

Abstract
Long non-coding RNAs (lncRNAs) are important for gene expression, but little is known about their structures. RepA is a 1.6 kb mouse lncRNA consisting of the same sequence found in the 5’ region of Xist, including A- and F-repeats. It is proposed to facilitate the initiation and spread of X-chromosome inactivation, although its exact role is poorly understood. To gain insight into the molecular mechanism of RepA/Xist, we determined a complete and phylogenetically validated secondary structural map of RepA using SHAPE and DMS chemical probing of a homogeneously folded RNA in vitro. UV crosslinking experiments were combined with RNA modeling methods to produce a three-dimensional model of RepA functional domains, demonstrating that tertiary architecture exists within lncRNA molecules and occurs within specific functional modules. This work provides a foundation for understanding the evolution and functional properties of RepA/Xist and offers a framework for exploring architectural features of other lncRNAs.
Introduction
Long non-coding RNAs (lncRNAs) are a rapidly growing class of cellular transcripts that exceed 200 nucleotides in length and lack protein coding potential. During the past decade, lncRNAs have been increasingly recognized as essential components of the mammalian transcriptome1. They are involved in various cellular processes and diseases, including signaling, embryonic stem cell differentiation, brain function, chromatin remodeling, and cancer2–7. Genome-wide analysis suggests that lncRNAs possess greater structural complexity than mRNA8–10. Recent experimental characterization of lncRNAs SRA and HOTAIR provide evidence that lncRNAs can adopt complex structures11,12.
There is particular interest in the X-inactivation specific transcript, Xist13,14, which functions in the dosage compensation pathway of eutherian mammals. Females silence one of two X-chromosomes to balance X-linked gene expression with monoallelic males15. The lncRNA Xist (>17 kb in mouse) is indispensible for initiating, establishing and maintaining X-chromosome inactivation (XCI)13,14. The proposed functional roles of Xist include the recruitment of silencing factors, and spreading over the inactive X-chromosome (Xi) in cis, causing transcriptional silencing of X-linked genes16.
Unlike some lncRNA genes that are conserved only in primates, the overall gene structure of Xist is conserved among all eutherian mammals. Its genetic architecture includes six regions that are composed of short tandem repeat sequences, termed A-F13,14,17 (Fig. 1a). A-repeat consists of 7.5 repeat-A units in mouse, and this region was shown to be crucial for initiation of XCI more than a decade ago18. Many protein factors that contribute to XCI have been proposed to directly interact with A-repeat, including polycomb repressive complex 2 (PRC2), ATRX, and Spen/Sharp19–24. F-repeat is located ~0.7 kb downstream of A-repeat and consists of 2 repeat-F units in mouse17. This region was recently demonstrated to directly interact with the Lamin B Receptor (LBR) and to be required for Xist-mediated silencing of distal genes25. However, the precise mechanism by which A-repeat and F-repeat recognize various protein partners and initiate X-chromosome silencing remains unclear.
Figure 1: RepA folds into a compact species upon addition of Mg2+.
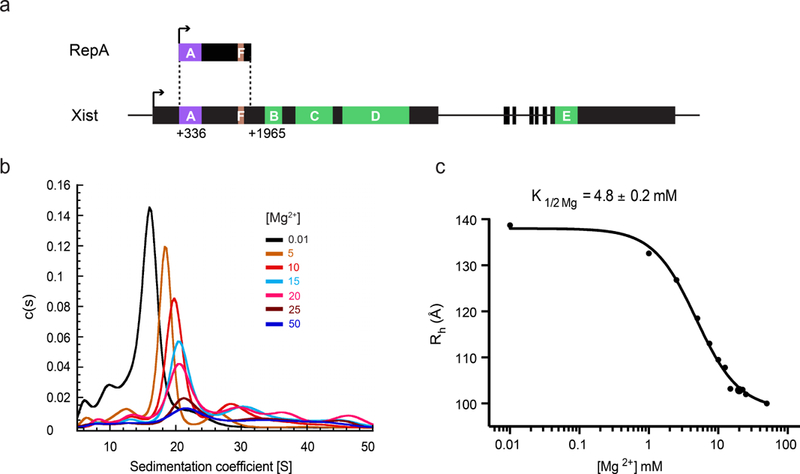
(a) RepA spans a region that extends from nucleotide 336 to 1965 in Xist, and includes A-repeat and F-repeat19. The transcription start sites in RepA and Xist are denoted with black arrows. All the repeat regions in RepA and Xist are highlighted and labeled. (b) SV-AUC profiles of RepA obtained under native conditions in the presence of increasing concentrations of Mg2+. The graph was obtained using SedFit43,44. (c) Hill plot of the hydrodynamic radii (Rh, in angstroms) derived from the SV-AUC experiment described in panel a. The Rh values are derived from one independent measurement. K1/2Mg is 4.8 ± 0.2 mM and Hill coefficient (nH) is 1.4 ± 0.1. Hill plots drawn for each set of measurements are shown in Supplementary Fig. 1.
The repeat-A and repeat-F units found within Xist are also embedded within a 1.6 kb murine transcript known as RepA. The RepA RNA (not to be confused with the short, repeat-A units themselves) is encoded from an internal promoter on the Xist gene sense strand19 (Fig. 1a), and has been proposed to recruit PRC2 histone methyltransferase to the future Xi prior to the expression of Xist. Additionally, RepA appears to upregulate the expression of Xist, which then initiates and spreads silencing across the inactive X-chromosome19.
Despite the availability of abundant functional data, structural work on this system has been limited. Early work focused on a single repeat-A unit or an isolated A-repeat containing sequence (427 nucleotides long). Most of these studies describe the entire A-repeat as a set of tandem dual stem-loop structures18, although some evidence suggests that isolated A-repeat sequences can adopt more complicated configurations26–28. A later study employing in vivo DMS probing provided the first in-cell secondary structural information for A-repeat sequence, along with other novel structural elements within Xist29. Recently, another study used in vivo RNA-RNA crosslinking to identify regions of human XIST involved in long-range interactions with near-nucleotide resolution30. This study revealed several conserved long-range interactions within Xist, but none involving the RepA region, indicating that A-repeat and F-repeat regions fold as isolated domains in the context of the full-length Xist. This finding suggests that structural data obtained on the RepA transcript is relevant to the RepA region in Xist. Altogether, previous studies have provided secondary structural information for less than 40% of the individual nucleotides in the RepA/Xist region due to technological limitations. This incomplete dataset, along with the use of truncated constructs, likely contributes to the conflicting models of Xist structure and our limited understanding of Xist function. A complete secondary structural map of RepA/Xist is needed for defining the details of its architectural units, setting the stage for phylogenetic analysis, and understanding the function of RepA/Xist at the molecular level.
In this work, we provide the first complete secondary structural map of the RepA RNA by generating a homogenous, monodisperse RNA sample and then mapping its secondary structure using SHAPE and DMS probing methodologies. The resultant map was subjected to a statistical and thermodynamic evaluation through jackknife31 and Shannon entropy analyses32. Taken together, the data demonstrate that RepA forms a complicated structure composed of three independently folding modules. This map provides landmarks for the construction of meaningful sequence alignments, which enabled us to conduct phylogenetic analyses that implicate regions of functional significance and provide functional validation for substructures in the map. The relative organization of RepA secondary structures in three-dimensional space was investigated using UV crosslinking experiments and computational 3D modeling, thereby providing tertiary structural information on a lncRNA and revealing regions of sequence conservation. The resultant features provide structural insights into the evolution and function of RepA, and the study lays the groundwork for exploring the three-dimensional architecture of other lncRNAs.
Results
RepA adopts a compact, monodisperse state upon folding
Functional RNAs, such as ribozymes, group II introns and riboswitch RNAs, can only exert their function as well-folded molecules33–35. Performing structural probing on less folded and compact RNA samples, which contain a mixture of RNA conformations, will prevent accurate structural predications. Therefore, the availability of a homogenous, monodisperse RNA sample is essential for meaningful structural characterization of any RNA. However, this is difficult to achieve for lncRNAs due to their length, as traditional RNA purification and refolding methods produce a mixture of lncRNA conformations12. To tackle this problem, we adopted an alternative native purification protocol that preserves the RNA secondary structure that is formed during in vitro transcription. Using this protocol (see Online Methods), we obtained a homogeneous and monodisperse sample of RepA suitable for in vitro structural characterization.
To identify optimal ionic conditions for promoting the homogeneous compaction of monomeric RepA molecules, we studied RepA compaction as a function of Mg2+ concentration. We conducted a series of sedimentation velocity experiments, using analytical ultracentrifugation (SV-AUC) at physiological K+ concentration (150 mM) to directly monitor the degree of molecular compaction as a function of Mg2+ concentration. As Mg2+ concentration increases, the sedimentation coefficient (s) of RepA increases (Fig. 1b) and the hydrodynamic radius (RH) decreases (Fig. 1c, Supplementary Results, Supplementary Fig. 1), indicating global compaction of the RepA molecule. Fitting the RH values at each Mg2+ concentration to the Hill equation yielded a K1/2 Mg value of 4.8 ± 0.2 mM, which is smaller than that of several highly structured RNAs, such as ai5γ group IIB intron (15 ± 2 mM)36 and lncRNA HOTAIR (8.6 ± 0.8 mM)12. This low K1/2 Mg for RepA compaction signifies a relatively high degree of structural stability.
The compaction of RepA upon addition of Mg2+ was also confirmed by size exclusion chromatography (SEC) (Supplementary Fig. 2). The SEC profiles demonstrate that the RepA population is stable and homogenous over a broad range of Mg2+ concentrations (0–20 mM). Both SV-AUC and SEC experiments indicate that a concentration of 15 mM Mg2+ (>3 times of the K1/2 Mg) is sufficient for promoting a completely compact and monodisperse form of RepA. This condition was therefore chosen for subsequent in vitro structural characterization.
Determination of RepA secondary structure
Having obtained a well-folded RNA sample, we examined its secondary structure using SHAPE and DMS probing. The SHAPE reagent 1M7 selectively acetylates the 2’-hydroxyl group of RNA nucleotides that have flexible backbones. DMS selectively methylates the heterocyclic nitrogen atoms on adenines and cytosines that are not base-paired. Because these two methods represent orthogonal approaches for interrogating secondary structure, good agreement between them suggests a robust secondary structural map.
SHAPE reactivity was monitored at single-nucleotide resolution and normalized SHAPE reactivity values were used as pseudo-free energy constraints to guide RepA structure prediction (see Online Methods). We evaluated the SHAPE-directed secondary structural models using jackknife resampling31 to estimate the confidence of each base pair in the predicted model. The majority of the helices (70%) can be described with high confidence, and most of the low-confidence helices are located adjacent to junctions (Supplementary Fig. 3). Next, we used normalized DMS reactivities to examine the low-confidence regions. The low DMS reactivity in these helices supported the formation of direct base pairs, which is consistent with the SHAPE-directed secondary structural model (Supplementary Fig. 3). Overall, the resulting map (Fig. 2) indicates that RepA is a highly structured RNA molecule in which more than 60% of the nucleotides are base-paired.
Figure 2: Secondary structure of RepA derived from SHAPE and DMS probing.
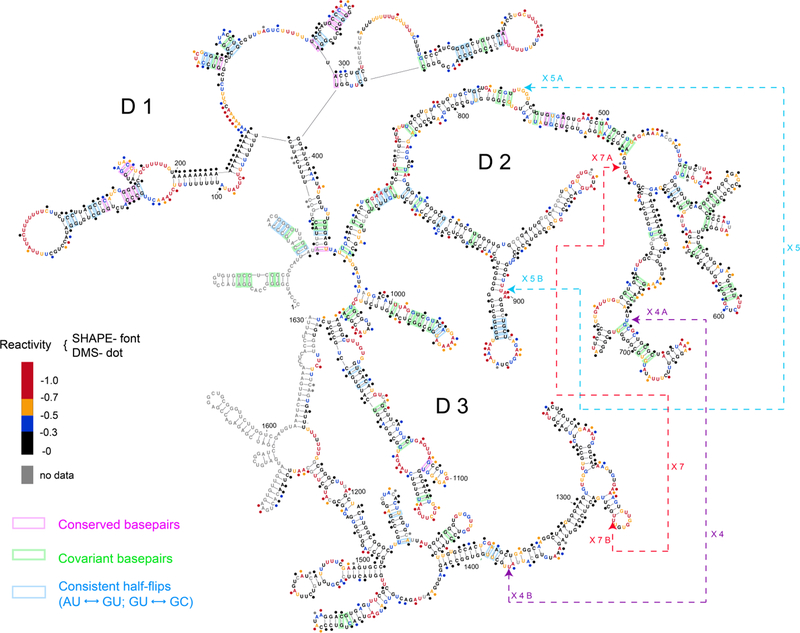
SHAPE reactivities are depicted by colored nucleotides. DMS reactivities are represented by colored dots over the nucleotides. SHAPE and DMS reactivities are denoted using the same color codes, as illustrated in the legend. Highly reactive nucleotides are indicated with red and orange, and nucleotides with low reactivities are displayed in black or blue according to their reactivity values. Covariant base-pairs in 56 mammalian sequences are highlighted in green, consistent half-flips pairs are highlighted in blue, and conserved base-pairs are highlighted in pink. UV-crosslinked nucleotide positions are indicated by arrows with dashed lines. Secondary structure was drawn using VARNA (http://varna-gui.software.informer.com/).
RepA consists of three independently folding modules
Our experimentally derived map suggests that RepA contains a set of secondary structural domains that radiate from a central junction (Fig. 2). To examine whether these individual sections are autonomous folding domains, we employed the 3S shotgun analysis method37 using two groups of fragments (Fig. 3a). One group contains fragments F1, F2, and F3, which together cover the span of full-length RepA. The boundaries of these fragments are designed to maintain all secondary structural elements identified in full-length RepA. The other group contains fragments F4, F5 and F6, which overlap with fragments F1-F3, but are expected to disrupt secondary structural domains that are predicted by the mapping and modeling procedure.
Figure 3: Fragment analysis reveals independently-folded domains in RepA.
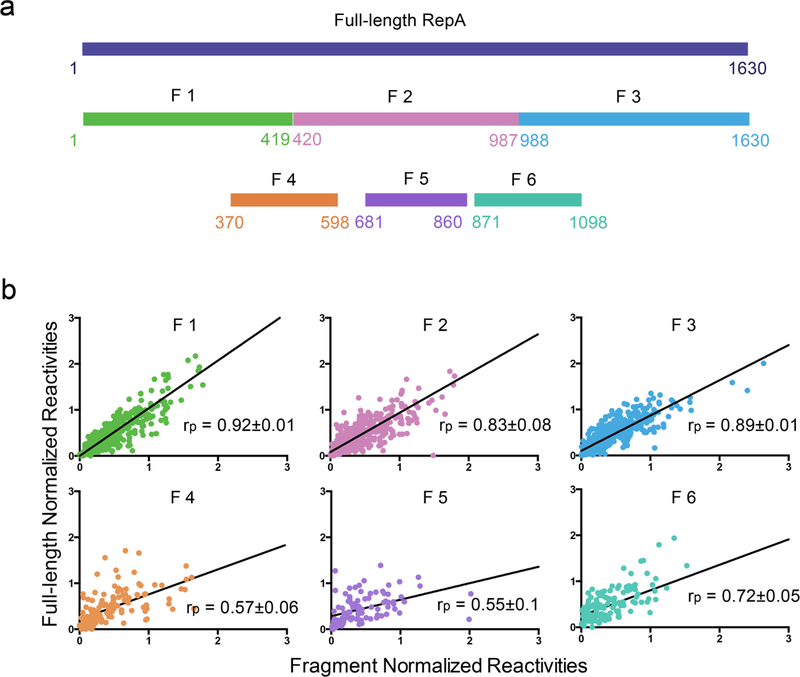
(a) Schematic representation of RepA fragments with respect to their position along the sequence of full-length RepA. (b) Scatterplots comparing SHAPE reactivity of each fragment with the corresponding region in full-length RepA. Pearson correlation values (rp) between SHAPE reactivities of each fragment and the corresponding regions in full-length RepA are indicated; the standard error in correlation coefficients was estimated by bootstrapping analysis.
SHAPE reactivities of fragments F1, F2, and F3 correlate with their corresponding regions in full-length RepA with Pearson’s correlation coefficients (rp) of 0.92±0.01 for F1, 0.83±0.08 for F2 and 0.89±0.01 for F3 (Fig. 3b). This indicates that fragments F1, F2, and F3 adopt the same structure in isolation (Supplementary Fig. 4) as in the full-length RepA, and are therefore independently folding domains. By contrast, SHAPE reactivities of fragment F4, F5, and F6 showed relatively poor correlation with the corresponding full-length RepA profiles (F4, F5, and F6 possess rp values of 0.57±0.06, 0.55±0.1 and 0.72±0.05, respectively) (Fig. 3b), indicating that they adopt structures different from the parent molecule (Supplementary Fig. 4). Our results suggest that RepA is composed of three independent structural modules, corresponding to fragments F1 (domain 1, D1), F2 (domain 2, D2), and F3 (domain 3, D3), which are connected through a central junction and assemble into a complex structure (Fig. 2).
To evaluate potential local structural heterogeneity, we calculated Shannon entropy (S) of each nucleotide using SHAPE-directed base-pair probabilities (Supplementary Fig. 5; Online Methods)32. Overall, the average S value of RepA is as low as 0.102, indicating an overall structural homogeneity in RepA.
D1 comprises 7.5 repeat-A units and forms an elaborate secondary structure different from the tandem dual stem-loop model18 (Fig. 4a). Only repeat 5 adopts the dual stem-loop structure, while repeat 1, 2 and 6 form diverse types of stem-loop motifs. Repeat 3 and 4 form an extended stem-loop, likely due to the long consecutive AU stem that lies adjacent. Similarly, repeat 7 and 8 base-pair with each other, forming a relatively long stem-loop. Additionally, one central six-way junction brings the repeat-A units closer in space and adds complexity to the overall structure of D1. D1 has the highest average Shannon entropy (0.122), compared to D2 (0.098) and D3 (0.093), suggesting the relative higher dynamics of D1. This observation is consistent with the recent RNA-RNA crosslinking results that suggested A-repeat exists in multiple confirmations in vivo30. Specifically, the nucleotides involved in the consecutive AU stem have the highest S values (>0.5), which contribute to the formation of multiple conformations. Although our RNA sample for chemical probing is globally homogeneous (Fig. 1b, Supplementary Fig. 1), our structural model for this region likely represents one of the A-repeat functional states.
Figure 4: D1 of RepA contains the functionally important A-repeat motif.
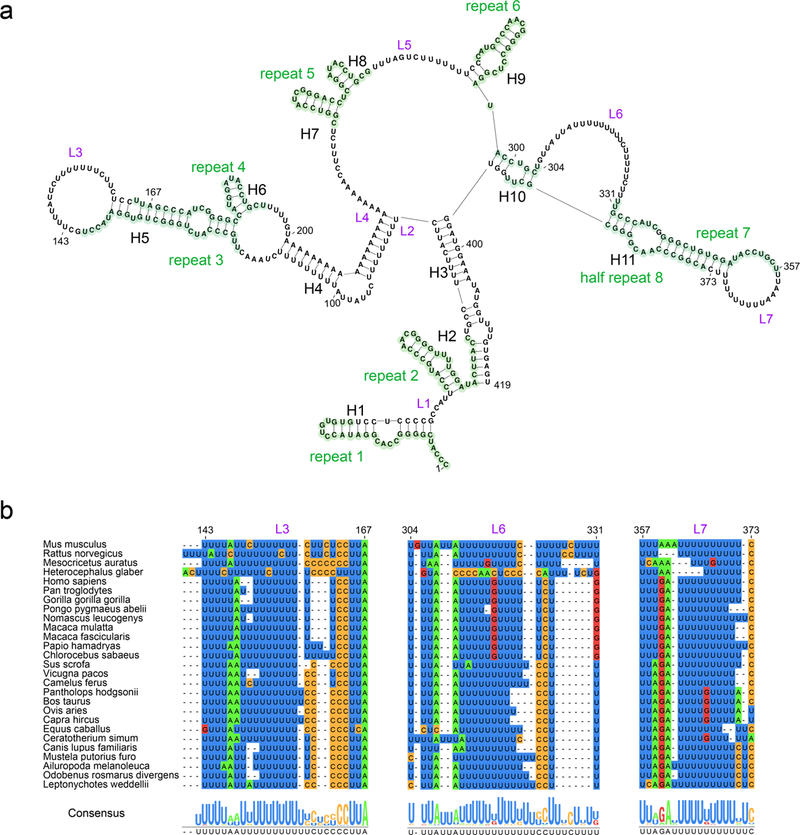
(a) Secondary structural map of D1. Repeat-A units are highlighted in green and labeled accordingly. Linker regions are indicated by L1-L7. Helices in D1 are indicated by H1-H11. (b) Sequence conservation of the linker regions L3 (nucleotides 143–167), L6 (nucleotides 304–331) and L7 (nucleotides 357–373) across 27 mammalian species. The alignment of the sequences is presented and color-coded by nucleotide type, and the consensus sequence is shown on the bottom.
D2 and D3 exhibit different structural features compared to D1, as they appear much more stable. The S values of most nucleotides in these domains are very low. Specifically, only 0.9% and 2.6% of nucleotides in D2 and D3, respectively, are located in very dynamic structural elements (S values over 0.4), compared to 10.5% in D1 (Supplementary Fig. 5). Moreover, D2 and D3 consist of more complicated structural motifs (Supplementary Fig. 6). D2 contains three 3-way junctions and two 4-way junctions. Three major helices, H12, H13, and H25, are linked by the central three-way junction, which may promote long-range contact. In D3, there is a 471-nucleotide long region, where fourteen helices assemble into a subdomain via three different junction segments. The 2 repeat-F units (highlighted in Supplementary Fig. 6b) are located in this region, participating in the base-pairing of the same helix, H34. Interestingly, internal loops and bulges are embedded within the majority of D2 and D3 helices, adding flexibility to these helices and potentially promoting tertiary contacts between structural motifs.
Covariation analysis of RepA secondary structure
Using our secondary structure map, we improved the sequence alignment of the RepA region and generated a covariance model (see Online Methods)38,39, which allowed us to identify the conserved structural motifs that may contribute to RNA function. Compared to previous studies29, our covariation analysis included a much broader range of species (56 mammalian species), which greatly increased its statistical power. Despite rapid changes in primary sequence through evolution, the RepA region displays a significant degree of covariation in base-pairing (Fig. 2), including the base pairs adjacent to the central junction connecting the three domains.
Most of the newly proposed structural elements in D1 can form in all mammalian species examined (Fig. 2). 10 out of 11 helices in D1 show a high degree of conservation across species. Previous phylogenetic analysis and functional studies have focused on the GC-rich repeat-A units, while we discovered that the sequences of some uridine-rich linker regions are highly conserved even in distant species (Fig. 4b). Interestingly, not only the uridines but also the other nucleotides in these regions are conserved. These linkers are all located in the loops or bulges adjacent to extended stem-loops. Our results suggest that the conserved linkers may be important in mediating specific interactions between repeat-A units and facilitating the formation of more complex structural elements. Additionally, the conservation of sequence and structure within these uridine-rich linkers indicates a potential binding site for poly-uridine or single-stranded RNA binding proteins.
We extended our phylogenetic analysis to the region downstream of A-repeat in Xist (D2 and D3 in RepA) and identified many conserved structural motifs for the first time (Fig. 2, Supplementary Fig. 6). The nucleotides participating in the central three-way junction in D2, which connects three important helices (H12, H13, and H25), are highly conserved in rodents, primates, and more distant mammals (Supplementary Fig. 7a), indicating that the overall architecture of D2 is likely conserved across species. The structure of the region containing nucleotides 498 to 771 is also highly conserved in rodents, suggesting its functional importance (Supplementary Fig. 7b). The corresponding regions in rat and Chinese hamster are capable of adopting similar intricate structures despite a somewhat modest sequence identity (85% and 74%, respectively). Similar structural motifs (H15–19) were also derived from in vivo DMS probing29.
Unlike the D1 and D2 regions, D3 exhibits relatively poor sequence conservation in mammals (Fig. 2, Supplementary Fig. 6b). Among all the structural motifs, only helices H28–30, H35, H37, and H41 show modest degree of conservation across species. Therefore, the D3 region in other species may display substantially different structural features.
Characterization of tertiary interaction sites in RepA
While secondary structure is informative, it is valuable to obtain information on the arrangement of helices in three dimensions. To determine which RepA subdomains are located in close proximity, we conducted UV crosslinking experiments on the folded RepA molecule.
We first examined the UV crosslinking pattern of RepA at different Mg2+ concentrations (0 to 15 mM, Fig. 5a and Online Methods). We observed that RepA forms crosslinks under all conditions tested, and the addition of Mg2+ changes the intensity of several crosslinked bands (Fig. 5a). Specifically, the crosslinked band CL1 becomes more prominent at higher Mg2+ concentration, while CL8 diminishes. The crosslinked bands CL4 and CL6 appear only when RepA is folded in the presence of Mg2+. All crosslinks become apparent at a relatively low Mg2+ concentration (5 mM); no additional crosslinks appear at higher concentrations. Co-migration of crosslinked and unirradiated monomeric RNA on a native agarose gel confirmed that all crosslinks were intramolecular. Altogether, these observations indicate that RepA undergoes a conformational change and tertiary structure stabilization upon Mg2+ addition, which is consistent with the Mg2+-dependent compaction of RepA observed by SV-AUC and SEC.
Figure 5: Characterization of the UV crosslinking sites within RepA.
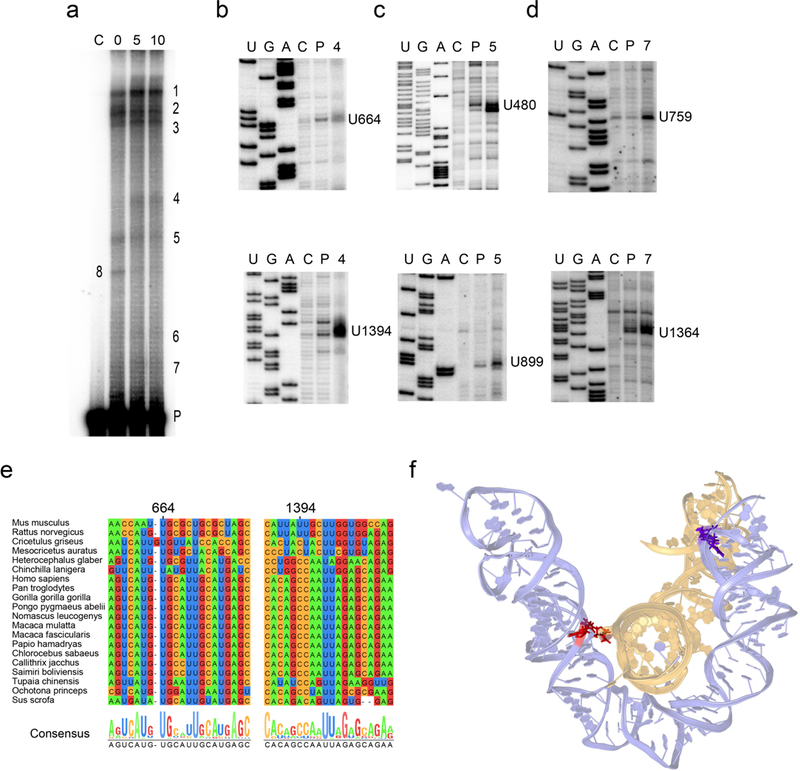
(a) UV crosslinking patterns of RepA. Unirradiated control RNA (C) is shown in the leftmost lane. UV-treated RNA samples in the presence of 0, 5 and 10 mM Mg are shown in lanes 0, 5 and 10, respectively. The crosslinked RNA bands are labeled 1 to 8. The un-crosslinked parent RNA band is denoted by P. (b-d) Primer extension mapping of the crosslinked nucleotides. Lanes U, G and A show dideoxy sequencing ladders. Lanes C, P, 4, 5 or 7 show reverse transcription (RT) products of corresponding RNA species. The nucleotide positions of identified RT stops are indicated to the right of the gels. Original gel images are shown in Supplementary Fig. 8. (e) Primary sequence conservation of 20 mammalian species corresponding to the regions containing crosslink CL4. The alignment of the sequences is presented and color-coded by nucleotide type, and the consensus sequence is shown on the bottom. (f) Three-dimensional model of the subdomains in D2 (nucleotides 494–508 and 632–776, colored in purple) and D3 (nucleotides 1272–1304 and 1349–1406, colored in yellow). Nucleotides participating in crosslink CL4 and CL7 are colored in purple and red, respectively.
Reverse transcription was used to identify the nucleotides involved in each crosslinked species. UV-crosslinked nucleotides generally interrupt primer extension one base prior to the crosslinked nucleotide residue40. Therefore, we assigned crosslinked nucleotides according to the position of strong reverse transcription stops that display substantially higher intensities than those in the unirradiated control (C) or the UV-treated but uncrosslinked parent RNA (P) (Fig. 5b-d). Three pairs of long-range crosslinks, CL4, CL5, and CL7 were identified within D2 and D3 (Fig. 2).
Specifically, crosslink CL5 between U480 (X 5A) and U899 (X 5B) is formed between the internal loops of helices H14 and H27 in D2 (Fig. 2, Supplementary Fig. 6a). This crosslink reflects probable interactions between helices H14 and H27, which bring U480 and U899 into proximity. In this way, D2 adopts a compact configuration and may serve as a scaffold for structural elements in other regions.
The other two crosslinks identified suggest long-range interactions between D2 and D3 (Fig.2, Supplementary Fig. 6). U664 (X 4A) is part of a three-way junction in D2 and crosslinks with U1394 (X 4B), a bulged nucleotide in helix H38 in D3. Importantly, U759 and U1364, which form CL7, are proximal to U664 and U1394 (CL4). These observations suggest that certain subdomains in D2 (nucleotides 494–508 and 632–776) and D3 (nucleotides 1272–1304 and 1349–1406) interact and form a specific tertiary architecture. The UV crosslinking results are consistent with the low Shannon entropy in this region, suggesting all the nucleotides involved in or adjacent to tertiary interactions are likely to stay in a single and stable conformation (Supplementary Fig. 5).
Phylogenetic analysis revealed that the crosslinked nucleotides and their surrounding primary sequences are well conserved, except for U1364 (X 7B) and surrounding sequences, which are only conserved in mouse and rat (Fig. 5e, Supplementary Fig. 9). Therefore, tertiary interactions suggested by the crosslinks appear to be conserved in the species we examined, although the specific arrangement of helices in three dimensions likely varies.
In order to visualize the tertiary contacts between D2 and D3, we modeled the spatially proximal subdomains with RNAComposer41 using the identified crosslinks as distance constraints (Fig. 5f). For comparison, we also modeled the subdomain of D2 alone. In the absence of D3, the corresponding subdomain in D2, is predicted to adopt a relatively extended and open conformation (Supplementary Fig. 10). In contrast, when constrained by tertiary interactions with D3, this subdomain forms a more compact structure (Fig. 5f). According to our 3D model, the D2 subdomain forms a platform for the docking of the distal stem-loop of D3, thereby placing U1364 in proximity to U759 and U664 in the vicinity of U1394.
Altogether, our UV crosslinking and 3D modeling results suggest a specific structural model for RepA. In this model, D2 and D3 interact and establish a relatively rigid scaffold. This structure may facilitate the proper orientation of bound protein partners, thereby facilitating the formation of functional machinery.
Discussion
In this study, we provide a complete and robust structural model for RepA, which is a functionally important lncRNA19 that is identical to a 5’ region of Xist. This phylogenetically validated secondary structure of RepA will serve as a roadmap for the future study of XCI so investigators can design targeted studies of specific structural elements and interactions that will lead to our understanding of the complex process of XCI. Furthermore, we defined specific tertiary contacts within RepA. This finding has important implications for RepA/Xist function and underscores the ability of lncRNAs to form complex and discrete secondary and tertiary structures.
In 2015, a secondary structural model for A-repeat was derived from in vivo DMS probing data29, suggesting an assembly of complex structural motifs within A-repeat that is more similar to our model than the dual stem-loop model. In fact, repeats 5–8 are predicted to form the same structure in both our model and the in vivo model. However, repeats 1–4 are dissimilar due to different predictions of which U-rich linker (L2 or L6) pairs with the A-rich region of L4. These discrepancies can be attributed to the fact that only the most DMS-reactive nucleotides (11% of the total nucleotides) were used to constrain RNA secondary structure modeling in the 2015 study29, while SHAPE and DMS probing data used in combination provide pseudo-free energy constraints on 85% of the nucleotides that were used for modeling of the secondary structure presented here. Due to its inability to report on the pairing status of uridines, DMS probing only provides information on 29% and 12% of nucleotides in uridine-rich linkers L2 and L6, respectively. Hence, DMS probing data alone is insufficient to determine which linker regions are base-paired. The SHAPE data presented here reflects the base-pairing status of all nucleotides and the low SHAPE-reactivity of linker L2 indicates that it is involved in base-pairing, while the high SHAPE-reactivity of linker L6 indicates a single-stranded segment. This rich dataset, provided by multiple chemical probes, combined with supporting phylogenetic covariation, provides a robust structural model for A-repeat.
While in vivo DMS probing is insufficient for generating a robust secondary structure, the raw data provide a valuable way to evaluate the biological relevance of our proposed structural map. We find that the majority of the structural elements in our model are consistent with the corresponding in vivo DMS probing data (Supplementary Fig. 11). One exception is helix H3, in which several nucleotides are highly DMS reactive in vivo, suggesting H3 may be unzipped upon protein binding in the cellular environment while other structural elements within A-repeat are maintained.
Many studies have focused on A-repeat after a systematic deletion analysis demonstrated its indispensible role during XCI18. One of the deletion constructs, ∆SX, was created by deleting 0.9 kb of the 5’ end of Xist. ∆SX lacks not only A-repeat but also 172 nucleotides immediately downstream (145 nucleotides in D2 of RepA). Because the ∆SX deletion completely abolishes silencing activity, A-repeat of Xist has been deemed essential for silencing. ∆SX has been used in later studies21,22,25,42, including one that focused on identifying proteins that interact directly with Xist and are required for silencing21. In this case, because ∆SX failed to recruit certain protein partners Spen/Sharp, Rnf20, and Wtap in vivo, it was deduced that A-repeat directly interacts with them21. However, our model of RepA suggests that the aforementioned protein partners are equally likely to interact with D2 since the deletion creating ∆SX disrupts the highly conserved structural motifs in D2, which is located immediately downstream of the A-repeat. Additionally, this deletion may trigger mis-folding of the RNA and disrupt tertiary interactions. Therefore, ∆SX is a suboptimal control for investigating the functional role of A-repeat. Our structural model will facilitate the design of new constructs that can be used in studies of XCI.
Recent work on Spen more conclusively identified a direct interaction with A-repeat, and our map suggests a precise structural motif that may be responsible. In vitro UV crosslinking between Spen and RepA (the same RNA construct used in this study) suggested that the uridine-rich linkers in A-repeat are Spen recognition sites30. However, Spen does not crosslink to the uridine-rich linkers equally. The dynamic inter-repeat duplex model previously proposed30 does not provide a conclusive explanation for this observation. Interestingly, our structural map correlates with the differential crosslinking efficiency. The strongest protein crosslinks are formed with the single-stranded uridine-rich linkers adjacent to helices H5 and H11 in our map (Fig. 4a). H5 and H11 are uniquely comprised of two repeat-A units that form an extended stem-loop, suggesting that Spen may preferentially bind to this structural motif. Moreover, these linker sequences are highly conserved in our phylogenetic analysis (Fig. 4b). In contrast, the linker regions between and upstream of the intra-repeat duplexes, which were inefficiently crosslinked to Spen30, have a lower degree of sequence conservation. Overall, the correlation between the functional data and the structural elements proposed in our work suggests that our structural map represents a functional state of RepA/Xist.
The region downstream of A-repeat (D2 and D3 in RepA), including F-repeat, has long been considered a functionally dispensable part of Xist18 and almost no functional or structural work has been done to characterize the role of this region. Very recently, an important player in XCI, protein LBR, was shown to directly interact with Xist through a LBR binding site (LBS), which spans the regions that we have designated as D2 and D325. However, deletion constructs that were created to test the LBS binding site (∆LBS, nucleotides 563–1347 in RepA) were designed without any knowledge of the potential significance of downstream substructures, and the ∆LBS construct is therefore predicted to disrupt some of the most important elements within D2 and D3, including the tertiary interaction sites. As a result, the actual LBS binding site remains undefined. Therefore, our structural model may serve as a guide when designing constructs for future mechanistic studies of interactions between RepA/Xist and protein partners.
Altogether, our findings show that RepA adopts specific, functional secondary structures and assembles into a set of three distinct domains that can be validated biochemically and phylogenetically. Our findings have important ramifications for the interpretation of previous studies on XCI and the silencing machinery. Additionally, we demonstrate that RepA and the 5’ end of Xist possess a defined tertiary architecture, thereby suggesting that lncRNAs can adopt three-dimensional structures. These structures can form autonomously in the absence of protein partners, which underscores a central role for RNA folding in the formation of lncRNP machinery. Our results complement the valuable information obtained using global methods for monitoring RNA and protein complexes in vivo, and provide precise, biophysical insights into the functional roles of RepA/Xist during XCI at the molecular level.
Online Methods
RNA synthesis and purification
DNA templates
Four different constructs of RepA were used in this work. A plasmid containing the full-length RepA sequence (GI|210076757) was purchased from Addgene (pCMV-Xist-PA, #26760). The RepA sequence and fragments F1 (1–419), F2 (420–987), F3 (988–1630), F4 (370–598), F5 (681–860), and F6 (871–1098) were amplified with PCR and cloned into pBlueScript vector immediately downstream of the T7 promoter and upstream of a XbaI (constructs RepA and F3) or BamHI (constructs F1, F2, F4, F5 and F6) restriction site.
In Vitro Transcription
Plasmids were linearized with the appropriate restriction enzyme (NEB, high fidelity), extracted twice with phenol-chloroform, precipitated with ethanol, washed with 70% ethanol, and dissolved in TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM Na-EDTA). In vitro transcription was carried out using 7 µg/mL template DNA in 15 mM MgCl2, 40 mM Tris-HCl pH 8.0, 2 mM spermidine, 10 mM NaCl, and 0.01% Triton X-100. Reactions were supplemented with 400 U/mL of RNase inhibitor (Roche) and T7 RNA polymerase, and incubated for 2 hours at 37 °C. Following transcription, 1.2 mM CaCl2 and DNase (20 U/mL; Ambion) were added sequentially and incubated for 30 min at 37°C. Finally, proteinase K (0.3 mg/mL; Ambion) was added and incubated for 30 min at 37 °C.
Purification by centrifugal filters and size exclusion chromatography
Immediately after transcription, reactions were diluted five times with HEK buffer (25 mM K-HEPES pH 7.0, 0.1 mM Na-EDTA, 150 mM KCl) and applied to Amicon Ultra-0.5 (Millipore) centrifugal filters (30, 50 or 100 kDa MW cut-off were selected depending on the size of the RNA transcript). Four consecutive filtrations were performed at 5400 rpm for four minutes at room temperature.
After purification on the centrifugal filters, size exclusion chromatography (SEC) was performed at room temperature using HEK as a running buffer. Fragment constructs F4-F6 were purified using Superdex 200 10/300 GL (GEHealthcare). The other constructs were purified using Tricorn columns (GE Healthcare) that were self-packed with Sephacryl S400 (for constructs F1-F3), or S500 (for full-length RepA). Throughout the entire duration of these experiments, RNA transcripts were purified in the aforementioned manner and stored at room temperature only.
Sedimentation velocity analytical ultracentrifugation (SV-AUC)
Sedimentation velocity analytical ultracentrifugation (SV-AUC) experiments were performed using a Beckman XL-1 centrifuge with An-60 Ti rotor (Beckman Coulter). Prior to centrifugation, RNA was supplemented with 25 mM K-HEPES pH 7.0, 150 mM KCl, 0.1 mM Na-EDTA, and appropriate MgCl2 concentrations as indicated in the results section, and incubated for 45 minutes at 37°C. RNA concentrations were adjusted to obtain an initial absorption value of 0.4 at 260 nm on the instrument. All experiments were performed at 20 °C at 25,000 rpm, and independently repeated once. Data were analyzed using the continuous c(s) distribution model as implemented in Sedfit43,44. Hydrodynamic radii (Rh) were calculated assuming a partial specific volume of 0.53 cm3/g and a hydration of 0.59 g/g in Sedfit36. Hill plots were drawn for each set of data in Prism (GraphPad).
It’s important to note that the homogeneity of RNA samples is not evaluated by the shape of the sedimentation curve itself, but by mathematically fitting the calculated molecule radii to the Hill equation in order to obtain a value of K1/2 Mg. At the Mg2+ concentration indicated by a particular K1/2 Mg value, 50% of the RNA molecules are compacted. Therefore, we chose 15 mM Mg2+ (more than 3 times of the K1/2 Mg value) to obtain a completely compact and monodisperse form of RepA.
Chemical probing
Prior to the chemical probing experiment, freshly purified RNA (20 pmol) was supplemented with HEMK buffer (25 mM K-HEPES pH 7.0, 0.1 mM Na-EDTA, 150 mM KCl, 15 mM MgCl2), and incubated at 37 °C for 45 minutes.
Chemical reagent titration
We performed reagent titration for both SHAPE and DMS probing prior to the actual structural characterization experiments. A range of final 1M7 or DMS concentrations (1–10 mM of 1M7 or 0.038% - 0.15% of DMS) was tested in the titration experiments. The 1M7/DMS lowest concentration that produced the saturated modification signals was chosen for the probing experiments.
Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE)
The folded RNA sample was divided equally into two tubes. The positive reaction was initiated by the addition of 1-methyl-7-nitroisatoic anhydride (1M7)45, which was synthetized in house and dissolved in anhydrous DMSO to a final concentration of 2.5 mM, and the negative control reaction was initiated by an equal amount of pure DMSO. Samples were incubated for 5 minutes at 37°C, precipitated with pure ethanol, and washed twice with 70% ethanol. Pellets were re-suspended in HEK buffer. Reverse transcription (RT) was performed with 1 pmol of RNA sample following the same protocol as previously described12. SHAPE reactivities of nucleotides within full-length RepA and fragments F1–6 were measured under the same reaction conditions.
Chemical probing by DMS
Methylation with dimethyl sulfate (DMS, SigmaAldrich) was conducted at room temperature for 10 minutes. Prior to reaction, 1 µl of DMS was diluted on ice with 129 µl ethanol and used as a 10X reaction buffer stock. For control samples, corresponding amounts of pure ethanol were used. Reactions were stopped by addition of 54.4 µl stop mix (5% 2-mercaptoethanol in ethanol). Samples were prepared for reverse transcription as previously mentioned.
Sequencing and structure mapping by capillary electrophoresis
Synthesis of fluorescent RT primers, preparation of sequencing ladders, and capillary electrophoresis were performed following the same protocol as previously described12.
Data processing, normalization and error assessment
All capillary data sets were analyzed using ShapeFinder as described previously46. For secondary structure prediction, the SHAPE reactivity profiles of all nucleotides were obtained by subtracting the peak areas of background (–) from the peak areas of the corresponding (+) reactions, and the data was then normalized as previously described47. DMS probing data was processed in the same manner, except that DMS reactivity of adenosines and cytosines were normalized separately due to their inherent reactivity difference48. All the SHAPE and DMS experiments in this study were independently repeated three times, on three different RNA samples. Reproducibility among triplicates was measured by Pearson’s correlation coefficient (rp).
For the 3S shotgun approach, normalized SHAPE reactivity profiles were obtained using the peak areas of (+) reactions without subtracting the background (–). Nucleotides with high peak areas in the background were identified as artificial reverse transcriptase stops and were eliminated from the analysis. The significance of structural similarities between each fragment and the corresponding region in full-length RepA was evaluated by Pearson’s correlation coefficient. The standard error in correlation coefficients was calculated using the ‘bootstrap’ function in MATLAB (Mathworks), with 1000 randomly generated datasets and removing 10% of the nucleotides.
Structure determination
To generate the RepA secondary structure map using the software RNAStructure (http://rna.urmc.rochester.edu/RNAstructureWeb), SHAPE reactivity was used to provide pseudo-energy constraints49. Resulting structures were manually evaluated for their match with DMS probing data. Resampling analysis and confidence estimation31 were performed using MATLAB (Mathworks). A total of 1000 ‘mock data sets’ were generated by randomly removing 10% of the nucleotides and setting them as ‘no data’. All the mock data sets were then used as pseudo-constraints to predict secondary structures with RNAStructure.
Conservation and covariance analysis
Multiple sequence alignments of 56 mammalian sequences comprising the full or partial RepA/Xist gene were downloaded from UCSC Genomic Browser38. Covariance analysis was performed with Infernal 1.139 in the following manner: first, a covariance model was built using cmbuild (Infernal 1.1) and a multiple sequence alignment of 10 homologous sequences (sequence similarity range: 95%−75%) that include murine Xist. Second, the covariance model was calibrated with cmcalibrate, followed by a homolog search using cmsearch on all the downloaded sequences. Covariance in the resulting alignment was calculated using R2R50 with 15% tolerance for non-canonical base pairs.
UV Crosslinking
Prior to crosslinking, full-length RepA was internally labeled by in vitro transcription in the presence of [α−32P] UTP, natively purified using Amicon filtration (100 kDa MW cut-off) and folded by incubating in HEMK buffer (25 mM K-HEPES pH 7.0, 0.1 mM Na-EDTA, 150 mM KCl and appropriate MgCl2 concentrations as indicated in the following text) at 37 °C for 45 minutes. Analytical UV crosslinking was carried out with 20 nM RNA in 30 ul of HEMK buffer (0 to 15 mM MgCl2 concentrations). At MgCl2 concentrations higher than 10 mM, poor resolution of corresponding RNA bands was observed on the gel. In preparation for downstream primer extension, crosslinking reactions were conducted on samples of 200 nM RNA in 450 ul HEMK buffer (5 mM MgCl2). Samples were distributed as drops of 30 ul each on a 96-well plate lid (Corning Costar) and then exposed to short wavelength UV light (UVP Handheld UV lamp, 6W) at a distance of 15 cm for 10 minutes. Immediately after irradiation, samples were mixed with equal volumes of 2X urea loading buffer (22.6 M Urea, 0.16% (w/v) xylene cyanol/bromophenol blue, 16% (w/v) sucrose, 80 mM Tris-HCl, pH 7.5, 1.6 mM EDTA, pH 8.0). Crosslinked RNAs were analyzed on a 4% polyacrylamide gel (29:1 acrylamide: bisacrylamide ratio) containing 8.3 M urea and TBE (90 mM Tris, 90 mM boric acid, and 2 mM EDTA, pH 8.2).
Primer extension mapping of intramolecular RNA crosslinks
To map crosslinking sites, labeled crosslinked RNA was isolated from gel slices using the ‘crush and soak’ method, and then precipitated with ethanol. The crosslinked nucleotide residues were mapped by primer extension with avian myeloblastosis virus (AMV) reverse transcriptase (ThermoFisher Scientific), using 16 different DNA primers (5’ end labeled with [γ−32P] ATP) complementary to different regions of RepA (Supplementary Table 1). For the primer extension assay, template RNA (i.e. RNA containing different RNA crosslinks or controls) was heated to 95 ºC for 1 min and then snap cooled on ice. DNA primer was mixed with the template and the mixture was allowed to incubate on ice for 10 min. The template/primer complex was then incubated with reverse transcriptase AMV and appropriate buffer and supplements that were provided along with the enzyme (ThermoFisher Scientific) at 46 ºC for 50 min. Dideoxy sequencing ladders were obtained using a cycle sequencing kit (Affymetrix) and plasmid-containing full-length RepA sequence. Reverse transcription products, along with corresponding sequencing ladders, were analyzed on a 8% polyacrylamide gel (29:1 acrylamide: bisacrylamide ratio) containing 8.3 M urea and TBE (90 mM Tris, 90 mM boric acid, and 2 mM EDTA, pH 8.2).
RNA Modeling
A tertiary structure model of subdomains in D2 and D3 was constructed with RNAComposer41 using the secondary structure mapped here and the crosslinks we identified as distance constraints (6±2Å). The input consists of the following nucleotides 494–508 and 632–776 in D2, and nucleotides 1272–1304 and 1349–1406 in D3. All the remaining nucleotides in D2 and D3 were removed and the two subdomains were connected using a polyA linker that was 20 nt in length (note: increasing the linker length to 30 nt or 40 nt didn’t affect the final model). Modeling was performed with ‘batch mode’ and the final model was chosen such that the distance between crosslinked nucleotides is < 6Å. Further, for comparison with the final model, a model of subdomain D2 was built in the absence of D3 using only secondary structure as the input (without any distance constraints).
Shannon Entropy Calculation
The SHAPE-directed base-pairing probabilities found by RNAstructure’s Partition Function RNA were used for calculations of Shannon entropy. The Shannon entropy of each nucleotide was calculated as described32:
where Si is the entropy of nucleotide i and and Pi,j is the probability of nucleotides i and j base-pairing (which is the probability of nucleotide i being unpaired when i = j).
Data availability
All chemical probing data and MATLAB scripts for data analyses are available upon request.
Supplementary Material
Acknowledgements
We acknowledge Dr. Matthew Simon (Yale University) for sharing the in vivo DMS probing data of Xist, and Dr. Erik Jagdmann for the synthesis of 1M7. We thank Dr. Thayne Dickey, Chen Zhao, Dr. Olga Fedorova, and all other members of the Pyle lab for constructive discussion and critical reading of the manuscript. This project was supported by the National Institute of Health (RO1GM50313). A.M.P. is an Investigator and F.L. is a Postdoctoral Fellow of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflicts of interest.
Competing financial interests
The authors declare no competing financial interests.
References:
- 1.Flicek P et al. Ensembl 2014. Nucleic Acids Res 42, D749–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wapinski O & Chang HY Long noncoding RNAs and human disease. Trends Cell Biol 21, 354–61 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Gutschner T & Diederichs S The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol 9, 703–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JT Epigenetic regulation by long noncoding RNAs. Science 338, 1435–9 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Sauvageau M et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2, e01749 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Froberg JE & Lee JT Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem Sci 39, 35–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinn JJ & Chang HY Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17, 47–62 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Wan Y et al. Genome-wide measurement of RNA folding energies. Mol Cell 48, 169–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark MB et al. Genome-wide analysis of long noncoding RNA stability. Genome Res 22, 885–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding Y et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Novikova IV, Hennelly SP & Sanbonmatsu KY Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res 40, 5034–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Somarowthu S et al. HOTAIR forms an intricate and modular secondary structure. Mol Cell 58, 353–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown CJ et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71, 527–42 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Brockdorff N et al. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71, 515–26 (1992). [DOI] [PubMed] [Google Scholar]
- 15.Lyon MF Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190, 372–3 (1961). [DOI] [PubMed] [Google Scholar]
- 16.Galupa R & Heard E X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev 31, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Nesterova TB et al. Characterization of the genomic Xist locus in rodents reveals conservation of overall gene structure and tandem repeats but rapid evolution of unique sequence. Genome Res 11, 833–49 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wutz A, Rasmussen TP & Jaenisch R Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30, 167–74 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Zhao JS, B.K.; Erwin JA.; Song JJ.; Lee JT Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 322, 7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarma K et al. ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 159, 869–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu C et al. Systematic discovery of Xist RNA binding proteins. Cell 161, 404–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHugh CA et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moindrot B et al. A Pooled shRNA Screen Identifies Rbm15, Spen, and Wtap as Factors Required for Xist RNA-Mediated Silencing. Cell Rep 12, 562–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monfort A et al. Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep 12, 554–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CK et al. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 354, 468–472 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Duszczyk MM, Zanier K & Sattler M A NMR strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a Xist RNA A-repeat. Nucleic Acids Res 36, 7068–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maenner S et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol 8, e1000276 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duszczyk MM, Wutz A, Rybin V & Sattler M The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA 17, 1973–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang R, Moss WN, Rutenberg-Schoenberg M & Simon MD Probing Xist RNA Structure in Cells Using Targeted Structure-Seq. PLoS Genet 11, e1005668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Z et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramachandran S, Ding F, Weeks KM & Dokholyan NV Statistical analysis of SHAPE-directed RNA secondary structure modeling. Biochemistry 52, 596–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathews DH Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 10, 1178–90 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takamoto K et al. Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol 343, 1195–206 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Pyle AM, Fedorova O & Waldsich C Folding of group II introns: a model system for large, multidomain RNAs? Trends Biochem Sci 32, 138–45 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Luna MT & Miranda-Rios J Riboswitch folding: one at a time and step by step. RNA Biol 5, 20–3 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Su LJ, Brenowitz M & Pyle AM An Alternative Route for the Folding of Large RNAs: Apparent Two-state Folding by a Group II Intron Ribozyme. Journal of Molecular Biology 334, 639–652 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Novikova IV, Dharap A, Hennelly SP & Sanbonmatsu KY 3S: shotgun secondary structure determination of long non-coding RNAs. Methods 63, 170–7 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Kent WJ et al. The human genome browser at UCSC. Genome Res 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nawrocki EP & Eddy SR Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris ME & Christian EL RNA crosslinking methods. Methods Enzymol 468, 127–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Popenda M et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res 40, e112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.da Rocha ST et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol Cell 53, 301–16 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–19 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown PH & Schuck P Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J 90, 4651–61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortimer SA & Weeks KM A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc 129, 4144–5 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Vasa SM, Guex N, Wilkinson KA, Weeks KM & Giddings MC ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA 14, 1979–90 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGinnis JL, Duncan CD & Weeks KM High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol 468, 67–89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouskin S, Zubradt M, Washietl S, Kellis M & Weissman JS Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Low JT & Weeks KM SHAPE-directed RNA secondary structure prediction. Methods 52, 150–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weinberg Z & Breaker RR R2R--software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics 12, 3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All chemical probing data and MATLAB scripts for data analyses are available upon request.