riz M, Galimberti D, Fenoglio C, Piccio LM, Scalabrini D, Venturelli E, Pietroboni A, Piola M, Naismith RT, Parks BJ, Fumagalli G, Bresolin N, Cross AH, and Scarpini E (2010) Cerebrospinal fluid progranulin levels in patients with different multiple sclerosis subtypes. Neurosci. Lett 469, 234–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, Martin JJ, Engelborghs S, Kretzschmar HA, Arzberger T, Van Broeckhoven C, Haass C, and Capell A (2014) Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 127, 845–860 [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, and Nishihara M (2014) Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun 2, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lui H, Zhang J, Makinson S, Cahill M, Kelley K, Huang H, Shang Y, Oldham M, Martens L, Gao F, Coppola G, Sloan S, Hsieh C, Kim C, Bigio E, Weintraub S, Mesulam M, Rademakers R, Mackenzie I, Seeley W, Karydas A, Miller B, Borroni B, Ghidoni R, Farese R Jr, Paz J, Barres B, and Huang E, (2016) Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165, 921–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, and Lewis J (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol 177, 311–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, Jiao J, Zhang Z, Huang EJ, Gao FB, Farese RV Jr., and Roberson ED (2013) Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J. Neurosci 33, 5352–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, Gelfand JM, Cruz-Herranz A, Cordano C, Alavi MV, Leslie S, Seeley WW, Miller BL, Bigio E, Mesulam MM, Bogyo MS, Mackenzie IR, Staropoli JF, Cotman SL, Huang EJ, Gan L, and Green AJ (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med 9, eaah5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, and Ding A (2010) Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 24, 4639–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, Gregg J, Lu G, Feldman HH, Mackenzie IR, Raymond LA, and Leavitt BR (2012) Synaptic dysfunction in progranulin-deficient mice. Neurobiol. Dis 45, 711–722 [DOI] [PubMed] [Google Scholar]
- 17.Arrant A, Filiano A, Warmus B, Hall A, and Roberson E (2016) Progranulin haploinsufficiency causes biphasic social dominance abnormalities in the tube test. Genes Brain Behav. 15, 588–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, and Ding A (2010) Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med 207, 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi T, Ebina K, Hirao M, Kawase R, Ohama T, Yamashita S, Morimoto T, Koizumi K, Kitaguchi K, Matsuoka H, Kaneshiro S, and Yoshikawa H (2015) Progranulin plays crucial roles in preserving bone mass by inhibiting TNF-α-induced osteoclastogenesis and promoting osteoblastic differentiation in mice. Biochem. Biophys. Res. Commun 465, 638–643 [DOI] [PubMed] [Google Scholar]
- 20.Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, and Berkovic SF (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet 90, 1102–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almeida M, Macario M, Ramos L, Baldeiras I, Ribeiro M, and Santana I (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol. Aging 41, 200 e201–205 [DOI] [PubMed] [Google Scholar]
- 22.Kojima Y, Ono K, Inoue K, Takagi Y, Kikuta K, Nishimura M, Yoshida Y, Nakashima Y, Matsumae H, Furukawa Y, Mikuni N, Nobuyoshi M, Kimura T, Kita T, and Tanaka M (2009) Progranulin expression in advanced human atherosclerotic plaque. Atherosclerosis 206, 102–108 [DOI] [PubMed] [Google Scholar]
- 23.Kawase R, Ohama T, Matsuyama A, Matsuwaki T, Okada T, Yamashita T, Yuasa-Kawase M, Nakaoka H, Nakatani K, Inagaki M, Tsubakio-Yamamoto K, Masuda D, Nakagawa-Toyama Y, Nishida M, Ohmoto Y, Nishihara M, Komuro I, and Yamashita S (2013) Deletion of progranulin exacerbates atherosclerosis in ApoE knockout mice. Cardiovasc. Res 100, 125–133 [DOI] [PubMed] [Google Scholar]
- 24.Matsubara T, Mita A, Minami K, Hosooka T, Kitazawa S, Takahashi K, Tamori Y, Yokoi N, Watanabe M, Matsuo E, Nishimura O, and Seino S (2012) PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab 15, 38–50 [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez EM, Mongiat M, Slater SJ, Baffa R, and Iozzo RV (2003) A novel interaction between perlecan protein core and progranulin: potential effects on tumor growth. J. Biol. Chem 278, 38113–38116 [DOI] [PubMed] [Google Scholar]
- 26.Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, and Farese RV Jr. (2012) Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest 122, 3955–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Chang J, Deng Q, Xu J, Nguyen TA, Martens LH, Cenik B, Taylor G, Hudson KF, Chung J, Yu K, Yu P, Herz J, Farese RV Jr., Kukar T, and Tansey MG (2013) Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci 33, 9202–9213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, and Herz J (1993) Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest 92, 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen AD, Nguyen TA, Zhang J, Devireddy S, Zhou P, Xu X, Karydas AM, Miller BL, Rigo F, Ferguson SM, Huang EJ, Walther TC, and Farese RV Jr.. (2018) Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc. Natl. Acad. Sci. U. S. A doi: 10.1073/pnas.1722344115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eberle D, Kim RY, Luk FS, de Mochel NS, Gaudreault N, Olivas VR, Kumar N, Posada JM, Birkeland AC, Rapp JH, and Raffai RL (2012) Apolipoprotein E4 domain interaction accelerates diet-induced atherosclerosis in hypomorphic Arg-61 apoe mice. Arterioscler. Thromb. Vasc. Biol 32, 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiller N, Kubo N, Boisvert W, and Curtiss L (2001) Effect of γ-irradiation and bone marrow transplantation on atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol 21, 1674–1680 [DOI] [PubMed] [Google Scholar]
- 32.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, and Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- 33.Haka AS, Grosheva I, Singh RK, and Maxfield FR (2013) Plasmin promotes foam cell formation by increasing macrophage catabolism of aggregated low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol 33, 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haka AS, Singh RK, Grosheva I, Hoffner H, Capetillo-Zarate E, Chin HF, Anandasabapathy N, and Maxfield FR (2015) Monocyte-derived dendritic cells upregulate extracellular catabolism of aggregated low-density lipoprotein on maturation, leading to foam cell formation. Arterioscler. Thromb. Vasc. Biol 35, 2092–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grosheva I, Haka AS, Qin C, Pierini LM, and Maxfield FR (2009) Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arteriosclerosis, Thrombosis & Vascular Biology 29, 1615–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, and Maxfield FR (2009) Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol. Biol. Cell 20, 4932–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh RK, Barbosa-Lorenzi VC, Lund FW, Grosheva I, Maxfield FR, and Haka AS (2016) Degradation of aggregated LDL occurs in complex extracellular sub-compartments of the lysosomal synapse. J. Cell Sci 129, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haka AS, Barbosa-Lorenzi VC, Lee HJ, Falcone DJ, Hudis CA, Dannenberg AJ, and Maxfield FR (2016) Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J. Lipid Res 57, 980–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown MS, and Goldstein JL (1983) Lipoprotein Metabolism in the Macrophage: Implications for Cholesterol Deposition in Atherosclerosis. Annu. Rev. Biochem 52, 223–261 [DOI] [PubMed] [Google Scholar]
- 40.Wang N, Tabas I, Winchester R, Ravalli S, Rabbani LE, and Tall A (1996) Interleukin 8 is induced by cholesterol loading of macrophages and expressed by macrophage foam cells in human atheroma. J. Biol. Chem 271, 8837–8842 [DOI] [PubMed] [Google Scholar]
- 41.Kim TW, Febbraio M, Robinet P, Dugar B, Greene D, Cerny A, Latz E, Gilmour R, Staschke K, Chisolm G, Fox PL, DiCorleto PE, Smith JD, and Li X (2011) The critical role of IL-1 receptor-associated kinase 4-mediated NF-κB activation in modified low-density lipoprotein-induced inflammatory gene expression and atherosclerosis. J. Immunol 186, 2871–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, and Strittmatter SM (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakr S, Eddy R, Barth H, Wang F, Greenberg S, Maxfield F, and Tabas I (2001) The uptake and degradation of matrix-bound lipoproteins by macrophages require an intact actin cytoskeleton, Rho family GTPases, and myosin ATPase activity. J. Biol. Chem 276, 37649–37658 [DOI] [PubMed] [Google Scholar]
- 44.Buton X, Mamdouh Z, Ghosh R, Du H, Kuriakose G, Beatini N, Grabowski GA, Maxfield FR, and Tabas I (1999) Unique cellular events occurring during the initial interaction of macrophages with matrix-retained or methylated aggregated low density lipoprotein (LDL). Prolonged cell-surface contact during which LDL-cholesteryl ester hydrolysis exceeds LDL protein degradation. J. Biol. Chem 274, 32112–32121 [DOI] [PubMed] [Google Scholar]
- 45.Stenbeck G (2002) Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol 13, 285–292 [DOI] [PubMed] [Google Scholar]
- 46.Jurdic P, Saltel F, Chabadel A, and Destaing O (2006) Podosome and sealing zone: specificity of the osteoclast model. Eur. J. Cell Biol 85, 195–202 [DOI] [PubMed] [Google Scholar]
- 47.Baron R, Neff L, Louvard D, and Courtoy PJ (1985) Cell-mediated extracellular acidification and bone resorption: evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J. Cell Biol 101, 2210–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao YP, Tian QY, Liu B, Cuellar J, Richbourgh B, Jia TH, and Liu CJ (2015) Progranulin knockout accelerates intervertebral disc degeneration in aging mice. Sci. Rep 5, 9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kao AW, Eisenhut RJ, Martens LH, Nakamura A, Huang A, Bagley JA, Zhou P, de Luis A, Neukomm LJ, Cabello J, Farese RV Jr, and Kenyon C (2011) A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U. S. A 108, 4441–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong S, Dissing-Olesen L, and Stevens B (2016) New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol 36, 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Similar levels of plasma lipids in Ldlr−/− mice transplanted with WT or Grn−/− bone marrow. (A–B) Plasma cholesterol (A) and triglyceride (B) levels in Tx-WT and Tx-KO mice after 10 weeks of western diet feeding (n=12–13 mice per group). (C) Cholesterol distribution in plasma lipoproteins separated by fast protein liquid chromatography (n=3 pools of 4 mice each). LDL, low-density lipoprotein; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; VLDL, very low-density lipoprotein. Values are mean ± SD.
Fig. S2. Grn−/− macrophages take up and store acLDL and oxLDL normally in vitro. (A) Similar neutral lipid levels in WT and Grn−/− BMDMs after treatment with 25 μg/ml oxLDL, 50 μg/ml acLDL, or 500 μM oleic acid for 14 h. Lipids were analyzed by imaging with the neutral lipid dye BODIPY. (B) Double-staining with LysoTracker (green) and LipidTOX (red, neutral lipids) indicates no accumulation of neutral lipids in lysosomes of Grn−/− macrophages. Scale bar, 5 μm. acLDL, acetylated LDL.
Fig. S3. agLDL does not trigger a pro-inflammatory response in cultured macrophages. WT and Grn−/− BMDMs were treated with agLDL (250 μg/ml) or acLDL (100 μg/ml) for 7 h, and gene expression was determined by qPCR. Values are mean ± SD. ****p<0.0001, as determined by two-way ANOVA with Tukey post hoc test. n.s., not significant.
Fig. S4. Lysosome loading of biotin-fluorescein-dextran is marginally decreased in Grn−/− macrophages. WT and Grn−/− BMDMs were incubated in media containing biotin-fluorescein-dextran (1 mg/ml) overnight, followed by a 2-h chase. Lysosome loading of biotin-fluorescein-dextran was assessed by confocal microscopy, and the fluorescence per field was quantified (right). Values are mean ± SD (n≥30 fields per genotype). ***p<0.001, as determined by student’s t test. Scale bar, 20 μm.