

Abstract
An organized series of complicated biological and molecular phenomena is required for normal skin wound healing. These processes depend on normal cellular responses to cytokines, growth factors, and other mediators, such as clotting factors, prostaglandins, free radicals, and nitric oxide. In diabetic ulcers, impaired responses to these molecules lead to abnormalities in vascularization, innervation, matrix reconstruction, and reepithelialization of wounds. keratinocyte migration and proliferation on an extracellular matrix is critical in reepithelialization, but the response to growth factors is blunted in diabetes, including the insulin/IGF–1 signaling axis. Ganglioside GM3, a sialylated epidermal glycosphingolipid, has been identified as a key mediator of the inhibition of insulin/IGF–1 signaling in response to factors, such as tumor necrosis factor-alpha (TNF–α) and hyperglycemia. Decreased expression of GM3 and the enzyme required for its synthesis, GM3 synthase (GM3S), leads to increased insulin/IGF-1 receptor signaling and accelerated keratinocyte migration, even in the presence of high glucose levels. GM3 depletion in GM3S knockout diabetic mice and diet-induced diabetic mice treated topically with nanoconstruct-mediated GM3S-targeting gene regulation also accelerates wound healing. These recent observations, coupled with evidence that GM3 depletion reverses distal innervation abnormalities in diabetic mice, suggest that GM3-depleting strategies are a promising new approach for human diabetic wounds.
1. INTRODUCTION
Disruption of the normal orderly progression of events in wound healing has been observed in wounds with preexisting pathophysiological abnormalities, such as diabetic ulcers.1,2 This impaired healing has been attributed to several intrinsic factors (e.g., neuropathy, vascular problems leading to ischemia, and signaling abnormalities in several wound cell types), as well as extrinsic factors (e.g., wound infection and trauma).3,4 Macrovascular5 and microvascular defects, which often occur in patients with diabetes, result in capillary size reduction and basement membrane thickening, limiting blood flow, and migration of dermal and epidermal cells into wounds.6,7 The slowed microcirculation is further exacerbated by denervation,8,9 with sensory nerve abnormalities that limit the patient’s intrinsic protection from external trauma3,4,10–12 and increase the risk of trauma-initiated ulceration.
2. GM3 MEDIATES INSULIN RESISTANCE IN DIABETES
Growing evidence suggests that GM3 mediates tumor necrosis factor-alpha (TNF–α) and glucose-induced development of insulin resistance and type 2 diabetes. Gangliosides are sialylated glycosphingolipids that mediate membrane-based growth factor receptor signaling in skin and other organs.13–15 Monosialylatcd GM3, a product of lactosylceramide by GM3 synthase (GM3S), is the most abundant ganglioside in skin and precursor for more complex gangliosides. Levels of GM3 and GM3S (also called SAT-I/ST3Gal-V and coded by the ST3GAL5 gene) are increased in the kidneys, liver, adipose tissue, and muscle of murine models of diabetes.16–18 Ganglioside levels in the serum of diabetic patients with microvascular complications are alsoincreased,19 and GM3S expression is higher in the kidneys of diabetics with nephropathy.20 Knocking out GM3S in mice21 and treating diabetic mice or rats with a glucosylceramide synthase inhibitor, which depletes GM3 precursors, improves insulin sensitivity,22–24 ameliorates hepatic steatosis,23 and suppresses the development of diabetic renal hypertrophy.17
3. GM3 IS A DRIVER OF IMPAIRED WOUND HEALING IN TYPE 2 DIABETIC MICE
The impaired wound healing of type 2 diabetes is associated with hyperglycemia and chronic, low-grade elevation of TNF-α, which is known to drive insulin resistance. We found that the expression of GM3S and GM3 is increased threefold in diet-induced obese (DIO) and genetic (ob/ob) diabetic mouse skin,14 and that GM3S and GM3 are three-and >twofold higher, respectively, in human diabetic plantar skin than in age- and site-matched controls.25 These observations suggested the possibility that regional increases in GM3S/GM3 might contribute to poor wound healing in diabetes. Indeed, DIO GM3S knockout (GM3S–/–) diabetic mice have normal wound healing, in contrast to the delayed healing of their DIO wild-type (WT) diabetic littermates,14 despite comparable obesity and often persistent abnormal glucose tolerance in the GM3S–/– mice (Fig. 1A–C). Furthermore, wound healing in DIO diabetic mice is normalized by nanoconstruct-mediated, skin-specific suppression of GM3S expression, and reduction of GM3 levels in skin.25 This regional in vivo inhibition of GM3S is achieved by the topical application to the wound area of siRNA spherical nucleic acids (SNA), in which the GM3S-targeting siRNA are densely arrayed on a gold nanoparticle surface, facilitating penetration of epidermis at the wound edge without adverse effects (Fig. 1D–F). In both GM3S–/– and GM3S SNA-treated diabetic mice, GM3 depletion reverses the suppression of insulin growth factor 1 receptor (IGF1R) phosphorylation in epidermis at the wound border,14,25 suggesting that reversing the suppressed IGF1R activation in diabetic skin is associated with improved healing.
Fig. 1.
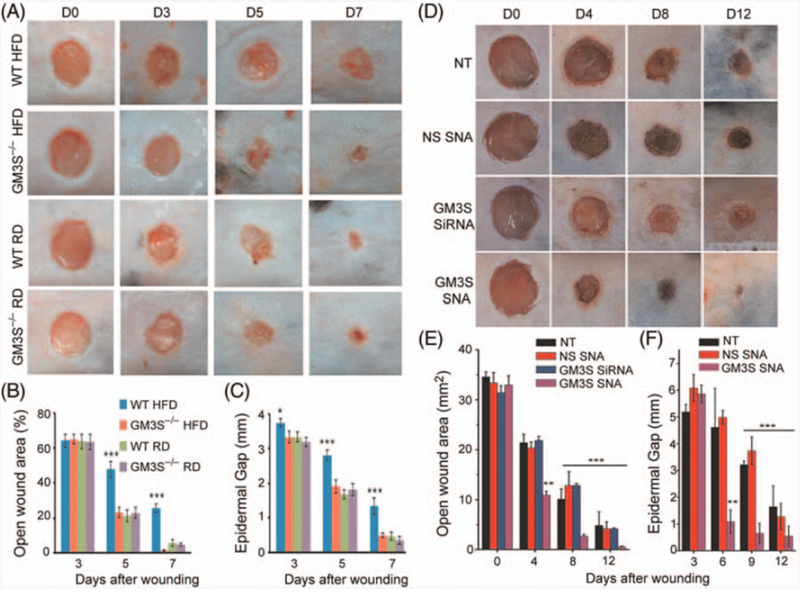
Accelerated wound healing in diabetic mice through depletion of GM3. (A–C) Complete deletion of GM3S (St3gal5) improved wound healing in diet-induced obese (DIO) GM3S knockout (GM3S–/–) diabetic mice, in contrast to the delayed healing of their DIO wild-type (WT) diabetic littermates.14 (D–F) Wound healing in DIO diabetic mice is normalized by nanoconstruct-mediated, skin-specific suppression of GM3S expression, and reduction of GM3 levels in skin through topical application. HFD, High fat diet; NS, nonsense; NT, nontreated; RD, regular diet; SNA, spherical nucleic acid. Part (A–C), reprinted with permission from Elsevier; Part (D–F), reprinted from Randeria PS, Seeger MA, Wang XQ, Wilson H, Shipp D, Mirkin CA, Paller AS. siRNA-based spherical nucleic acids reverse impaired wound healing in diabetic mice by ganglioside GM3 synthase knockdown. Proc Natl Acad Sci USA. 2015;112:5573–5578,25 with permission.
Furthermore, diabetic mouse models with GM3 depletion display improvement in wound healing parameters beyond the keratinocytes (KC), suggesting a multitissue impact of GM3 depletion. An impact on fibroblasts and the vasculature is suggested by the increase in granulation tissue and vasculature in GM3-depleted wounds.25 The small fiber neuropathy in the mouse footpad is thought to be responsible for neuropathic pain in diabetic mouse models and is representative of the neuropathy associated with acral impaired wound healing in humans.26 Depletion of GM3S and resultant decrease in GM3 expression rescued the denervation in mouse footpad skin, promoted sprouting of small fiber nerves into the epidermal layer, and fully ceased neuropathic nerve pain in DIO mice with biochemical confirmation of diabetes. Reduction of GM3 in dorsal root ganglia derived from diabetic mice also prevented hyperexcitability of calcium influx or calcium toxicity, which contributes to small fiber degeneration in diabetes.26 These observations suggest a more global role for regional GM3 depletion in accelerating diabetic wound healing and deserve further investigation.
4. THE INSULIN/IGF1 SIGNALING AXIS IN DIABETES AND DIABETIC WOUNDS
Insulin resistance is a cardinal feature of type 2 diabetes. Insulin and insulin-like growth factor (IGF–1) bind to both the insulin receptor (IR) and IGF–1 receptor (IGF1R) and trigger activation of the IGF–l/insulin signaling axis (with insulin the major ligand for IR and IGF–1 for IGF1R).27,28 Impaired IGF–l–/insulin-induced signaling impairs endothelial repair in type 2 diabetes-related atherosclerosis29 and diabetic retinopathy.30 Activation of IGF1R by IGF–1 stimulates hypoxia-inducible factor 1a protein synthesis and enhances the expression of vascular endothelial growth factor (VEGF) in human retinal pigment epithelial cells.30,31 This induction of VEGF by IGF–1 plays a critical role during the development of proliferative diabetic retinopathy. IGF–1 also stabilizes nascent blood vessels by ERK activation.32 IGF–1 also improves neuromuscular recovery and nerve regeneration after nerve injury by acting primarily on the axon, Schwann cells, and indirectly on neuromuscular junctions.33 Furthermore, IGF–1 acts during development of the peripheral nervous system, while promoting axon regeneration of motor and sensory axons in a nerve crush model.34
Importantly, IGF–1 and, to a lesser extent, insulin are growth factors that stimulate KC migration through activation primarily of IGF1R and IR, respectively, during wound closure.27,35 Indeed, studies in IR– and IGFIR-null mice have shown that deletion of IGF1R profoundly suppresses KC migration and proliferation, whereas deletion of IR has little effect.36,37 IGF–1 stimulates lamellipodial protrusion and cell spreading,28 and promotes KC migration through activation of the phosphatidylinositol-3-kinase and Rac1 signaling pathways.28,38 Decreased IGF1R phosphorylation has been described at the wound edge epidermis of DIO diabetic mice with impaired healing, suggesting the role of suppressed IGF1R signaling in the impaired diabetic wound healing.14 In addition, basal KCs from human diabetic foot ulcer skin are known to be deficient in IGF–1 expression. Topical supplementation of IGF–1 to the wound edge of diabetic rats has been shown to accelerate wound closure,40,41 and topical application of insulin has similarly been shown to encourage diabetic wound closure by enhancing protein kinase B(Akt) and extracellular signal-regulated kinases (ERK) signaling pathways (Fig. 2).42
Fig. 2.
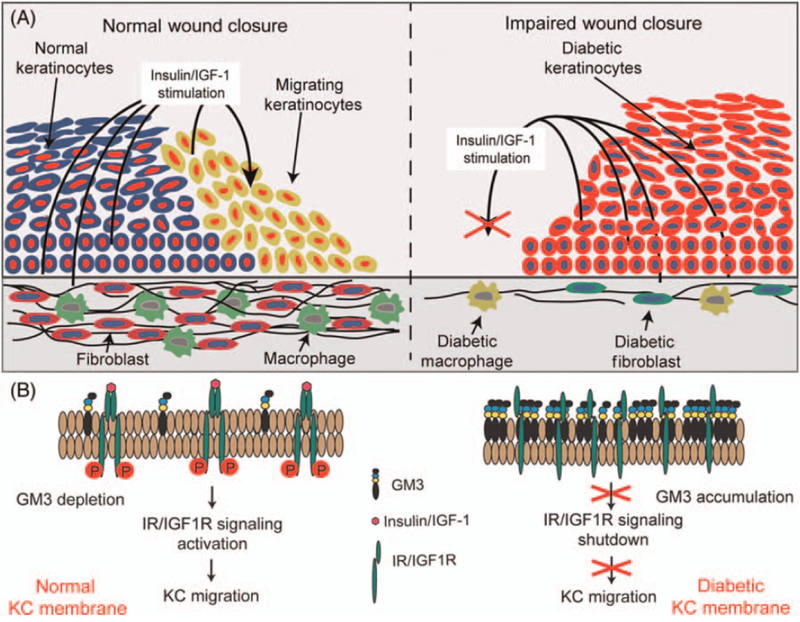
Increased keratinocyte GM3 expression is associated with reduction in the insulin/IGF–1 signaling axis and impaired healing in diabetes. (A) Diabetic keratinocytes at the edge of the wound have increased GM3 expression and blunted responses to growth factors, particularly of the insulin/IGF–1 signaling axis. GM3 also affects the responses of fibroblasts, macrophages, vessels, and nerves. The result is an impairment in wound closure. (B) GM3 accumulation in the keratinocyte membrane shuts down IR/IGF1R signaling, leading to reduction in keratinocyte migration and scratch wound reepithelialization.
5. GANGLIOSIDES REGULATE IGF1R SIGNALING AXIS IN CULTURED KCS
In vitro, TNF-α and glucose supplementation increase GM3S and GM3 expression in canonical insulin target cells (adipocytes, myocytes, and hepatocytes),16,18 and directly suppress IR and IR substrate-1 (IRS–1) tyrosine phosphorylation in cultured adipocytes, decreasing glucose uptake.16 In normal human epidermal KCs, high ambient glucose (e.g., a 2.5-fold increase to simulate hyperglycemia) and chronic exposure to low-dose TNF-α (e.g., 100 pM for 4 days) slow cell migration and inhibit proliferation.43,44 Similarly, TNF-α and glucose supplementation elevate expression of GM3S and GM3 in cultured KCs,14,38 leading to inhibition of bothinsulin- and IGF–l–induced receptor phosphorylation.14,43,44 As noted, IGF1R activation is thought to play a more important role than IR activation in KCs migration, proliferation, and survival. Indeed, increases in GM3 by pharmacological supplementation, such as exposure to excess glucose or blockade of GM3 metabolism with accumulation of GM3, inhibit IGF–l–induced IGF1R phosphorylation and dramatically slow cell movement to a much greater extent than its effect on IR activation. The mechanism of this inhibition of migration has been shown to involve prevention of lamellipodial protrusion through suppression of Rac1 signaling and activation of RhoA.38
The trisialylated epidermal ganglioside GT1b45 has been shown in scratch assays to inhibit KC migration through a variety of mechanisms, including its attachment to fibronectin,46 its binding to integrin α5β1 to block the integrin-fibronectin interaction,46 and its inhibition of the urokinase-type plasminogen activator (uPA)-integrin interaction.15 Whether GT1b is increased in diabetic skin has not been studied; however, KC migration is slowed despite GT1b depletion when GM3 accumulates from gene knockdown of the enzyme needed to synthesize GT1b from GM3. This finding further suggests that GM3, not GT1b, plays the predominant role in blocking KC migration, including in diabetes.38
The ganglioside-specific impact on KC migration in vitro is further confirmed by its reversal in the face of GM3 depletion through treatment with GM3S SNAs or glucosylceramide synthase inhibitors. GM3 depletion leads to ligand-independent and increased ligand-dependent receptor activation, which accelerates KC migration, promotes directional lamellipodial protrusion, and increases cell velocity, displacement and persistence.38 GM3 depletion activates IGFR and Rac1 signaling38 and reverses the glucose- and TNF-α-induced suppression of p-IGFIR and p-IR.14,25 In the presence of glucose or chronic low-dose TNF-α, this increase in KC movement from GM3 depletion further suggests that GM3 is the required intermediate in glucose-induced exacerbation of insulin resistance. Depletion of GM3 has been shown to promote glucose uptake into cells and GLUT1 transport to the membrane of KCs, suggesting that the accelerated migration in the face of GM3 depletion and glucose excess relates to increased availability of this carbon source.47 GM3 supplementation or overexpression has also been shown to inhibit epidermal growth factor receptor (EGFR) activation both directly and through preventing in the matrix metallopeptidase 9-integrin interaction in squamous carcinoma cells48 and normal human KCs, leading to inhibition of both cell proliferation and migration.13 It is possible that GM3-induced inhibition of EGFR activation or increased phosphorylation of EGFR by GM3 depletion also plays a role in the wound healing impairment in diabetes and its reversal, respectively. However, the gene knockdown of IGF1R or inhibition of its activation dramatically suppresses the migration in scratch assays induced by GM3 depletion, and migration ceases with the combination of inhibition of both IGF1R and IR.38 This finding suggests that EGFR inhibition plays a lesser role in GM3-modulatcd inhibition of KC migration, but further investigation into the role of GM3-induced EGFR inhibition is warranted.
6. CONCLUSIONS
The wound healing process requires a carefully orchestrated series of events. Diabetic ulcers affect a variety of cells and are multifactorial in pathogenesis. Central to poor healing is the dampened response to growth factors, including activators of the insulin/IGF–1 signaling axis. Ganglioside GM3 has been shown to be a mediator of TNF-α and glucose-induced insulin resistance and directly suppresses the cutaneous IGF–1/insulin signaling axis. The reduced IGF–1 levels in diabetic wounds, improved diabetic wound epidermal gap closure with IGF–1 overexpression, and critical role of IGF1R signaling in wound neovascularization all suggest that activating insulin/IGF–1 signaling to accelerate reepithelialization in diabetic wounds can be a novel means of intervention. Furthermore, reduction of GM3 through topically administered gene therapy at the wound edge in diabetic mice reduces their excess cutaneous GM3S and GM3, leading to normalization of wound healing. These findings reinforce a pivotal role for GM3–induced insulin resistance in impairing diabetic reepithelialization and wound closure and suggest GM3-depleting strategies as a novel approach for human diabetic wounds.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health Grants R01AR44619 and R21AR062898 (AP), and the Postgraduate Training in Cutaneous Biology T32 AR060710 (DD).
REFERENCES
- 1.Loots MAM, Kenter SB, Au FL, van Galen WJM, Middelkoop E, Bos JD, Mekkes JR. Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. EurJ Cell Biol 2002;81: 153–160. [DOI] [PubMed] [Google Scholar]
- 2.Loots MAM, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol 1998;111:850–857. [DOI] [PubMed] [Google Scholar]
- 3.Jeffcoate WJ, Harding KG. Diabetic foot ulcers. Lancet 2003;361:1545–1551. [DOI] [PubMed] [Google Scholar]
- 4.Jeffcoate WJ, Price P, Harding KG. Wound heating and treatments for people with diabetic foot ulcers. Diabetes Metab Res 2004;20:S78–S89. [DOI] [PubMed] [Google Scholar]
- 5.Ferrier TM. Comparative study of arterial disease in amputated lower limbs from diabetics and non-diabetics (with special reference to feet arteries). MedJAust 1967;1:5–11. [PubMed] [Google Scholar]
- 6.Dinh T, Veves A. Microcirculation of the diabetic foot. Curr Pharm Des 2005; 11: 2301–2309. [DOI] [PubMed] [Google Scholar]
- 7.Dinh TL, Veves A . A review of the mechanisms implicated in the pathogenesis of the diabetic foot. Int J Low ExtremWounds 2005;4:154–159. [DOI] [PubMed] [Google Scholar]
- 8.LoGerfo FW, Coffman JD. Current concepts. Vascular and microvascular disease of the foot in diabetes. Implications for foot care. N Engl J Med 1984;311:1615–1619. [DOI] [PubMed] [Google Scholar]
- 9.Veves A, Akbari CM, Primavera J, Donaghue VM, Zacharoulis D, Chrzan JS, DeGirolami U, LoGerfo FW, Freeman R. Endothelial dysfunction and the expression of endothelial nitric oxide synthetase in diabetic neuropathy, vascular disease, and foot ulceration. Diabetes 1998;47:457–463. [DOI] [PubMed] [Google Scholar]
- 10.Boulton AJ, Kirsner RS, Vileikyte L. Clinical practice. Neuropathic diabetic foot ulcers. N Engl J Med 2004;351:48–55. [DOI] [PubMed] [Google Scholar]
- 11.Pecoraro RE, Ahroni JH, Boyko EJ, Stensel VL. Chronology and determinants of tissue repair in diabetic lower-extremity ulcers. Diabetes 1991;40:1305–1313. [DOI] [PubMed] [Google Scholar]
- 12.Brem H, Sheehan P, Boulton AJ. Protocol for treatment of diabetic foot ulcers. AmJSurg 2004;187:1S–10S. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Rahman Z, Sun P, Meuillet E, George D, Bremer EG, Al-Qamari A, Paller AS. Ganglioside modulates ligand binding to the epidermal growth factor receptor. J Invest Dermatol 2001;116:69–76. [DOI] [PubMed] [Google Scholar]
- 14.Wang XQ, Lee S, Wilson H, Seeger M, Iordanov H, Gatla N, Whittington A, Bach D, Lu JY, Paller AS. Ganglioside GM3 depletion reverses impaired wound healing in diabetic mice by activating IGF–1 and insulin receptors. J Invest Dermatol 2014;134: 1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang XQ, Sun P, Paller AS. Gangliosides inhibit urokinase-type plasminogen activator (uPA)-dependent squamous carcinoma cell migration by preventing uPA receptor/alphabeta integrin/epidermal growth factor receptor interactions. J Invest Dermatol 2005;124:839–848. [DOI] [PubMed] [Google Scholar]
- 16.Tagami S, Inokuchi Ji J, Kabayama K, Yoshimura H, Kitamura F, Uemura S, Ogawa C, Ishii A, Saito M, Ohtsuka Y, Sakaue S, Igarashi Y. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem 2002;277:3085–3092. [DOI] [PubMed] [Google Scholar]
- 17.Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest 1993;91:797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Memon RA, Holleran WM, Uchida Y, Moser AH, Ichikawa S, Hirabayashi Y, Grunfeld C, Feingold KR. Regulation of glycosphingolipid metabolism in liver during the acute phase response. JBiolChem 1999;274:19707–19713. [DOI] [PubMed] [Google Scholar]
- 19.Hamed EA, Zakary MM, Abdelal RM, Abdel Moneim EM. Vasculopathy in type 2 diabetes mellitus: role of specific angiogenic modulators. J Physiol Biochem 2011. ;67: 339–349. [DOI] [PubMed] [Google Scholar]
- 20.Wei D, Tao R, Zhang Y, White MF, Dong XC. Feedback regulation of hepatic gluconeogenesis through modulation of SHP/Nr0b2 gene expression by Sirt1 and FoxO1. Am J Physiol Endocrinol Metab 2011. ;300:E312–E320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita T. Glycosphingolipid modification: structural diversity, functional and mechanistic integration of diabetes. Diabetes MetabJ 2011;35:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, Sethi JK, O’Rahilly S, Overkleeft HS. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 2007;56:1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Przybylska M, Wu IH, Zhang J, Maniatis P, Pacheco J, Piepenhagen P, Copeland D, Arbeeny C, Shayman JA, Aerts JM, Jiang C, Cheng SH, Yew NS. Inhibiting glycosphingolipid synthesis ameliorates hepatic steatosis in obese mice. Hepatology 2009;50: 85–93. [DOI] [PubMed] [Google Scholar]
- 24.Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 2007;56:1210–1218. [DOI] [PubMed] [Google Scholar]
- 25.Randeria PS, Seeger MA, Wang XQ, Wilson H, Shipp D, Mirkin CA, Paller AS. siRNA-based spherical nucleic acids reverse impaired wound healing in diabetic mice by ganglioside GM3 synthase knockdown. Proc Natl Acad Sci USA 2015;112:5573–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menichella DM, Jayaraj ND, Wilson HM, Ren D, Flood K, Wang XQ, Shum A, Miller RJ, Paller AS. Ganglioside GM3 synthase depletion reverses neuropathic pain and small fiber neuropathy in diet-induced diabetic mice. Mol Pain 2016;12: doi: 10.1177/1744806916666284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Fan J, Chen M, Li W, Woodley DT. Transforming growth factor-alpha: a major human serum factor that promotes human keratinocyte migration. J Invest Dermatol 2006;126:2096–2105. [DOI] [PubMed] [Google Scholar]
- 28.Haase I, Evans R, Pofahl R, Watt FM. Regulation of keratinocyte shape, migration and wound epithelialization by IGF-1- and EGF-dependent signalling pathways. J Cell Sci 2003;116:3227–3238. [DOI] [PubMed] [Google Scholar]
- 29.Cubbon RM, Kearney MT, Wheatcroft SB. Endothelial IGF-1 receptor signalling in diabetes and insulin resistance. Trends Endocrinol Metab 2016;27:96–104. [DOI] [PubMed] [Google Scholar]
- 30.Sall JW, Klisovic DD, O’Dorisio MS, Katz SE. Somatostatin inhibits IGF-1 mediated induction ofVEGF in human retinal pigment epithelial cells. ExpEyeRes 2004;79:465–476. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. JBiolChem 2002;277:38205–38211. [DOI] [PubMed] [Google Scholar]
- 32.Jacobo SM, Kazlauskas A. Insulin-like growth factor 1 (IGF-1) stabilizes nascent blood vessels. JBiol Chem 2015;290:6349–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apel PJ, Ma J, Callahan M, Northam CN, Alton TB, Sonntag WE, Li Z. Effect of locally delivered IGF-1 on nerve regeneration during aging: an experimental study in rats. Muscle Nerve 2010;41:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabinovsky ED. The multifunctional role of IGF-1 in peripheral nerve regeneration. Neurol Res 2004;26:204–210. [DOI] [PubMed] [Google Scholar]
- 35.Ando Y, Jensen PJ. Epidermal growth factor and insulin-like growth factor I enhance keratinocyte migration. J Invest Dermatol 1993;100:633–639. [DOI] [PubMed] [Google Scholar]
- 36.Sadagurski M, Yakar S, Weingarten G, Holzenberger M, Rhodes CJ, Breitkreutz D, Leroith D, Wertheimer E. Insulin-like growth factor 1 receptor signaling regulates skin development and inhibits skin keratinocyte differentiation. Mol Cell Biol 2006;26:2675–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stachelscheid H, Ibrahim H, Koch L, Schmitz A, Tscharntke M, Wunderlich FT, Scott J, Michels C, Wickenhauser C, Haase I, Bruning JC, Niessen CM. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. EmboJ 2008;27:2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dam DH, Wang XQ, Sheu S, Vijay M, Shipp D, Miller L, Paller AS. Ganglioside GM3 mediates glucose-induced suppression of IGF-1 receptor-Rac1 activation to inhibit keratinocyte motility. J Invest Dermatol 2017;137:440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blakytny R, Jude EB, Martin Gibson J, Boulton AJ, Ferguson MW. Lack of insulin-like growth factor 1 (IGF1) in the basal keratinocyte layer of diabetic skin and diabetic foot ulcers. J Pathol 2000;190:589–594. [DOI] [PubMed] [Google Scholar]
- 40.Bitar MS. Insulin-like growth factor-1 reverses diabetes-induced wound healing impairment in rats. Horm Metab Res 1997;29:383–386. [DOI] [PubMed] [Google Scholar]
- 41.Korolkiewicz RP, Tashima K, Fujita A, Kato S, Takeuchi K. Exogenous insulin-like growth factor (IGF)-l improves the impaired healing of gastric mucosal lesions in diabetic rats. Pharmacol Res 2000;41:221–229. [DOI] [PubMed] [Google Scholar]
- 42.Lima MH, Caricilli AM, de Abreu LL, Araujo EP, Pelegrinelli FF, Thirone AC, Tsukumo DM, Pessoa AF, dos Santos MF, de Moraes MA, Carvalheira JB, Velloso LA, Saad MJ. Topical insulin accelerates wound healing in diabetes by enhancing the AKT and ERK pathways: a double-blind placebo-controlled clinical trial. PLoSOne 2012;7:e36974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lan CC, Liu IH, Fang AH, Wen CH, Wu CS. Hyperglycaemic conditions decrease cultured keratinocyte mobility: implications for impaired wound healing in patients with diabetes. BrJDermatol 2008. [DOI] [PubMed]
- 44.Spravchikov N, Sizyakov G, Gartsbein M, Accili D, Tennenbaum T, Wertheimer E. Glucose effects on skin keratinocytes: implications for diabetes skin complications. Diabetes 2001;50:1627–1635. [DOI] [PubMed] [Google Scholar]
- 45.Paller AS, Arnsmeier SL, Robinson JK, Bremer EG. Alteration in keratinocyte ganglioside content in basal cell carcinomas. J Invest Dermatol 1992;98:226–232. [DOI] [PubMed] [Google Scholar]
- 46.Paller AS, Arnsmeier SL, ChenJD, Woodley DT. Ganglioside GT1b inhibits keratinocyte adhesion and migration on a fibronectin matrix. J Invest Dermatol 1995;105:237–242. [DOI] [PubMed] [Google Scholar]
- 47.Shehu A, Lu JY, Wilson H, Bach D, Shipp D, Randeria PS, Mirkin CA, Paller AS. Cytoplasmic sequestion of keratinocyte GLUT1 by ganglioside GM3 mediates impaired diabetic wound healing. J Invest Dermatol 2013;133:S249. [Google Scholar]
- 48.Wang XQ, Sun P, Paller AS. Ganglioside GM3 inhibits matrix metalloproteinase-9 activation and disrupts its association with integrin. JBiolChem 2003;278:25591–25599. [DOI] [PubMed] [Google Scholar]