

Abstract
The Ca2+-independent phospholipases, designated as group VI iPLA2s, also referred to as PNPLAs due to their shared homology with patatin, include the β, γ, δ, ε, ζ, and η forms of the enzyme. The iPLA2s are ubiquitously expressed, share a consensus GXSXG catalytic motif, and exhibit organelle/cell-specific localization. Among the iPLA2s, iPLA2β has received wide attention as it is recognized to be involved in membrane remodeling, cell proliferation, cell death, and signal transduction. Ongoing studies implicate participation of iPLA2β in a variety of disease processes including cancer, cardiovascular abnormalities, glaucoma, and peridonditis. This review will focus on iPLA2β and its links to male fertility, neurological disorders, metabolic disorders, and inflammation.
Keywords: iPLA2β-derived lipids, signaling, membrane remodeling, immune responses, diabetes
I. iPLA2β: The Beginning
Phospholipase enzymes catalyze hydrolysis of phospholipid substrates and Figure 1 illustrates the site of action of various classes of phospholipases. Phospholipases C cleave the diglyceride moiety from the phospho-headgroup. Phospholipases D cleave the polar head group from the phosphatidic acid moiety. Phospholipases A1 hydrolyze the sn-1 fatty acid substituent from the glycerol backbone to yield a free fatty acid and a 1-lysophospholipid, and phospholipases A2 (PLA2) cleave the sn-2 substituent from the glycerol backbone to yield a free fatty acid and a 2-lysophospholipid. The PLA2s include (s)ecretory, (c)ytosolic and Ca2+-(i)ndependent PLA2s and their products of action on the PC species are a fatty acid, such as arachidonic acid, and 2-lysophosphatidylcholine (LPC). Downstream effects of PLA2 action may arise from intrinsic actions of the fatty acid product or of its metabolites, such as oxygenated eicosanoids, or from actions of the lysophospholipid products or its metabolites. In the case of LPC, such metabolites could include the bioactive product lysophosphatidic acid (LPA), and an alkyl ether LPC species could also be acetylated to yield the lipid mediator platelet activating factor (PAF), e.g., 1-O-hexadecyl-2-acetyl-sn-glycero-3-phosphocholine. Other downstream effects of PLA2 action can arise from remodeling or loss of the phospholipid substrate. With Group VI PLA2, examples of downstream effects attributable to each of these possibilities are thought to occur.
Figure 1. The phospholipase A2 reaction and actions of its products.
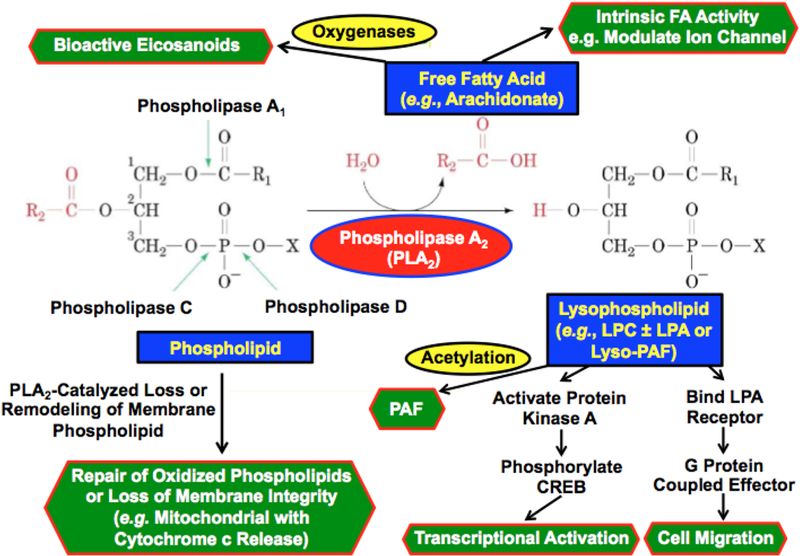
Phospholipids have a glycerol backbone and an esterified phosphate moiety at the sn-3 position that may also be esterified to a polar head group, such as choline, ethanolamine, glycerol, serine, inositol or phosphatidylglycerol. In the sn-2 position, a fatty acid moiety is esterified to the glycerol backbone. In the sn-1 position, there may be a second esterified fatty acid residue or there may be a saturated ether linkage to a fatty alcohol residue or a vinyl ether linkage to a fatty aldehdye residue. Phospholipases C cleave the diglyceride moiety from the phospho-headgroup. Phospholipases D cleave the polar head group from the phosphatidic acid moiety. Phospholipases A1 hydrolyze the sn-1 fatty acid substituent from the glycerol backbone to yield a free fatty acid and a 1-lysophospholipid, and Phospholipases A2 (PLA2) cleave the sn-2 substituent from the glycerol backbone to yield a free fatty acid and a 2-lysophospholipid. The products of PLA2 action on the phosphatidylcholine (PC) species illustrated in the figure are arachidonic acid and 2-lysophosphatidylcholine (LPC). Downstream effects of PLA2 action may arise from intrinsic actions of the fatty acid product or of its metabolites, such as oxygenated eicosanoids, or from actions of the lysophospholipid products or its metabolites. In the case of LPC, such metabolites could include the bioactive product lysophosphatidic acid (LPA), and an alkyl ether LPC species could also be acetylated to yield the lipid mediator Platelet Activating Factor (PAF), e.g., 1-O-hexadecyl-2-acetyl-sn-glycero-3-phosphocholine. Other downstream effects of PLA2 action can arise from remodeling or loss of the phospholipid substrate. With Group VIA PLA2 (iPLA2β) examples of downstream effects attributable to each of these possibilities are thought to occur. The free fatty acid arachidonic acid derived from iPLA2β action in pancreatic islet β-cells is thought to activate voltage-operated K+ channels83,153. A 12/15lipoxygenase product from iPLA2β-derived free arachidonic acid is thought to activate a RhoA kinase pathway to vascular smooth muscle cell activation in diabetic vasculopathy123. LPC derived from iPLA2β action is thought to participate in CREB-mediated activation of transcriptional responses to viral infection in macrophages154, and LPA derived from iPLA2β action is thought to stimulate macrophage migration into sites of vascular inflammation in diabetes111. Ether linked LPC derived from iPLA2β action appears to provide substrate for PAF biosynthesis in inflamed endothelial cells122,131,155. iPLA2β also appears to participate in the repair of oxidized cardiolipin species in mitochondria by excising oxygenated fatty acid residues, and with overwhelming mitochondrial injury this may lead to cytochrome c release and initiation of apoptosis67–69.
Ca2+-independent PLA2 (iPLA2) enzymes.
Although mammalian Groups I, II, III, V, X and XII sPLA2 enzymes all require Ca2+ for catalysis, and the Group IV cPLA2 requires Ca2+ for membrane-association, Ca2+-independent PLA2 activities had been described in some cells and tissues before the proteins responsible for these activities were identified1,2 (Table 1). The Greek letter designations for the iPLA2 isoforms were first suggested by Mancuso et al. in 20003 but have subsequently come into wide usage, including in recent reviews of the PLA2 area4–7. All of the iPLA2 isoforms are members of the Group VI PLA2 family in the numbering system proposed by Dennis et al.4,7, and distinct family members are designated by letters of the Roman alphabet in the order in which they were discovered in mammals. Group VIA PLA2 was discovered first in 19978−10, and Group VIB was described next in 20023. By that time, Greek letter designations for cPLA2 isoforms of the Group IV PLA2 family has come into widespread usage, and the first discovered was designated cPLA2α11 and corresponded to Group IVA PLA24,7. The next two recognized cPLA2 paralogs were designated cPLA2β and cPLA2γ11, respectively, and corresponded to Group IVB and Group IVC PLA24,7. Subsequently discovered Group IV paralogs were designated cPLA2 δ, ε, and ζ12 and corresponded to Group IVD, IVE, and IVF PLA2. Mancuso et al.3 proposed Greek letter designations of the Group VI PLA2 family members to mimic the conventions that had been applied to the Group IV PLA2 members11. The proposal of Mancuso et al. was that these designations be based on the first recognized phospholipase with both a GXSXG lipase consensus motif and a GXGXXG nucleotide-binding motif, which was the potato enzyme patatin13. The first recognized mammalian Group VI PLA2, i.e. Group VIA PLA2, was thus designated iPLA2β, and Group VIB PLA2 was designated iPLA2γ3. Subsequently recognized Group VI PLA2 family members14,15 were designated iPLA2δ, iPLA2ε, iPLA2ζ, and iPLA2η14, and corresponded to Group VI C, D, E, and F PLA24,7, respectively. Because the plant enzyme patatin was considered the founding enzyme of the iPLA2 family but is not assigned a letter designation in the Group VI PLA2 naming conventions, there is a discrepancy in the ordinal positions of the letter designations of the mammalian iPLA2/Group VI PLA2 family members in the Greek and Roman alphabets, respectively. The relationships between these designations and the patatin-like phospholipase domain-containing protein numbering system16,17 are specified in Table 1.
Table 1.
Patatin-Like Phospholipase (PNPLA) Protein Family Members
|
PNPLA
Member |
PLA2
Group |
iPLA2
Greek Letter |
Other
Designations |
Tissues | Possible Functions | References |
|---|---|---|---|---|---|---|
| PNPLA1 | Not Applicable |
Not Applicable |
Not Applicable |
Epidermal Keratinocytes |
Acylceramide Synthase | 144,145 |
| PNPLA2 | Group VIE PLA2 |
iPLA2ζ | TTS-2.2, Adipose Triglyceride Lipase (ATGL), Desnutrin |
Adipose, Heart | First step of TAG hydrolysis | 6 |
| PNPLA3 | Group VID PLA2 |
iPLA2ε | Adiponutrin | Adipose, Liver | May Regulate Hepatic Fat Content |
6,146 |
| PNPLA4 | Group VIF PLA2 |
iPLA2η | GS2 | Keratinocytes, Adipose, Liver, Muscle, Heart |
May Regulate Cell all- trans retinoic acid levels |
6 |
| PNPLA5 | NA | NA | GS2-like | Adipose, Lung, Brain, Pituitary |
Unknown | 6 |
| PNPLA6 | Group VIC PLA2 |
iPLA2δ | Neuropathy Target Esterase (NTE) |
Nervous System |
Lysophospholipase Activity Maintains Axonal Integrity |
6 |
| PNPLA7 | NA | NA | NTE-Related Esterase (NRE) |
Adipose, pancreas, muscle, prostate | Lysophospholipase Activity May Modulate Energy Metabolism |
6 |
| PNPLA8 | Group VIB PLA2 |
iPLA2γ | NA | Heart Predominant, Nervous System, Adipose, Other Tissues |
Mitochondrial Bioenergetics Modulation |
6,147–152 |
| PNPLA9 | Group VIA PLA2 |
iPLA2β | NA | Ubiquitous, Especially Testes, Nervous System, Pancreatic Islets |
Spermatozoa Motility, Acrosome Exocytosis, Insulin Secretion, Apoptosis, Autophagy, Modulating Ion Channels, Axon Maintenance, Mitochondrial Repair |
6 |
Ca2+-independent Group VIA PLA2 in rodents.
In myocardium, P388D1 macrophage-like cells, and pancreatic islet β-cells, such Ca2+-independent PLA2 activities shared the properties of activation by ATP and susceptibility to inhibition by a bromoenol lactone suicide substrate designated BEL18–23. This led Jones et al. at Genetics Institute to survey a variety of cells for Ca2+-independent PLA2 activity10. This activity was designated iPLA2 to denote independence of Ca2+ for catalysis, and such iPLA2 activity was found to be widely distributed10.
The Genetics Institute group then focused on Chinese Hamster Ovary (CHO) cells10, which expressed abundant iPLA2 activity and could be readily grown in large quantities. The protein responsible for iPLA2 activity was purified from the cytosolic fraction of 500 L of CHO cells by sequential chromatographic analyses involving ion exchange, hydrophobic interaction, heparin affinity, chromatofocusing, and gel filtration steps to yield an 85 kDa protein upon SDS-PAGE analyses, although catalytic activity migrated with an apparent molecular mass of 250–450 kDa on gel filtration chromatography. This was taken to suggest that the active form of iPLA2 might be a multimer. The 85 kDa SDS-PAGE band was excised and digested with trypsin, and tryptic peptides isolated by reverse-phase HPLC, were sequenced by Edman degradation. Their sequences were used to design degenerate oligonucleotide probes with which to screen a CHO cell cDNA library to obtain full-length clones that were then sequenced. The cDNA encoded a protein with a calculated molecular mass of 85 kDa containing 752 amino acid residues that included a GXSXG serine lipase consensus motif (GTS465TG) and eight strings of an ankyrin-like repetitive motif. The iPLA2 sequence lacked homology with cPLA2 or sPLA2 enzymes. Northern blotting analyses revealed ubiquitous tissue expression of iPLA2 mRNA, with the highest levels in testis and liver.
The iPLA2 cDNA was subcloned into a mammalian expression plasmid and transiently expressed in COS (monkey kidney-derived fibroblast-like) cells, which resulted in over a 300-fold rise in Ca2+-independent PLA2 activity. A truncated form of iPLA2 that lacked the N-terminal 150 amino acids and the ankyrin-repeat (AR) region lacked iPLA2 activity upon expression as a FLAG epitope fusion protein, as did fusion proteins lacking C-terminal sequence from residue 416 to 752. An S465A mutant lacked catalytic activity when expressed as a FLAG fusion protein, but an S252A mutant was fully active, consistent with S465 of the GTS465TG sequence representing the active site nucleophile. Studies with model substrates indicated that iPLA2 was selective for the sn-2 over sn-1 fatty acid substituent. Apparent selectivity for the identity of the sn-2 substituent depended on the mode of substrate presentation, but there was no significant preference for any particular fatty acid. Recombinant iPLA2 was found to hydrolyze sn-2 acyl linkages in phospholipid substrates with sn-1 O-alkyl ether linkages readily, including platelet activating factor (PAF) with its short chain sn-2 acetyl substituent10, including platelet activating factor (PAF) with its short chain sn-2 acetyl substituent. In contrast, cPLA2 and Group IB, Group IIA, Group V sPLA2 enzymes do not exhibit PAF acetylhydrolase activity, although PAF is a substrate for Group X sPLA224. Expressed iPLA2 did not exhibit a preference for choline over ethanolamine substrate head-groups, but PI substrates were hydrolyzed 5-fold more slowly than the corresponding PC species, while PA substrates were hydrolyzed 20-fold more rapidly than the corresponding PC species.
Reports of cloning of iPLA2 from mouse P388D1 cells8 and from rat pancreatic islet β-cells9 appeared virtually simultaneously with the report on cloning of the hamster iPLA2 and exploited the CHO cell sequence10. An antiserum generated against the CHO cell iPLA2 recognized a purified P388D1 cell protein of corresponding molecular mass on SDS-PAGE analysis, and the BEL concentration-dependences for inhibition of the CHO cell and P388D1 cell iPLA2 activities were identical8. Both iPLA2 proteins were also labeled by [3H]BEL. Using PCR primers designed from the CHO cell iPLA2 sequence and cDNA prepared from P388D1 cell mRNA as template, the nucleotide sequence of the P388D1 cell iPLA2 mRNA was determined, and the mouse nucleotide sequence exhibited 92% homology with the hamster sequence. There was 95% sequence identity at the amino acid level between the CHO cell and P388D1 cell iPLA2 proteins.
To clone the iPLA2 cDNA from a rat pancreatic islet cDNA library, an oligonucleotide probe was generated by PCR using HIT-T15 insulinoma cell RNA as template9. HIT-T15 cells are a proliferating β-cell line of hamster origin that also express an iPLA2 similar to that in pancreatic islets25. A [32P]-labeled form of the resultant 820 bp PCR product was generated by randomly primed labeling and used to screen an islet cDNA library that yielded five clones that were sequenced from both 5’- and 3’-ends and found to contain a single long open reading frame predicted to encode a protein with 751 amino acid residues and a calculated molecular mass of 83,591 daltons. The islet iPLA29 exhibited 90% identity of nucleotide sequence and 95% identity of amino acid sequence with the CHO cell iPLA210. The deduced amino acid sequence included a region (residues 150–414) containing 8 strings of a repetitive sequence motif of about 33 residues in length similar to that in ankyrin-related proteins9. One absolutely conserved region of sequence is that between His421 and Glu551, which is identical to the corresponding region in the CHO cell iPLA2 and flanks and includes the GXSXG serine lipase consensus sequence of GTS464TG. The islet iPLA2 cDNA was subcloned into a mammalian expression vector and transiently expressed in COS-7 cells and CHO cells, which resulted in the appearance of Ca2+-independent PLA2 activity that was sensitive to inhibition by BEL with a concentration-dependence identical to that for the native iPLA2 activity in HIT cell and islet cytosol. A similar pH dependence and stimulation by ATP were also observed for the iPLA2 activity expressed from islet iPLA2 cDNA and that in native HIT cell or islet cytosol. Studies with FACS-purified populations of islet β-cells and non-β- (predominantly α-) cells indicated that iPLA2 mRNA resided specifically in β-cells20.
These observations8–10 established that the iPLA2 enzymes cloned from CHO cells, P388D1 cells, and pancreatic islets represented species homologs of the same Ca2+-independent PLA2 enzyme from hamster, mouse, and rat, respectively, and these cytosolic enzymes were designated Group VI PLA2 members4,6,7. They are now designated Group VIA PLA2 or iPLA2β enzymes, distinguishing them from subsequently described membrane-associated Ca2+-independent PLA2 activity3, designated Group VIB PLA2 or iPLA2γ enzymes. Later observations indicated that the R-BEL enantiomer selectively inhibited Group VIB PLA2 (iPLA2γ) and the S-BEL enantiomer selectively inhibited Group VIA PLA2 (iPLA2β)5, providing means to distinguish the role of each in biological processes.
Human Group VIA PLA2 enzymes, splice variants, and the Group VIA PLA2 gene.
Two groups independently cloned the human Group VIA PLA2 through different approaches26,27. In one, PCR experiments were performed using total RNA from two monoclonal B-lymphocyte cultured cell lies as template26. Sequences used for design of the RT-PCR primers were obtained from a TBLASTN database search using amino acid sequence derived from the CHO cell Group VIA PLA2 sequence. Two human EST clones (46450 and 30643) thought to represent the human form of Group VIA PLA2 exhibited 65–84% identify with the CHO cell sequence. The Group VIA PLA2 primers amplified at least three distinct products representing splice variants. To obtain a full-length cDNA clone, 5’-RACE experiments were performed with cDNAs from several human tissues, including testes, which resulted in amplification of a 2.2 kb DNA fragment that was then subcloned. Five different clones were obtained a sequenced, and three of them had identical sequence and contained an open reading frame encoding the human Group VIA PLA2 sequence. One clone had an insertion before the catalytic domain that resulted in a frame-shift that changed the reading frame and yielded a truncated sequence without the catalytic center designated ankyrin-iPLA2-1 to denote that it coded only for the AR domain (Fig. 2). Another clone contained two inserts that again resulted in a truncated sequence without the catalytic domain and was designated ankyrin-iPLA2-2. The full-length human Group VI PLA2 cDNA sequence obtained by combining the 3’-end of EST 46450 with the appropriate 5’-RACE fragment exhibited 90% overall identity to the hamster, rat, and mouse sequences with the major difference that the human sequence had a 54 amino acid insert that interrupted the last AR in the rodent sequences. This insert was also present in the ankyrin-iPLA2-1 and −2 sequences. An additional isoform contained the ARs and the catalytic domain but had a truncated C-terminus, and all four human isoforms appeared to represent variants produced by alternative splicing of the transcript. Expression of the full-length human Group VIA PLA2 cDNA in COS cells resulted in a substantial increase in Ca2+-independent PLA2 activity, while expression of ankyrin-iPLA2-1 cDNA alone did not. Co-expression of full-length human Group VIA PLA2 cDNA with ankyrin-iPLA2-1 cDNA resulted in less Ca2+-independent PLA2 activity than with the full-length cDNA alone, which was taken to suggest that hetero-oligomer formation might represent a means to regulate and diminish Group VIA PLA2 activity. All of the observed human Group VIA PLA2 cDNA sequences contained the 54 amino acid insert interrupting the last AR, and no short human isoform corresponding to the rodent Group VIA PLA2 cDNA sequences was observed.
Figure 2. Schematic diagram of iPLA2β pre-mRNA and transcripts derived from it by alterative splicing.
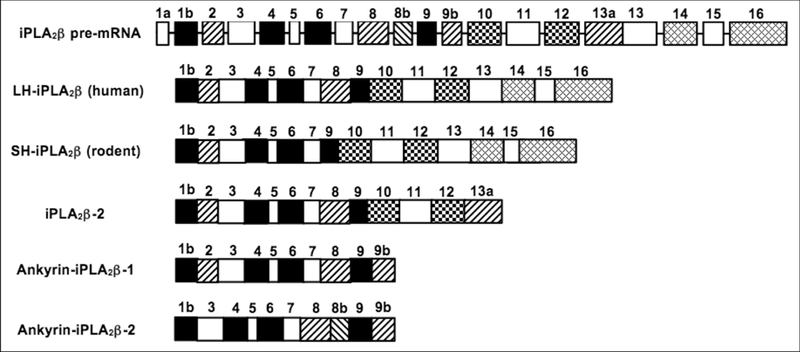
Ma et al. cloned the human iPLA2β gene by screening a human Lambda FIX II genomic library and determined the gene structure by combining sequencing and PCR approaches. Larsson-Forsell et al.28 analyzed the human iPLA2β gene with two genomic clones (HS228A9 and HS447C4). Both groups found that the human iPLA2β gene spans about 70 kb and consists of at least 17 exons, ranging from 74 to 811 bp in size, and 16 introns, ranging from 0.2 kb to 23 kb. Ma et al. designated the 5′-untranslated region (UTR) as exon 1a and considered the coding region to begin with exon 1b27. Larsson-Forsell designated the 5’-UTR as exon 1 and considered the coding region to begin with exon 228. Exons 1b-16 in the report by Ma et al.27 thus correspond to exons 2–17 in that by Larson-Forsell et al.28. The convention of Ma et al. 27 is used here. The human genomic clone HS447C4 contains exon 1a, and clone HS228A9 contains exon lb through exon 16. Exon 16 contains the iPLA2β translational stop codon, the 3’-UTR, and the polyadenylation signal. At exon/intron boundaries, the 5’-donor and 3’-acceptor sequences conform to splicing sites consensus sequences27,28. Human islets and human U937 promonocytic cells express mRNA species encoding two iPLA2β isoforms, designated LH-iPLA2β and SH-iPLA2β, respectively27. The latter corresponds to iPLA2β isoforms cloned from rodent species8–10. A 162-bp in-frame insertion in the eighth AR of SHiPLA2β corresponds to exon 8 of the human iPLA2β gene, indicating that mRNA encoding the SHiPLA2β isoform arises from an exon-skipping mechanism of alternative splicing. Splicing variants of human iPLA2β have been identified from EST clones that reflect insertions of 52, 53, and 168 bp, respectively, that do not occur in the transcripts encoding LH- and SH-iPLA2β isoforms26. These insertions arise from introns designated E8b, E9b, and E13a. The exon/intron boundary sequences of these alternative-splice sites demonstrate consensus splicing site sequences in the corresponding introns27,28. These alternative splicing events may yield three putative transcripts designated iPLA2β2, Ankyrin-iPLA2β−1, and Ankyrin-iPLA2β−2 in the figure. These putative transcripts contain a polyadenylation signal encoded within exon 16 and therefore acquire a poly(A) tail required for translational competence. The iPLA2β−2 transcript includes E13a that introduces a premature translational stop codon that results in a C-terminally truncated protein that retains the GXSXG lipase consensus sequence. The Ankyrin-iPLA2β−1 transcript includes E9a, which introduces a premature translational stop codon that results in a C-terminally truncated protein that lacks the GXSXG lipase consensus sequence. The Ankyrin-iPLA2β−2 transcript results from skipping exon 2 and inclusion of E8a and E9a. This transcript encodes a truncated protein that has a deletion near the N terminus and a C-terminal truncation from a premature stop codon within E9a and also lacks the GXSXG lipase consensus sequence28.
In the second approach to cloning the human Group VIA PLA2 cDNA, a human insulinoma cell cDNA library was screened with a [32P]-labeled rat Group VIA PLA2 cDNA probe27. Two clones (NS0C1 and INS-C2) of 1.59 kb and 1.80 kb, respectively, hybridized to the probe and were sequenced. Both clones contained identical 3’-sequences that included a presumptive polyadenylation sequence and a poly(A) tail, and their sequences were identical except for additional 5’-sequence in the longer clone not contained in the shorter clone. Alignment of the rat Group VIA cDNA sequence indicated that both clones contained the 3’-end of the human Group VIA cDNA but that neither contained the full 5’-coding sequence. RNA from human islets was then used as template in PCR experiments with primers designed from the 5’-sequence of rat Group VIA cDNA and from sequence in the INS-C1 and INS-C2 clones. The primer sets were designed to amplify cDNA from the initiator methionine codon at the 5’-end through the region of sequence at the 3’-end recognized by primers designed from the INS-C1 and INS-C2 sequences. A nested primer approach was used with the 3’-end primers to verify the specificity of amplification products. PCR with a given set of primers using human pancreatic islet RNA as template yielded two products. With one primer set, a 1.65 kb PCR band was observed upon agarose gel electrophoresis that was expected based on the rat Group VIA cDNA sequence. In addition, a longer product of 1.85 kb was observed27. The 1.65 kb and 1.85 kb human islet PCR products were subcloned and sequenced, and each contained a 5’-coding sequence that specified an amino acid sequence highly homologous to the N-terminal portion of rat Group VIA PLA2. The 1.65 kb and 1.85 kb sequences were identical except for a 162 bp insert in the longer product that was not contained in the shorter one. Similar PCR experiments using RNA from human U937 promonocytic cells as template indicated that U937 cells also express two distinct Group VIA PLA2 mRNA species, and the lengths of the PCR products corresponded exactly to those from human islet RNA. Upon subcloning and sequencing, the U937 and islet products were found to be identical. Those findings indicate that human islets and U937 cells express mRNA species encoding two distinct isoforms of Group VIA PLA2. No evidence for truncated Group VIA PLA2 species such as those observed in human lymphocyte lines26 was observed with human islet or U937 cell RNA.
The nucleotide and deduced amino acids of the long and short isoforms of human Group VI PLA2 cloned from islets indicated that the shorter isoform was highly homologous to the hamster, mouse, and rat Group VIA PLA2 sequences and that the longer isoform contained a 54 amino acid residue insert interrupting the eighth AR27. This insert is proline-rich, and a BLAST search revealed similarities to the proline-rich middle linker domain of the DAF-3 Smad protein from Caenorhabditis elegans, which is most closely related to mammalian Smad 4. The proline-rich middle linker domains of Smad4, DAF-3, and the human Group VIA insert share a PX5PX8HHPX12Q motif, and this region mediates protein interactions of Smad4 with signaling partners.
The 5’-fragments of human islet Group VIA PLA2 cDNA obtained from RT-PCR experiments with human islet RNA overlapped the 3’-fragments obtained from screening the insulinoma cell cDNA library, and within the region of overlap was an NcoI restriction endonuclease site, although there were no other NcoI sites in the sequences. To obtain cDNA species with the full coding sequences for the long and short human islet Group VIA PLA2, appropriate 5’- and 3’- fragments were digested with NcoI, and ligation reactions were performed. The resultant plasmids were used to transform bacterial host cells and sequenced, and the cDNA species contained the full coding sequences of the human Group VIA PLA2 isoforms and were inserted into vectors for expression in bacteria and in Sf9 insect cells and used to prepare a [32P]-labeled Group VIA PLA2 cDNA to generate a probe for genomic screening. Upon expression in bacteria the short and long isoform cDNA species yielded proteins of the expected 85 kDa and 88 kDa sizes, respectively. Uninfected Sf9 cells exhibited no measurable Ca2+-independent PLA2 activity, but expression of either short or long isoform human islet Group VIA PLA2 resulted in appearance of such activities in cytosol and membranes that were sensitive to inhibition by the iPLA2 suicide substrate BEL. PLA2 activity of the long isoform in both cytosol and membranes was stimulated by ATP, but neither cytosolic nor membranous activity of the short isoform was affected by ATP. Both human Group VIA isoforms were thus catalytically active Ca2+-independent PLA2 enzymes inhibitable by BEL, but they exhibited differential sensitivity to ATP.
To determine the structure of the human Group VIA PLA2 gene a [32P]-labeled cDNA of the long isoform was prepared and used to screen a human genomic DNA library27. Eight genomic clones with overlapping regions of sequence were isolated and analyzed by Southern blotting and restriction endonuclease digestion. The cloned sequence spanned over 60 kb of DNA and included 16 exons representing 5’-untranslated region, the entire coding sequence, and 3’-untranslated region. Intron sizes were estimated from the length of PCR fragments produced by using genomic DNA as template and primers that hybridize with sequences in adjacent exons. Sequences of intron-exon boundaries were determined by comparing the sequences of genomic DNA and cDNA. In each case, the intron sequence at the 5’-boundary of the exon ended in the dinucleotide AG and that at the 3’-boundary of the exon began with the dinucleotide GT, conforming to recognized rules for sequences at such junctions. The 54 amino acid-insert interrupting the last AR in the long isoform of human Group VIA PLA2 was found to correspond exactly to the amino acid sequence encoded by exon 8 of the human gene, indicating that the mRNA encoding the short isoform arises by an exon-skipping mechanism of alternative splicing. Different mechanisms of alternative splicing account for the Group VIA PLA2 truncation variants observed in human B-lymphocyte lines, which contain additional sequence arising from introns that result in premature stop codons that yield proteins that contain the AR region but lack the catalytic domain26. To determine the chromosomal location of the human Group VIA PLA2 gene, a clone identified in screening the human genomic DNA library with long isoform cDNA was biotinylated to generate a probe for Fluorescence In Situ Hybridization (FISH) experiments with human lymphocyte chromosomes27. Human chromosomes were identified from their DAPI (4′,6-diamidine-2′-phenylindole)-banding pattern, which were correlated with the sites of fluorescent signal from the biotinylated probe and indicated that the human Group VIA PLA2 gene resides on chromosome 22. Detailed positional assignment from analyses of multiple photographs indicated that the gene resides in region q13.1 of chromosome 22.
This assignment was confirmed by a different approach based on computational gene identification28. A PAC human genomic library was screened with human Group VIA PLA2 cDNA, and a parallel BLASTN computational search of the GenBank database was performed using the Group VIA PLA2 cDNA sequence. The comparison revealed segments of identical sequences in two genomic clones (HS228A9 and HS447A4). The total coding sequence and the 3’-UTR of the Group VIA PLA2 mRNA was contained in clone HS228A9, and the 5’-UTR was contained in clone HS447C4, both of which had been localized to chromosome 22q13.1 between genetic markers DS426 and DS272. Ad additional non-coding exon was identified in the 5’-untranslated region that had not been appreciated in the earlier study27, resulting in the assignment of 17 exons that included the 162 bp sequence encoding the 54 amino acid-insert in the long human isoform of Group VI PLA2 in exon 928.
II. iPLA2β: Early Links to Male Infertility and Neurological Disorders
iPLA2β and male fertility.
In early studies of iPLA2β physiological functions, its gene was disrupted by homologous recombination to generate mice that do not express iPLA2β29. Heterozygous iPLA2β+/− breeding pairs yielded a Mendelian 1:2:1 ratio of iPLA2β+/+, iPLA2β+/−, and iPLA2β−/− pups and a 1:1 male:female gender distribution of iPLA2β−/− pups. Several tissues of wild-type mice were found to express iPLA2β mRNA, immunoreactive protein, and activity, and testes expressed the highest levels. Testes or other tissues of iPLA2β−/− mice expressed no iPLA2β mRNA or protein. The most striking phenotype initially observed with iPLA2β−/− mice was impaired male fertility. Spermatozoa from iPLA2β−/− mice exhibited reduced motility and impaired ability to fertilize mouse oocytes in vitro and in vivo. Mating iPLA2β−/− male mice with iPLA2β+/+, iPLA2β+/−, or iPLA2β−/− female mice yielded only about 7% of the number of pups produced by mating pairs with an iPLA2β+/+ or iPLA2β+/− male, but iPLA2β−/− female mice had nearly normal fertility29. These findings indicate that iPLA2β plays an important functional role in spermatozoa and stand in contrast to earlier reports that disruption of the Group IVA PLA2 (cPLA2α) gene impairs female but not male reproductive ability30,31.
More recent observations indicate that iPLA2β participates in the acrosome reaction of spermatozoa32. The roles of two intracellular (iPLA2β and cytosolic PLA2α) and one secreted (group X) PLA2s were examined in spontaneous and progesterone-induced acrosome reactions by using knock-out mice and specific inhibitors32. These studies revealed that iPLA2β is critical for the spontaneous acrosome reaction and that both iPLA2β and Group X sPLA2 are involved in progesterone-induced acrosomal exocytosis32. In contrast, cytosolic PLA2α was not required for either type of acrosome reaction. Progesterone-induced acrosomal exocytosis was found to spread over 30 min in the mouse, and kinetic analyses suggested the presence of different subpopulations of spermatozoa that utilized, separate PLA2 pathways to achieve acrosomal exocytosis32. At progesterone concentrations below 2 μM, spermatozoa undergoing early acrosome reactions (0–5 min) relied on iPLA2β, while those undergoing late acrosome reactions (20–30 min) relied on Group X sPLA232.
iPLA2β and neurological disorders.
Despite readily apparent phenotypic abnormalities in male fertility29,32 and glucose tolerance33 in iPLA2β-null mice, initial experience with these mice revealed no obvious neurologic impairment, but in 2006 mutations in the human iPLA2β gene were reported in patients with childhood genetic disorders categorized as neuroaxonal dystrophies34,35. A locus for infantile neuroaxonal dystrophy (INAD), neurodegeneration with brain iron accumulation (NBIA), and Karak syndrome was mapped to human chromosome 22q12-q13 and associated with iPLA2β mutations34,35. INAD and NBIA share the distinctive pathologic feature of axonal degeneration with distended axons (spheroid bodies) throughout the central nervous system (CNS)36. INAD is characterized by progressive motor and sensory impairment, and spheroids are also found in peripheral nerves35. NBIA comprises a clinically and genetically heterogeneous group of disorders with high basal ganglia iron. MRI imaging in INAD typically shows cerebellar atrophy with signal hyperintensity in the cerebellar cortex and hypointensity in the pallida and substantia nigra34. The designation PLA2G6-associated neurodegeneration (PLAN) was proposed for these disorders in 200837, and accumulated experience by that date indicated that 80% of INAD patients and 20% of NBIA patients had associated iPLA2β mutations38. Mutations predicted to lead to absent protein were associated with the INAD profile of early onset and rapid progression, while compound heterozygous missense mutations correlated with the less severe phenotype of NBIA, consistent with residual function in the mutant protein38. iPLA2β mutations were subsequently recognized to be associated with adult-onset dystonia-Parkinsonism39, and PARK14-linked Parkinsonism associated with iPLA2β mutations is clinically heterogeneous with respect to age of onset and association with fronto-temporal dementia and other features40–43. Hereditary spastic paraplegia is also associated with iPLA2β mutations44. A current perspective is that the PLAN spectrum comprises a continuum of three predominant human phenotypes with overlapping clinical and radiologic features: 1.) INAD usually begins between ages 6 months and 3 years with psychomotor regression or delay, hypotonia, and progressive spastic tetraparesis. Disease progression is rapid, and survival past age 10 years infrequent38. 2.) Atypical NAD has more phenotypic variability and onset can be as late as the end of the second decade with presenting signs of gait instability, ataxia, speech delay, and autistic features. The course is a fairly stable static encephalopathy initially that after several years is followed by neurologic deterioration38. 3.) PLA2G6-related dystonia parkinsonism has a variable age of onset but most individuals present in early adulthood with gait disturbance or neuropsychiatric manifestations. Dystonia and parkinsonism typically develop in late teens to early twenties and are accompanied by rapid cognitive decline38.
Recognition of the association of human neurologic disorders with iPLA2β prompted reexamination of iPLA2β-null mice for evidence of neurologic impairment, and this revealed that such mice did in fact exhibit an INAD-like syndrome45. iPLA2β-null mice were found to develop age-dependent neurological impairment that was evident in rotarod, balance, and climbing tests by 13 months of age45. The primary abnormality underlying this neurological impairment was the formation of spheroids containing tubulovesicular membranes remarkably similar to human INAD45. Spheroids were strongly labeled with anti-ubiquitin antibodies. Accumulation of ubiquitinated protein in spheroids was evident in some brain regions as early as 4 months of age, and the onset of motor impairment correlated with a dramatic increase in ubiquitin-positive spheroids throughout the neuropil in nearly all brain regions. Furthermore accumulating ubiquitinated proteins were observed primarily in insoluble fractions of brain tissue, implicating protein aggregation in this pathogenic process45. These results indicate that loss of iPLA2β causes age-dependent impairment of axonal membrane homeostasis and protein degradation pathways, leading to age-dependent neurological impairment45. This iPLA2β-null mouse line was also studied by another group who demonstrated that these mice develop cerebellar atrophy by the age of 13 months46. Atrophied cerebella exhibited significant loss of Purkinje cells, as well as reactive astrogliosis, the activation of microglial cells, and pronounced upregulation of the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). Glial cell activation and the elevation in TNF-α and IL-1β expression occurred before apparent cerebellar atrophy46
Another group who independently generated a separate iPLA2β-null mouse line using a different targeting construct to generate the iPLA2β-null allele also reported that those mice developed similar neurologic abnormalities47. They developed normally and grew to maturity, but all showed evidence of severe motor dysfunction, including a hind limb clasping reflex during tail suspension, abnormal gait, and poor performance in the hanging wire grip test. Neuropathological examination of the nervous system revealed widespread degeneration of axons and/or synapses, accompanied by the presence of numerous spheroids (swollen axons) and vacuoles. Those findings provided additional, independent evidence that lack of iPLA2β causes neuroaxonal degeneration is mice47. A third group reported development of a mouse line with a point mutation in the AR domain of iPLA2β that had been generated by N-ethyl-N-nitrosourea mutagenesis48. These mutant mice developed severe motor dysfunction, including abnormal gait and poor performance in the hanging grip test, as early as 7 to 8 weeks of age. Neuropathological examination revealed widespread formation of spheroids containing tubulovesicular membranes similar to human INAD. Molecular and biochemical analysis revealed that the mutant mice expressed iPLA2β mRNA and protein, but the mutated iPLA2β protein had no glycerophospholipid-hydrolyzing enzyme activity48. The significantly earlier onset of disease in this model compared to those reported earlier45,46 was suggested to be related to production of iPLA2β protein, albeit catalytically inactive as a PLA248.
A fourth group independently developed an additional iPLA2β-deficient mouse strain with an INAD-like neurologic disorder49. This strain carried a hypomorphic allele of iPLA2β that reduced transcript levels to 5–10% of that observed in wild-type mice. Homozygous mice from this strain developed pathology analogous to that observed in INAD patients49. In addition to its PLA2 activity in phospholipid hydrolysis, iPLA2β had been suggested to have non-canonical functions, including regulating store-operated Ca2+ entry into cells and modulation of mitochondrial function, and deficiency in this functions was postulated to contribute to INAD development49. This group studied changes in ATP-induced Ca2+ signaling in astrocytes derived from the mouse strain with the hypomorphic iPLA2β allele and from the strain developed earlier with an inactivating point mutation48 because ATP is an important transmitter inducing Ca2+ signals in astroglial networks49. Severe disturbances in Ca2+ responses to ATP were observed in astrocytes derived from both mutant mouse strains. The durations of the Ca2+ responses in mutant astrocytes were significantly reduced when compared with values observed in control cells and the reduced Ca2+ responses appeared to be attributable to a 2.3-fold reduction in capacitative Ca2+ entry. Others have reported that in a mouse model in which exon 2 of the iPLA2β gene is deleted, there is defective store-operated Ca2+ entry that mimics the defects observed in cells from human subjects with idiopathic Parkinson’s disease50.
That iPLA2β plays an evolutionarily conserved role in neuronal function is also supported by the observations that: 1.) Mutations in the Drosophila homolog of human iPLA2β results in age-dependent loss of psychomotor activity and neurodegeneration51; 2.) An iPLA2β missense mutation near the active site that is associated with human INAD also results in neuroaxonal dystrophy in the Papillon dog52; and 3.) iPLA2β-deficiency in Zebrafish leads to dopaminergic cell death, axonal degeneration, increased β-synuclein expression, and defects in brain functions53. Taken together with the human and mouse data, these observations point to important and highly conserved functions of iPLA2β in the nervous system.
Comparison of the PLA2 catalytic activity of wild type iPLA2β and of various iPLA2β mutants that had been expressed and assayed in vitro revealed that human wild type iPLA2β enzyme hydrolyzes both phospholipids and lysophospholipids to release free fatty acids from radiolabeled phospholipid substrates, as expected54. Mutations associated with different disease phenotypes were found to have different effects on catalytic activity. Mutations associated with INAD/NBIA caused loss of enzyme activity, with mutant proteins exhibiting less than 20% of the specific activity of WT protein in both lysophospholipase and phospholipase assays. These findings were taken to suggest that INAD/NBIA is caused by loss of iPLA2β-catalyzed fatty acid release from phospholipids that results in accumulation of phospholipid substrates and provides a mechanistic explanation for the accumulation of membranes in neuroaxonal spheroids54. In contrast, mutations associated with dystonia-parkinsonism did not impair catalytic activity, and two mutations produced a significant increase in specific activity for phospholipid but not lysophospholipid substrates54. Although dystonia-parkinsonism mutations did not appear to directly impair catalytic function, such mutations may modify substrate preferences or iPLA2β regulatory mechanisms54. These findings54 stand in contrast to another report55 in which iPLA2β sequence variants were screened in 250 patients with Parkinson’s Disease and 550 controls in Chinese Han populations. Four iPLA2β sequence mutants were observed that included a coding synonymous 1959T>A transition of exon 13 in one patient and two missense mutations c.1966C>G in exon 13 and c.2077C>G in exon 14 in two different patients, which resulted in the amino acid changes Leu656Val and Leu693Val respectively55. A frame-shift mutation P.His597fx69 in exon 12 was also observed in one patient. These four rare variants were not observed in 550 control individuals. Upon expression and assay in vitro wild type iPLA2β hydrolyzed phospholipid substrates, but the frame-shift P.His597fx69 iPLA2β mutant exhibited less than 6% of wild type phospholipase specific activity The Leu656Val and Leu693Val iPLA2β mutants exhibited 45% and 35% reductions in phospholipase assay, respectively. Thus, in this Chinese population iPLA2β mutations associated with Parkinson’s Disease did result in diminished or absent PLA2 activity55.
How alterations in iPLA2β might affect neuronal function is not clearly established. Loss of iPLA2β activity has been reported to result in an increase in ceramide levels in Drosophila central nervous system with adverse consequences56, although iPLA2β activity has been found to promote ceramide accumulation in mammalian cells57. In iPLA2β-null mice decreased brain docoshexaenoic acid metabolism and signaling have been observed that may play a role in neuronal dysfunction58,59. It has also been suggested that neuroaxonal dystrophy in iPLA2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes60, and there is evidence of mitochondrial injury and dysfunction in iPLA2β-null mice61,62 and of increased mitochondrial lipid peroxidation and dysfunction in brains of iPLA2β-null Drosophila and in fibroblasts of human subjects with iPLA2β gene mutations that cause INAD63. iPLA2β PARK14 mutants are also defective in preventing rotenone-induced mitochondrial dysfunction, generation of reactive oxygen species, and activation of the mitochondrial apoptotic pathway under conditions where WT iPLA2β does prevent these events64. There is also growing evidence that iPLA2β participates in the repair of oxidatively damaged mitochondrial membranes65–69.
III. iPLA2β: Current Understanding of its Role in Metabolic Disorders and Inflammation
Metabolic Disorders.
Among the roles ascribed to iPLA2β is its function in signal transduction 6. Early studies revealed that iPLA2β participates in insulin secretion, where glucose-stimulated insulin secretion (GSIS) is attenuated by iPLA2β-deficiency and amplified by overexpression of iPLA2β in β-cells33. The impact of iPLA2β on GSIS has been linked to iPLA2β-mediated accumulation of arachidonic acid, which negatively regulates the activity of the delayed rectifier potassium channel Kv2.1, thereby prolonging glucose-induced depolarization of the β-cell membrane and allowing for greater Ca2+ influx to support higher insulin secretion. Imposition of stress (i.e. diabetogenic agent streptozotocin or high fat diet) on iPLA2β-knockout mice leads to greater impairment in glucose tolerance, relative to WT mice33. However, this was not found to be associated with reduced insulin sensitivity, suggesting that the glucose intolerance is a consequence of reduced insulin secretory capacity.
Another role proposed for iPLA2β is its functioning as a housekeeping enzyme to regulate cellular levels of 2-lysophosphatidylcholine (LPC) and arachidonate incorporation into PC70. Type 2 diabetes (T2D) is characterized by peripheral insulin resistance and the ultimate failure of islet β-cells is typically associated with elevations in plasma free fatty acids71. Various lipid-related factors are reported to contribute to the development of insulin resistance, including triglycerides, ceramides, and diacylglycerols72–75. Fatty acid-induced insulin resistance is coupled to activation of C-Jun N-terminal kinase (JNK) and subsequent phosphorylation of the insulin receptor substrate 1 (IRS-1)76. Utilizing L6 myotubes and the db/db mouse, which develops obesity, hyperlipidemia, and diabetes due to deficiency in leptin receptor activity, Han et al77 identified a role for LPC in the development of insulin resistance. They determined that palmitate-induced increase in intracellular LPC in the myotubes was inhibited by BEL, an iPLA2-selective inhibitor, as were JNK activation and IRS-1 phosphorylation. Further, administration of BEL to the db/db mice reduced LPC abundance in both the liver and muscle along with JNK activation and IRS-1 phosphorylation. These changes were accompanied by a reduction in plasma insulin levels and an increase in β-cell volume, most likely due to BEL-mediated decrease in β-cell stress. While exogenous addition of LPC increased JNK activation and IRS-1 phosphorylation and rescued their decreases from siRNA-mediated reduction in iPLA2β, inhibition of ceramide synthesis did not prevent palmitate-induced insulin resistance. These observations coupled with the finding that BEL increased diacyglycerides, while mitigating insulin resistance, highlighted a potential role of LPC in insulin resistance. A caveat in this study is that the BEL used was a racemic mixture of S- and R-enantiomers, which inhibit iPLA2β and iPLA2γ, respectively. In fact, siRNA knockdown of both iPLA2β and iPLA2γ reduced LPC and JNK activation. As other studies have reported that genetic ablation of iPLA2γ counters development of obesity, insulin resistance, and metabolic abnormalities78,79, it is likely that both iPLA2β and iPLA2γ generate lipid signals that contribute to the development of insulin resistance. Collectively, these studies reveal that iPLA2 (β and γ)-derived lipids (iDLs) can impair the ability to preserve glucose tolerance in the presence of added stress.
Genome wide meta-analysis revealed a significant association between iPLA2β and plasma lipids that are recognized to be important mediators of metabolic syndrome80. Surprisingly, neither arachidonate-containing PC species of islets, brain, and testis81,82 nor the rate of incorporation of arachidonate into PC29,83 in iPLA2β-deficient mice maintained on a normal chow diet are altered. Further, the content of triacylglycerides and total cholesterol in plasma, liver, muscle, and adipose tissue was similar in iPLA2β-deficient mice fed a normal chow diet or high fat diet84. In contrast, when iPLA2β-deficiency was introduced into ob/ob mice, homozygous for leptin mutation, these mice were protected against becoming obese and developing hepatic steatosis85. This improved phenotype was associated with reductions in serum lipid, insulin resistance, and de novo lipogenesis. It was suggested that iPLA2β and cPLA2α, by providing mono- and poly-unsaturated fatty acid substrates that are directed towards incorporation into triacylglycerides participate in the development of hepatic steatosis in the ob/ob. With iPLA2β-deficiency, however, recovery of arachidonate- and DHA-containing PC and PE species is observed85.
A more recent genome wide meta-analysis86 identified novel genetic loci that were linked with body mass index (BMI) and body fat percentage (BF%). Among these was a loci in or near PLA2G6 that associated with BF%, rather than BMI, and this effect was more pronounced in men (2-fold) than in women. The BF% increasing allele was found to be linked with lower insulin and triglyceride levels and a reduced T2D risk. Further analyses of single nucleotide polymorphisms in PLA2G6 loci identified a strong association between BF% and higher birth weight and greater pre-pubertal height. While this study identified novel linkage between PLA2G6 and adiposity, the authors noted the potential relevance of other genes present in the PLA2G6 locus. Candidate genes included PICK1, which encodes a membrane sculpting BAR domain protein that facilitates storage and secretion of hormones (i.e., insulin from beta-cell) and SOX10, which encodes SRY-related HMG-box family of transcription factors and is an important regulator of embryonic development. The similarity in phenotypes resulting from mutations in PICK1 and SOX10 suggested that they may be the driving force of the observed PLA2G6-associations.
Studies performed by the Gross group using transgenic mice, selectively overexpressing iPLA2β in cardiac myocytes87, reveal that myocardial ischemia promotes activation of iPLA2β leading to accumulation of acylcarnitines in the ischemic heart. Similarly, diabetic hearts also accumulate long chain aclycarnitines (LCACs) due to higher expression and activity of iPLA2β, which provides free fatty acids that can be used to generate LCACs. The increases in LCACs are amplified by overexpression of iPLA2β in the heart and reduced by selective inhibition of iPLA2β88. LCACs are generated via carnitine palmitoyl transferase I (CPT-I), which is regulated through cytosolic malonyl CoA, and the combination of increased iPLA2β and CPT-1 and reduced malony CoA in the diabetic myocardium appears to be central to the LCAC accumulation. This may be expected to alter membrane properties and give rise to diabetes-induced arrhythmias.
The development of diabetes is associated with β-cell death (type 1 diabetes, T1D) or β-cell dysfunction preceding their death (T2D). Participation in apoptotic processes is another role suggested for iPLA2β6 and we have reported that iPLA2β participates in β-cell death due to ER stress and pro-inflammatory cytokines, which are critical contributors to β-cell death leading to T1D. Our collection of work57,89–91 reveals that iPLA2β participation in mediating β-cell death is through (a) induction of ceramides generation via neutral sphingomyelinase-catalyzed hydrolysis of sphingomyelins, which leads to disruption of mitochondrial integrity, caspse-3 activation and ultimately β-cell death and by (b) modulating alternate splicing of Bcl-x that promotes a decrease in the ratio of anti-apoptotic variant Bcl-x(L)/pro-apoptotic variant Bcl-x(s), that occurs via a nonceramide dependent pathway, possible involving arachidonic acid.
iPLA2β and inflammation.
As recognition of the roles for iPLA2β is expanding, there are now a number of studies that demonstrate a critical role for iPLA2β-derived lipids (iDLs) in promoting inflammatory responses, which include chemokine-mediated recruitment of leukocytes to inflamed sites, and the production of cytokines and reactive oxygen species (ROS). An early-demonstrated signaling role for iDLs in this process is their impact on monocyte migration92,93. The chemokine, monocyte chemoattractant protein (MCP-1), plays a critical role in inflammatory disorders by stimulating chemotaxis to promote migration of monocytes towards inflamed sites94. The Cathcart group linked monocyte migration with PLA2 activity and demonstrated that the chemotactic response to MCP-1 involved both iPLA2β and cPLA2α. Utilizing chemical and siRNA approaches, they identified different roles for the two PLA2s. With MCP-1 stimulation, cPLA2α was found to translocate to the endoplasmic reticulum where its activity leads to the generation of arachidonic acid, which functions to regulate the speed of monocyte migration. In contrast, iPLA2β mobilizes to the membrane to increase the generation of lysophosphatidic acid (LPA), whose function appears to be to provide migration directionality by targeting F-actin polymerization. These findings led to their suggestion that iPLA2β functions as a “cellular compass” to affect monocyte migration in response to MCP-1.
Production of ROS is a critical step in the propagation of the inflammatory response. The NADPH oxidases (NOX) catalyze ROS generation in macrophages95, which express both NOX2 and NOX496. The NOX is comprised of cytosolic components Rac2, and p47phox, p67phox, and p40phox, which are tightly associated, and membrane flavocytochrome b56897. Activation of NOX requires translocation of the cytosolic components to the membrane, facilitating transfer of electrons from NOX to O2 to generate superoxide anion (O2−)98,99. Moreover, the activation and superoxide generation steps appear to require the presence of arachidonic acid98,100,101. It has been reported that in neutrophils from diabetic subjects and when exposed to high glucose, there is a prominent translocation of the NADPH complex to the membrane resulting in increased superoxide generation102. It has seen been shown that neutrophils from diabetic subjects manifest greater expression and activity of iPLA2β. Superoxide production from these neutrophils was found to require iPLA2β-mediated generation of arachidonic acid. This was supported by inhibition of superoxide production with an inhibitor and siRNA directed against iPLA2β and subsequent rescue of production with addition of arachidonic acid103. While not directly assessed in this study, the role of arachidonic acid in this process has been proposed to include promoting NOX binding and translocation of cytosolic components to the membrane104.
The activity of NOX2 is regulated by [Ca2+]i105 and S100 Ca2+-binding proteins, S100A8 and S100A9, are thought to participate106. This process requires translocation of S100A8/9 complex to the membrane that is promoted following p38 MAPK-mediated phosphorylation of S100A9107. Previously, it was reported that p38 MAPK activity can be modulated by iPLA2β108, giving rise to studies demonstrating that iPLA2β-mediated phosphorylation and translocation of S100A8/9 through activation of p38 MAPK109. This led to regulation of NOX2 activity and subsequent ROS production. These outcomes were prevented by chemical inhibition of iPLA2β or siRNA directed against iPLA2β. While iPLA2β activity was implicated in this process, the study did not assess the relevant iDL required to cause affect.
While the major isoforms of NOX in macrophages are NOX2 and NOX496, the latter has been reported to promote myocytes chemotaxis and macrophage recruitment during diabetic metabolic stress110. Lipoprotein receptor-knockout mice (LDLR−/−), a model of diet-induced atherosclerosis, exhibit temporal increases in iPLA2β and aortic atherosclerotic lesions111. In this model, MCP-1-induced macrophage migration in response to high glucose or low-density lipoprotein (LDL) was mitigated by an inhibitor or siRNA directed against iPLA2β. Inhibiting iPLA2β prevented subsequent H2O2 production and this was associated with reduction in NOX4, but not NOX2, expression. These outcomes were rescued by LPA, suggesting that iPLA2β-mediated generation of LPA was required for NOX4 activation it macrophages.
When the above observations are considered along with findings linking cPLA2α with NOX activation101,112, a scenario evolves where coordinated activities of iPLA2β and cPLA2α in concert promote NOX activation. In those studies, the Levy group utilizing neutrophils and granulocyte-like PLB-385 cells and employing immunoprecipitation and microscopy approaches reported that upon stimulation, cPLA2α is recruited to the plasma membrane after assembly of NOX. The association of cPLA2α with NOX is thought to require the phosphorylated form of cPLA2α. Subsequent generation of cPLA2α -mediated hydrolysis of arachidonic acid coincided with the production of superoxide. It was suggested that the arachidonic acid facilitates association of the NOX complex resulting in optimal oxidase activation. They went on further to determine using blot overlay and Forster resonance energy analyses that the binding between cPLA2α and NOX at the membrane is mediated by p47phox and that the anchoring of cPLA2α is facilitated by the cPLA2α -C2 domain, which exhibits specificity for phosphatidylserine-rich plasma membranes 113. Collectively, it might be surmised that iPLA2β activity induces translocation of the NOX components to the membrane and that cPLA2α activity promotes optimal membrane-associated oxidase activity.
Other instances of interplay between iPLA2β and cPLA2α have also been suggested. An earlier report had suggested that thapsigargin-triggered activation of store-operated channels and capacitative Ca2+ influx is mediated by lysophospholipids generated via iPLA2β activation114. This study relied exclusively on chemical inhibition of iPLA2β to discern its role. Subsequently, utilizing iPLA2β-deficient mice to preclude non-specific effects of chemical inhibitors, and stimulants (thapsigargin or ionophore A231987), the Gross group described sequential roles of iPLA2β and cPLA2α in promoting arachidonic acid release and eicosanoid generation115, and contributing to vascular tone, signaling, and in smooth muscle cell proliferation and migration. Based on the difference in their mode of action, where thapsigargin promotes Ca2+-mediated depletion of internal stores and A23187-mediated transmembrane Ca2+ influx can lead to activation of cPLA2α, they proposed that an initial iPLA2β-mediated release of arachidonic acid is followed by a later induction of cPLA2α via iPLA2β-mediated activation of capacitative Ca2+ influx.
An underlying process in atherosclerosis is foam cell formation and ROS-mediated oxidation of low density lipoproteins are critical for the conversion of macrophages to foam cells116. The generation of ROS appears to be dependent on toll-like receptor (TLR), in particular TLR9 signaling117,118 through induction of NOX. A study done by the Lee group116 revealed that S-BEL (selective for iPLA2β) and iPLA2β siRNA, but not R-BEL (selective for iPLA2γ) or iPLA2γ siRNA, more strongly reduced lipopolysaccharide (LPS)-induced ROS production, NOX1 expression, and inhibited foam cell formation. They further observed that inhibition of iPLA2β reduced LPS-stimulated Akt phosphorylation. These studies identified a role for iPLA2β in mediating the atherosclerosis process via Akt signaling to induce NOX1 expression and generation of ROS by a TLR4-dependent pathway. However, this study did not extend to discerning the specific lipids generated by iPLA2β activity and are critical in promoting foam cell formation.
Macrophage adhesion to modified extracellular matrix (ECM) components and their retention is another requisite for the inflammatory process. The Class A Scavenger Receptors (SR-A) present on macrophages are homotrimeric membrane glycoproteins and they function in Ca2+-independent macrophage adhesion to modified ECM proteins119. This also involves intracellular signaling cascades and the assembly and organization of the actin cytoskeleton to allow for spreading and firm cell adhesion. These processes are regulated by Rac and Cdc42, members of the Rho-like GTPase family120. In vitro studies with peritoneal macrophages revealed that inhibition of iPLA2β, but not cPLA2α, decreased cell adhesion and spreading121. Such inhibition was recapitulated by inactivation of 12/15-lipoxygenase (LOX), but not of cyclooxygenase or CytP450 epoxygenase, and rescued by arachidonic acid addition. Further, Rac and Cdc42 activation were also mitigated by inhibitors of iPLA2β and 12/15-LOX. These findings suggest that 12/15-LOX metabolites of arachidonic acid, derived from iPLA2β activation, participate in macrophage adhesion and spreading by coupling SR-A to Rac and Cdc42 activation. A subsequent study demonstrated that Trypanosoma cruzi infection, which results in inflammation of myocardial tissue, increases polymorphonuclear (PMN) leukocyte adhesion that was associated with increased iPLA2β and iPLA2γ activity, arachidonic acid release, and PAF production122. While Inhibition of iPLA2β or iPLA2γ reduced arachidonic acid release, only inhibition of iPLA2β decreased PAF production and PMN adhesion. These outcomes were recapitulated by the inability of RAW 264.7 murine macrophage/monocyte cell adherence to cardiac endothelia cells obtained from iPLA2β-deficient mice. These findings suggest a role for PAF derived from iPLA2β activity in promoting cell adhesion during tissue inflammation.
An additional link between iPLA2β and a GTP binding protein was identified in aortic and mesenteric arteries of type 1 and type 2 diabetic mouse models123. Diabetic vascular complications encompass increase in vasoconstrictive and decrease in vasodilatory responses and G-protein coupled receptor agonism can modulate vascular smooth muscle (VSM) contraction by eliciting Ca2+-sensitization124. Dysregulation of this process can lead to severe cardiovascular complications. The GTP-binding protein RhoA, RHO kinase (ROCK), protein kinase C (PKC), and CPI-17 (protein kinase C-potentiated phosphatase inhibitor) function as Ca2+-sensitizers. Elevations in glucose, as in T1D and T2D, were found to induce iPLA2β via PKC activation leading to activation of RhoA/ROCK/CPI-17 and diabetes-induced hypercontractility of VSM, which was ameliorated with inhibition or knockout of iPLA2β, but not iPLA2γ. Inhibition of 12/15-LOX was also effective in reducing VSM hypercontractility, suggesting that iPLA2β, through generation of 12/15-LOX products, participates in the evolution of VSM abnormalities that are prevalent in diabetes.
Proinflammatory cytokines produced by immune cells are integral to the inflammatory process. We reported that a selective inhibitor of iPLA2β reduces T1D incidence in the non-obese diabetic (NOD) mouse model125. This was associated with reduced infiltration of leukocytes into the islets and decreased generation of TNFα from CD4+ T-cells and antibodies from B-cells isolated from NOD. Further, iPLA2β-deficiency favored macrophage polarization towards an M2 anti-inflammatory phenotype126, concurrent with reduced macrophage IFNγ+LPS-activated production of TNFα and IL-1β. Decreased production of the pro-inflammatory cytokines in iPLA2β-deficient macrophages was recapitulated in WT macrophages exposed to inhibitors of iPLA2β, COX, or 12-LOX. These inhibitors also mitigated M1 polarization of WT macrophages with IFNγ+LPS activation. An impact of iPLA2β on cytokine production was also recognized in a neointima formation, which can lead to development of atherosclerosis127. That study revealed an increase in iPLA2β expression in arteries following coronary artery ligation that preceded neointima formation. Inhibition, knockdown, or genetic deletion of iPLA2β inhibited and overexpression of iPLA2β exacerbated macrophage vascular infiltration and neointima formation. Activation of iPLA2β was also linked to higher production of inflammatory markers TNFα, IL-6, IL-1β, and MCP-1. These outcomes were again reduced in iPLA2β-deficient macrophages and by inhibition of 12/15-LOX, but not COX, 5-LOX, or cytochrome P450. Collectively, these findings suggest that COX and 12-LOX products generated downstream of iPLA2β activation affect immune cell function and responses under different disease settings.
Other studies suggest protective effects of iPLA2β, as its deletion exacerbated experimental colitis128, autoimmune hepatitis129, and age-related liver and intestinal disorders130. These were noted to be related to defective membrane remodeling, and reductions in LPC, which provides the eat-me signal for recruitment of phagocytes to facilitate clearance of apoptotic cellular debris, and in LPA, which promotes macrophage migration. iPLA2β-deficiency also has been reported to result in higher abundance of parasite pseudocysts in the hearts of mice infected with Trypanosoma cruzi131. In WT hearts, the infection induced NO production to facilitate parasite clearance by macrophages. With iPLA2β deficiency or inhibition of iPLA2β, but not iPLA2γ, NO production was reduced leading to accumulation of the pseudocysts.
These reports suggest that iDLs are important contributors to inflammation and as such could be critical in the development of autoimmune disorders125,129,132. While the focus of this review is iPLA2β, it must be recognized that other PLA2s, working in concert with iPLA2β, may also be required to produce the inflammatory sequela. In this regard, a study of pleurisy provides insight into involvement of multiple PLA2s that may apply to the etiology of other inflammatory landscapes as well133. They report that the inflammatory process encompasses two stages of arachidonic acid release. The release at onset leads to generation of pro-inflammatory eicosanoids and release at the later stage to generation of resolving eicosanoids. At the onset of pleurisy, iPLA2β activity is predominant and leads to increased production of PGE2, LTB4, PAF, and IL-1β to drive the inflammatory process. Subsequently, a resolution phase evolves during which sPLA2s (Groups IIA and V) mediate generation of PAF and lipoxin A4, which in turn induce cPLA2α and the second stage of arachidonic acid release. This pool of arachidonic acid is metabolized to anti-inflammatory PGD2 and its breakdown product 15deoxy delta 12–14 PGJ2, as a means to continue the resolution phase. Interestingly, corticosterone is released at the beginning of inflammation to limit inflammatory responses. It inhibits cPLA2α, but not iPLA2β, and its levels decrease during resolution, which could lead to an increase in cPLA2α and the emergence of a more dynamic resolution phase.
IV. iPLA2β – Moving Forward
Ongoing studies are greatly increasing our understanding of the multitude of roles that iPLA2β and iDLs have in a variety of biological processes and diseases. Continued studies are facilitated by the availability of genetically-modified mice with iPLA2β-deficiency29 and the significant advances recently reported in the following areas:
Crystal structure of iPLA2β.
The iPLA2β protein sequence consists an N-terminal domain, an AR domain, and a catalytic domain (CAT)134. To examine the structure of the short isoform iPLA2β, its cDNA cloned from CHO cells was used to express the enzyme with a C-terminal 6XHisTag, and the recombinant enzyme was purified and crystalized for selenomethione single-wavelength anomalous diffraction (SAD) studies combined with molecular replacement (MR) studies using two different protein models. The model proteins were patatin (32% sequence identity to the CAT domain) and four ARs (20% sequence identity to the four C-terminal ARs of iPLA2β. The core secondary structure elements of the CAT domain are similar to patatin and the fold structure resembles that of iPLA2β. The active site is located within the globular domain as in patatin, but the catalytic residues are more solvent accessible. The active site cavity is wide open and can accommodate phospholipids with long polyunsaturated fatty acid chains. A long C-terminal α-helix is kinked and participates in dimer formation. The electron density map reveals nine ARs in the ANK domain, and its orientation surprisingly reflects attachment to the CAT domain on the side opposite to the membrane-binding surface. The outer helices of AR7 and AR8 form an extensive hydrophobic interface with the CAT domain. ATP was found to bind near Trp293 rather than in the previously suspected glycine-rich domain. Trp293 lies within AR6 (residues 282–308), which contains a helix two amino acids shorter than a conventional AR helix that creates a kink of the ANK domain. Potential ATP binding at this location suggests that nucleotides can regulate ANK domain conformation. The crystalized iPLA2β is a dimer formed through association of the CAT domains, and the ANK domains previously suspected to be involved in oligomerization are oriented outwards in opposite directions for form an elongated structure. The CAT domains interact through an extended hydrophobic dimerization interface formed by three α-helices, several loops (including residues 554–570), and part of the central β-sheet. Trp695 forms extensive hydrophobic interactions with the opposite monomer, including a stacking interaction with its counterpart, and substitution of Glu at this position (W695E) by mutagenesis causes the resultant protein to behave as a monomer in solution. In the dimer, two active centers and the predicted membrane binding loops are oriented in the same direction. The active sites are close to the dimerization interface and to each other, and the catalytic Asp598 is at the beginning of the π-helical loop (residues 599–603). Two leucines of this loop form contacts with the long α-helix (residues 604–624) of the opposite monomer134. These features suggest strong allosteric association of the two active sites and dependence of the catalytic activity on the dimer conformation. Binding studies indicate that calmodulin (CaM) 135–137 associates with the iPLA2β dimer rather than a monomer, and, consistent with this, two 1–9-14 CaM binding motifs lie on the same side of the dimer in close proximity.
Surprising features of these findings include the open conformation of the active site in the absence of membrane interaction with both sites of the dimer providing sufficient space for phospholipids to access the catalytic center134. In addition, the dimer is formed by interaction of the CAT domains, while the ANK domains are oriented outwards and do not mediate interactions of the monomers. Disruption of the dimer in the W695E mutant yields an inactive enzyme. Intimate allosteric interaction of the active sites and the dimerization interface also provides a potential mechanism for inhibition by CaM, the binding of which stabilizes a closed conformation of the active sites. The outward-facing ANK domains of the model suggest potential interaction sites for cytosolic Ca2+/calmodulin-dependent protein kinase IIβ (CaMKIIβ) and for the cytosolic C-terminus of transmembrane calnexin, both of which are known to interact with iPLA2β10,138. The knowledge of the crystal structure for iPLA2β will allow further studies on how iPLA2β may be regulated in the absence and presence of potential binding partners.
Substrate specificity and molecular dynamics of iPLA2β.
The Dennis group utilizing lipidomics-based LC/MS protocols assessed substrate preference of iPLA2β versus cPLA2α and Group V sPLA2 and noted several features that were unique to iPLA2β139. In the presence of equimolar mixture of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphate (PAPA), 1-palmitoyl-2-arachidonoyl-sn-phosphatidylcholine (PAPC), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine (PAPE), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (PAPG), and 1-palmitoyl-2arachidonoyl-sn-glycero-3-phospho-L-serine (PAPS), in contrast to cPLA2α which showed no preference and Group V sPLA2 was more active towards PAPG, iPLA2β was found to have the greatest activity towards PAPE. They further demonstrated that with respect to the sn-2 substituent on membrane phospholipids, cPLA2α preferred arachidonic acid, Group V sPLA2 was active towards most fatty acids with linoleic and myristic acids slightly better preferred, but that iPLA2β was much more selective for linoleate and myristate. Also noted were differences in the preference for sn-1 fatty acid, with cPLA2α and Group V sPLA2 preferring steric acid, while iPLA2β was more active with phospholipids containing palmitic acid in the sn-1 position. The differences in substrate preference were very nicely rationalized with atomistic molecular dynamics analyses. Using simulations with each enzyme in the presence of optimal substrates, they were able to describe distinguishing features about the active site for each enzyme. The active site was described as containing two binding regions, a hydrophobic one where the headgroup binds and a hydrophilic one where the two acyl chains bind. Substrate specificity is conferred by the recruitment of the optimal substrate (phospholipid + sn-2 fatty acid) to its select hydrophobic binding subsite. Their simulations revealed that the active site of cPLA2α is a channel that is deep and rigid and able to take up the whole phospholipid molecule, lending to its specificity for arachidonate-containing phospholipids. However, the active site for Group V sPLA2 is a shallow cavity that can only take up the sn-2 fatty acid, reflected by its similar activity towards various sn-2 fatty acids. The active site of iPLA2β on the other hand is more flexible and is capable of changing volume, thus facilitating activity towards different phospholipids containing different sn-2 fatty acids. Analyses such as these can increase our understanding of how enzyme activity might be affected by changes in the enzyme molecular structure in a diseased state, relative to a healthy environment.
Development of novel and more selective inhibitors of iPLA2β.
In view of the similar functions of the PLA2s, selective inhibitors of the enzymes would significantly enhance our understanding of the specific role of each PLA2 in a biological event. In this regard, the work done by the Kokotos group has made major strides in generating more potent and selective inhibitors of the PLA2s. Utilizing computational chemistry and organic synthesis in conjunction with SAR and in vitro activity assays, they identified fluoroketone compounds with high potency and selectivity towards iPLA2β, as compared against cPLA2α or Group V sPLA2140. Additionally, one such compound, designated FKGK18, was demonstrated to be more selective for iPLA2β than iPLA2γ141. Unlike BEL, FKGK18 is a reversible inhibitor without significant cytotoxic effects and has proven to be effective in reducing T1D in a spontaneous diabetes rodent model125. Analogously, the earlier generation FKGK11 was able to ameliorate clinical scores associated with experimental autoimmune encephalomyelitis 132. Studies such as these highlight the potential of utilizing these inhibitors to mitigate complications associated with autoimmune- and inflammation-based disorders. More recently, modification of the fluoroketone backbone has led to identification of even more potent and selective iPLA2 inhibitors (in revision). Moreover, integration of molecular dynamics stimulations, SAR assays, and hydrogen/deuterium exchange mass spectrometry has led to identification of new fluoroketone compounds and one based on a keto-1,2,4 cadaizole functionality with a thioether that manifested selective potency against iPLA2β, relative to cPLA2α or Group V sPLA2142,143. Those studies were able to further establish that the sulfur atom in the beta-thioether analogues is vital for the potency and that the hydrophobic chain for the selectivity. In the future, coupling these approaches with the now available crystal structure of iPLA2β would most definitely lead to the generation of very selective and potent inhibitors of iPLA2β that will be amenable for therapeutics.
Supplementary Material
Figure 3. Participation of iPLA2β-derived lipids in metabolic and inflammatory processes.
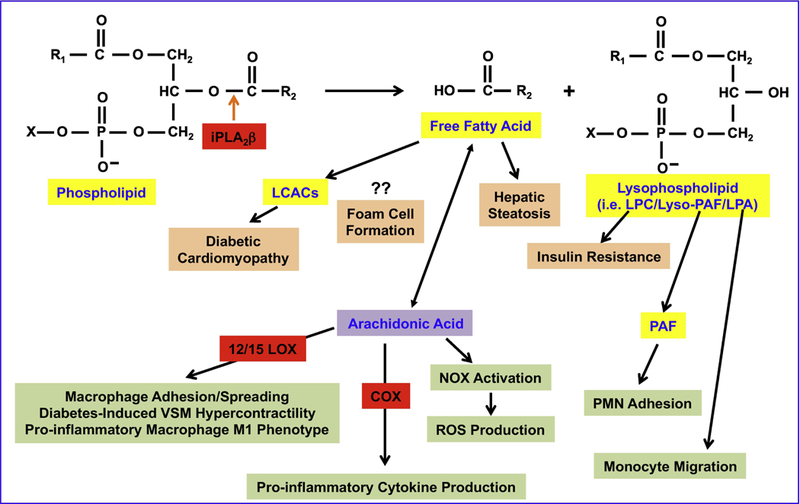
Activation of iPLA2β leads to the release of the sn-2 fatty acid and and production of a lysophospholipid. The free fatty acids and LPC have been linked to metabolic disorders including cardiac abnormalities associated with diabetes, hepatic steatosis, foam cell formation, and insulin resistance. Further, arachidonic acid and 12/15-LOX- and COX-derived metabolites of arachidonic acid, along with PAF and LPA, have been implicated in several inflammatory processes.
Acknowledgements
This work was supported in part by grants from R01DK110292 (SR) and P41GM103422, P30DK020579, P30DK056341, and R37DK34388 (JT) from the United States Public Health Service and by a bridge grant from the Washington University Dean’s Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross MI, Deems RA, Jesaitis AJ, Dennis EA, and Ulevitch RJ (1985) Phospholipase activities of the P388D1 macrophage-like cell line.. Archives of biochemistry and biophysics 238, 247–258 [DOI] [PubMed] [Google Scholar]
- 2.Wolf RA, and Gross RW (1985) Identification of neutral active phospholipase C which hydrolyzes choline glycerophospholipids and plasmalogen selective phospholipase A2 in canine myocardium. J Biol Chem 260, 7295–7303 [PubMed] [Google Scholar]
- 3.Mancuso DJ, Jenkins CM, and Gross RW (2000) The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J Biol Chem 275, 9937–9945 [DOI] [PubMed] [Google Scholar]
- 4.Burke JE, and Dennis EA (2009) Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 50 Suppl, S237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenkins CM, Han X, Mancuso DJ, and Gross RW (2002) Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J Biol Chem 277, 32807–32814 [DOI] [PubMed] [Google Scholar]
- 6.Ramanadham S, Ali T, Ashley JW, Bone RN, Hancock WD, and Lei X (2015) Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J Lipid Res 56, 1643–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaloske RH, and Dennis EA (2006) The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761, 1246–1259 [DOI] [PubMed] [Google Scholar]
- 8.Balboa MA, Balsinde J, Jones SS, and Dennis EA (1997) Identity between the Ca2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. J Biol Chem 272, 8576–8580 [DOI] [PubMed] [Google Scholar]
- 9.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, and Turk J (1997) Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J Biol Chem 272, 11118–11127 [PubMed] [Google Scholar]
- 10.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, and Jones SS (1997) A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem 272, 8567–8575 [DOI] [PubMed] [Google Scholar]
- 11.Pickard RT, Strifler BA, Kramer RM, and Sharp JD (1999) Molecular cloning of two new human paralogs of 85-kDa cytosolic phospholipase A2. J Biol Chem 274, 8823–8831 [DOI] [PubMed] [Google Scholar]
- 12.Ohto T, Uozumi N, Hirabayashi T, and Shimizu T (2005) Identification of novel cytosolic phospholipase A2s, murine cPLA2{delta}, {epsilon}, and {zeta}, which form a gene cluster with cPLA(2){beta}. J Biol Chem 280, 24576–24583 [DOI] [PubMed] [Google Scholar]
- 13.Andrews DL, Beames B, Summers MD, and Park WD (1988) Characterization of the lipid acyl hydrolase activity of the major potato (Solanum tuberosum) tuber protein, patatin, by cloning and abundant expression in a baculovirus vector. Biochem J 252, 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, and Gross RW (2004) Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279, 48968–48975 [DOI] [PubMed] [Google Scholar]
- 15.van Tienhoven M, Atkins J, Li Y, and Glynn P (2002) Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem 277, 20942–20948 [DOI] [PubMed] [Google Scholar]
- 16.Kienesberger PC, Oberer M, Lass A, and Zechner R (2009) Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res 50 Suppl, S63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson PA, Gardner SD, Lambie NM, Commans SA, and Crowther DJ (2006) Characterization of the human patatin-like phospholipase family. J Lipid Res 47, 1940–1949 [DOI] [PubMed] [Google Scholar]
- 18.Ackermann EJ, Conde-Frieboes K, and Dennis EA (1995) Inhibition of macrophage Ca(2+)-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem 270, 445–450 [DOI] [PubMed] [Google Scholar]
- 19.Ackermann EJ, Kempner ES, and Dennis EA (1994) Ca(2+)-independent cytosolic phospholipase A2 from macrophage-like P388D1 cells. Isolation and characterization. J Biol Chem 269, 9227–9233 [PubMed] [Google Scholar]
- 20.Gross RW, Ramanadham S, Kruszka KK, Han X, and Turk J (1993) Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 32, 327–336 [DOI] [PubMed] [Google Scholar]
- 21.Hazen SL, and Gross RW (1991) ATP-dependent regulation of rabbit myocardial cytosolic calcium-independent phospholipase A2. J Biol Chem 266, 14526–14534 [PubMed] [Google Scholar]
- 22.Hazen SL, Zupan LA, Weiss RH, Getman DP, and Gross RW (1991) Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem 266, 7227–7232 [PubMed] [Google Scholar]
- 23.Ramanadham S, Gross RW, Han X, and Turk J (1993) Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry 32, 337–346 [DOI] [PubMed] [Google Scholar]
- 24.Gora S, Lambeau G, Bollinger JG, Gelb M, Ninio E, and Karabina SA (2006) The proinflammatory mediator Platelet Activating Factor is an effective substrate for human group X secreted phospholipase A2. Biochim Biophys Acta 1761, 1093–1099 [DOI] [PubMed] [Google Scholar]
- 25.Ramanadham S, Wolf MJ, Jett PA, Gross RW, and Turk J (1994) Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: a possible molecular component of the beta-cell fuel sensor. Biochemistry 33, 7442–7452 [DOI] [PubMed] [Google Scholar]
- 26.Larsson PK, Claesson HE, and Kennedy BP (1998) Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem 273, 207–214 [DOI] [PubMed] [Google Scholar]
- 27.Ma Z, Wang X, Nowatzke W, Ramanadham S, and Turk J (1999) Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J Biol Chem 274, 9607–9616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsson Forsell PK, Kennedy BP, and Claesson HE (1999) The human calcium-independent phospholipase A2 gene multiple enzymes with distinct properties from a single gene. European journal of biochemistry 262, 575–585 [DOI] [PubMed] [Google Scholar]
- 29.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, and Turk J (2004) Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem 279, 38194–38200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, and Sapirstein A (1997) Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 390, 622–625 [DOI] [PubMed] [Google Scholar]
- 31.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, and Shimizu T (1997) Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390, 618–622 [DOI] [PubMed] [Google Scholar]
- 32.Abi Nahed R, Martinez G, Escoffier J, Yassine S, Karaouzene T, Hograindleur JP, Turk J, Kokotos G, Ray PF, Bottari S, Lambeau G, Hennebicq S, and Arnoult C (2016) Progesterone-induced acrosome exocytosis requires sequential involvement of calcium-independent phospholipase A2beta (iPLA2beta) and Group X secreted phospholipase A2 (sPLA2). J Biol Chem 291, 3076–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, and Turk J (2006) Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J Biol Chem 281, 20958–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, and Birk OS (2006) PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet 79, 942–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, and Hayflick SJ (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38, 752–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayflick SJ, Kurian MA, and Hogarth P (2018) Neurodegeneration with brain iron accumulation. Handb Clin Neurol 147, 293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupta R, Philip SG, Hendriksz C, Morton JE, Kingston HM, Rosser EM, Wassmer E, Gissen P, and Maher ER (2008) Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70, 1623–1629 [DOI] [PubMed] [Google Scholar]
- 38.Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, and Hayflick SJ (2008) Neurodegeneration associated with genetic defects in phospholipase A2. Neurology 71, 1402–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, and Schneider SA (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65, 19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, Chataway J, Bhatia KP, Lees AJ, Hardy J, Revesz T, and Houlden H (2012) Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 33, 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomiyama H, Yoshino H, and Hattori N (2011) Analysis of PLA2G6 in patients with frontotemporal type of dementia. Parkinsonism Relat Disord 17, 493–494 [DOI] [PubMed] [Google Scholar]
- 42.Tomiyama H, Yoshino H, Ogaki K, Li L, Yamashita C, Li Y, Funayama M, Sasaki R, Kokubo Y, Kuzuhara S, and Hattori N (2011) PLA2G6 variant in Parkinson’s disease. J Hum Genet 56, 401–403 [DOI] [PubMed] [Google Scholar]
- 43.Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, Hashimoto T, Takashima S, and Hattori N (2010) Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 75, 1356–1361 [DOI] [PubMed] [Google Scholar]
- 44.Ozes B, Karagoz N, Schule R, Rebelo A, Sobrido MJ, Harmuth F, Synofzik M, Pascual SIP, Colak M, Ciftci-Kavaklioglu B, Kara B, Ordonez-Ugalde A, Quintans B, Gonzalez MA, Soysal A, Zuchner S, and Battaloglu E (2017) PLA2G6 mutations associated with a continuous clinical spectrum from neuroaxonal dystrophy to hereditary spastic paraplegia. Clin Genet 92, 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malik I, Turk J, Mancuso DJ, Montier L, Wohltmann M, Wozniak DF, Schmidt RE, Gross RW, and Kotzbauer PT (2008) Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol 172, 406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Z, Wang J, Zhao C, Bi W, Yue Z, and Ma ZA (2011) Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. PLoS One 6, e26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, and Tsujimoto Y (2008) Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci 28, 2212–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wada H, Yasuda T, Miura I, Watabe K, Sawa C, Kamijuku H, Kojo S, Taniguchi M, Nishino I, Wakana S, Yoshida H, and Seino K (2009) Establishment of an improved mouse model for infantile neuroaxonal dystrophy that shows early disease onset and bears a point mutation in PLA2G6. Am J Pathol 175, 2257–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strokin M, Seburn KL, Cox GA, Martens KA, and Reiser G (2012) Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated PLA2G6 . Hum Mol Genet 21, 2807–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Q, Yen A, Rymarczyk G, Asai H, Trengrove C, Aziz N, Kirber MT, Mostoslavsky G, Ikezu T, Wolozin B, and Bolotina VM (2016) Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson’s disease. Nat Commun 7, 10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iliadi KG, Gluscencova OB, Iliadi N, and Boulianne GL (2018) Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci Rep 8, 2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsuboi M, Watanabe M, Nibe K, Yoshimi N, Kato A, Sakaguchi M, Yamato O, Tanaka M, Kuwamura M, Kushida K, Ishikura T, Harada T, Chambers JK, Sugano S, Uchida K, and Nakayama H (2017) Identification of the PLA2G6 c.1579G>A Missense Mutation in Papillon Dog Neuroaxonal Dystrophy Using Whole Exome Sequencing Analysis. PLoS One 12, e0169002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanchez E, Azcona LJ, and Paisan-Ruiz C (2018) Pla2g6 Deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased beta-synuclein expression, and defects in brain functions and pathways. Mol Neurobiol 55, 6734–6754 [DOI] [PubMed] [Google Scholar]
- 54.Engel LA, Jing Z, O’Brien DE, Sun M, and Kotzbauer PT (2010) Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 5, e12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, and Hu XY (2013) Four novel rare mutations of PLA2G6 in Chinese population with Parkinson’s disease. Parkinsonism Relat Disord 19, 21–26 [DOI] [PubMed] [Google Scholar]
- 56.Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin WW, Wang L, and Bellen HJ (2018) Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab 28, 605618 e606 [DOI] [PubMed] [Google Scholar]
- 57.Lei X, Zhang S, Bohrer A, and Ramanadham S (2008) Calcium-independent phospholipase A2 (iPLA2 beta)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem 283, 34819–34832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Basselin M, Rosa AO, Ramadan E, Cheon Y, Chang L, Chen M, Greenstein D, Wohltmann M, Turk J, and Rapoport SI (2010) Imaging decreased brain docosahexaenoic acid metabolism and signaling in iPLA2beta (VIA)-deficient mice. J Lipid Res 51, 3166–3173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, Greenstein D, Wohltmann M, Turk J, Rapoport SI, and Taha AY (2012) Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A2-VIA (iPLA2beta)-knockout mice. Biochim Biophys Acta 1821, 1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y, Sakoda S, and Sumi-Akamaru H (2011) Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 31, 11411–11420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strokin M, and Reiser G (2016) Mitochondria from a mouse model of the human infantile neuroaxonal dystrophy (INAD) with genetic defects in VIA iPLA2 have disturbed Ca2+ regulation with reduction in Ca2+ capacity. Neurochem Int 99, 187–193 [DOI] [PubMed] [Google Scholar]
- 62.Sumi-Akamaru H, Beck G, Shinzawa K, Kato S, Riku Y, Yoshida M, Fujimura H, Tsujimoto Y, Sakoda S, and Mochizuki H (2016) High expression of alpha-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol Commun 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kinghorn KJ, Castillo-Quan JI, Bartolome F, Angelova PR, Li L, Pope S, Cocheme HM, Khan S, Asghari S, Bhatia KP, Hardy J, Abramov AY, and Partridge L (2015) Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 138, 1801–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiu CC, Yeh TH, Lu CS, Huang YC, Cheng YC, Huang YZ, Weng YH, Liu YC, Lai SC, Chen YL, Chen YJ, Chen CL, Chen HY, Lin YW, and Wang HL (2017) PARK14 PLA2G6 mutants are defective in preventing rotenone-induced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway. Oncotarget 8, 79046–79060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma ZA (2012) The role of peroxidation of mitochondrial membrane phospholipids in pancreatic beta -cell failure. Curr Diabetes Rev 8, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seleznev K, Zhao C, Zhang XH, Song K, and Ma ZA (2006) Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem 281, 22275–22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma ZA, Zhao Z, and Turk J (2012) Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Exp Diabetes Res 2012, 703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song H, Wohltmann M, Tan M, Ladenson JH, and Turk J (2014) Group VIA phospholipase A2 mitigates palmitate-induced beta-cell mitochondrial injury and apoptosis. J Biol Chem 289, 14194–14210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K, Turk J, and Ma ZA (2010) Protection of pancreatic beta-cells by group VIA phospholipase A2-mediated repair of mitochondrial membrane peroxidation. Endocrinology 151, 3038–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balsinde J (2002) Roles of various phospholipases A2 in providing lysophospholipid acceptors for fatty acid phospholipid incorporation and remodelling. Biochem J 364, 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, and Chen YD (1988) Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37, 1020–1024 [DOI] [PubMed] [Google Scholar]
- 72.Eitel K, Staiger H, Rieger J, Mischak H, Brandhorst H, Brendel MD, Bretzel RG, Haring HU, and Kellerer M (2003) Protein kinase C delta activation and translocation to the nucleus are required for fatty acid-induced apoptosis of insulin-secreting cells. Diabetes 52, 991–997 [DOI] [PubMed] [Google Scholar]
- 73.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, and Summers SA (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5, 167–179 [DOI] [PubMed] [Google Scholar]
- 74.Petan T, and Krizaj I (2015) Is iPLA2beta a Novel Target for the Development of New Strategies to Alleviate Inflammatory Bowel Disease? Dig Dis Sci 60, 3504–3506 [DOI] [PubMed] [Google Scholar]
- 75.Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, and Unger RH (1998) Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem 273, 32487–32490 [DOI] [PubMed] [Google Scholar]
- 76.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, and Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 77.Han MS, Lim YM, Quan W, Kim JR, Chung KW, Kang M, Kim S, Park SY, Han JS, Park SY, Cheon HG, Dal Rhee S, Park TS, and Lee MS (2011) Lysophosphatidylcholine as an effector of fatty acid-induced insulin resistance. J Lipid Res 52, 1234–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F, Schappe T, Moore K, Han X, Abumrad NA, and Gross RW (2010) Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J Biol Chem 285, 36495–36510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song H, Wohltmann M, Bao S, Ladenson JH, Semenkovich CF, and Turk J (2010) Mice deficient in group VIB phospholipase A2 (iPLA2gamma) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am J Physiol Endocrinol Metab 298, E1097–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee JY, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJ, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BW, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PK, Lucas G, Luben R, Loos RJ, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, Konig IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens AC, Ingelsson E, Igl W, Kees Hovingh G, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Doring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJ, de Faire U, Crawford G, Collins FS, Chen YD, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA Jr., Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, and Kathiresan S (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, and Turk J (2006) Effects of stable suppression of Group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. J Biol Chem 281, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, and Turk J (2001) Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2beta) indicate a signaling rather than a housekeeping role for iPLA2beta. J Biol Chem 276, 13198–13208 [DOI] [PubMed] [Google Scholar]
- 83.Bao S, Jacobson DA, Wohltmann M, Bohrer A, Jin W, Philipson LH, and Turk J (2008) Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2beta in pancreatic beta-cells and in iPLA2beta-null mice. Am J Physiol Endocrinol Metab 294, E217–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang L, Zhong S, Li Y, Ji G, Sundaram M, and Yao Z (2013) Global inactivation of the PLA2G6 gene in mice does not cause dyslipidemia under chow or high-fat diet conditions. J Cancer Prev 18, 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deng X, Wang J, Jiao L, Utaipan T, Tuma-Kellner S, Schmitz G, Liebisch G, Stremmel W, and Chamulitrat W (2016) iPLA2beta deficiency attenuates obesity and hepatic steatosis in ob/ob mice through hepatic fatty-acyl phospholipid remodeling. Biochim Biophys Acta 1861, 449–461 [DOI] [PubMed] [Google Scholar]
- 86.Lu Y, Day FR, Gustafsson S, Buchkovich ML, Na J, Bataille V, Cousminer DL, Dastani Z, Drong AW, Esko T, Evans DM, Falchi M, Feitosa MF, Ferreira T, Hedman AK, Haring R, Hysi PG, Iles MM, Justice AE, Kanoni S, Lagou V, Li R, Li X, Locke A, Lu C, Magi R, Perry JR, Pers TH, Qi Q, Sanna M, Schmidt EM, Scott WR, Shungin D, Teumer A, Vinkhuyzen AA, Walker RW, Westra HJ, Zhang M, Zhang W, Zhao JH, Zhu Z, Afzal U, Ahluwalia TS, Bakker SJ, Bellis C, Bonnefond A, Borodulin K, Buchman AS, Cederholm T, Choh AC, Choi HJ, Curran JE, de Groot LC, De Jager PL, Dhonukshe-Rutten RA, Enneman AW, Eury E, Evans DS, Forsen T, Friedrich N, Fumeron F, Garcia ME, Gartner S, Han BG, Havulinna AS, Hayward C, Hernandez D, Hillege H, Ittermann T, Kent JW, Kolcic I, Laatikainen T, Lahti J, Mateo Leach I, Lee CG, Lee JY, Liu T, Liu Y, Lobbens S, Loh M, Lyytikainen LP, Medina-Gomez C, Michaelsson K, Nalls MA, Nielson CM, Oozageer L, Pascoe L, Paternoster L, Polasek O, Ripatti S, Sarzynski MA, Shin CS, Narancic NS, Spira D, Srikanth P, Steinhagen-Thiessen E, Sung YJ, Swart KM, Taittonen L, Tanaka T, Tikkanen E, van der Velde N, van Schoor NM, Verweij N, Wright AF, Yu L, Zmuda JM, Eklund N, Forrester T, Grarup N, Jackson AU, Kristiansson K, Kuulasmaa T, Kuusisto J, Lichtner P, Luan J, Mahajan A, Mannisto S, Palmer CD, Ried JS, Scott RA, Stancakova A, Wagner PJ, Demirkan A, Doring A, Gudnason V, Kiel DP, Kuhnel B, Mangino M, McKnight B, Menni C, O’Connell JR, Oostra BA, Shuldiner AR, Song K, Vandenput L, van Duijn CM, Vollenweider P, White CC, Boehnke M, Boettcher Y, Cooper RS, Forouhi NG, Gieger C, Grallert H, Hingorani A, Jorgensen T, Jousilahti P, Kivimaki M, Kumari M, Laakso M, Langenberg C, Linneberg A, Luke A, McKenzie CA, Palotie A, Pedersen O, Peters A, Strauch K, Tayo BO, Wareham NJ, Bennett DA, Bertram L, Blangero J, Bluher M, Bouchard C, Campbell H, Cho NH, Cummings SR, Czerwinski SA, Demuth I, Eckardt R, Eriksson JG, Ferrucci L, Franco OH, Froguel P, Gansevoort RT, Hansen T, Harris TB, Hastie N, Heliovaara M, Hofman A, Jordan JM, Jula A, Kahonen M, Kajantie E, Knekt PB, Koskinen S, Kovacs P, Lehtimaki T, Lind L, Liu Y, Orwoll ES, Osmond C, Perola M, Perusse L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Rivadeneira F, Rudan I, Salomaa V, Sorensen TI, Stumvoll M, Tonjes A, Towne B, Tranah GJ, Tremblay A, Uitterlinden AG, van der Harst P, Vartiainen E, Viikari JS, Vitart V, Vohl MC, Volzke H, Walker M, Wallaschofski H, Wild S, Wilson JF, Yengo L, Bishop DT, Borecki IB, Chambers JC, Cupples LA, Dehghan A, Deloukas P, Fatemifar G, Fox C, Furey TS, Franke L, Han J, Hunter DJ, Karjalainen J, Karpe F, Kaplan RC, Kooner JS, McCarthy MI, Murabito JM, Morris AP, Bishop JA, North KE, Ohlsson C, Ong KK, Prokopenko I, Richards JB, Schadt EE, Spector TD, Widen E, Willer CJ, Yang J, Ingelsson E, Mohlke KL, Hirschhorn JN, Pospisilik JA, Zillikens MC, Lindgren C, Kilpelainen TO, and Loos RJ (2016) New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun 7, 10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mancuso DJ, Abendschein DR, Jenkins CM, Han X, Saffitz JE, Schuessler RB, and Gross RW (2003) Cardiac ischemia activates calcium-independent phospholipase A2beta, precipitating ventricular tachyarrhythmias in transgenic mice: rescue of the lethal electrophysiologic phenotype by mechanism-based inhibition. J Biol Chem 278, 22231–22236 [DOI] [PubMed] [Google Scholar]
- 88.Su X, Han X, Mancuso DJ, Abendschein DR, and Gross RW (2005) Accumulation of long-chain acylcarnitine and 3-hydroxy acylcarnitine molecular species in diabetic myocardium: identification of alterations in mitochondrial fatty acid processing in diabetic myocardium by shotgun lipidomics. Biochemistry 44, 5234–5245 [DOI] [PubMed] [Google Scholar]
- 89.Barbour SE, Nguyen PT, Park M, Emani B, Lei X, Kambalapalli M, Shultz JC, Wijesinghe D, Chalfant CE, and Ramanadham S (2015) Group VIA Phospholipase A2 (iPLA2beta) Modulates Bcl-x 5′-Splice Site Selection and Suppresses Anti-apoptotic Bcl-x(L) in beta-Cells. J Biol Chem 290, 11021–11031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lei X, Zhang S, Barbour SE, Bohrer A, Ford EL, Koizumi A, Papa FR, and Ramanadham S (2010) Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem 285, 6693–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lei X, Zhang S, Bohrer A, Barbour SE, and Ramanadham S (2012) Role of calcium-independent phospholipase A2beta in human pancreatic islet beta-cell apoptosis. Am J Physiol Endocrinol Metab 303, E1386–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carnevale KA, and Cathcart MK (2001) Calcium-independent phospholipase A2 is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J Immunol 167, 3414–3421 [DOI] [PubMed] [Google Scholar]
- 93.Mishra RS, Carnevale KA, and Cathcart MK (2008) iPLA2beta: front and center in human monocyte chemotaxis to MCP-1. The Journal of experimental medicine 205, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luster AD (1998) Chemokines--chemotactic cytokines that mediate inflammation. The New England journal of medicine 338, 436–445 [DOI] [PubMed] [Google Scholar]
- 95.Lambeth JD, Kawahara T, and Diebold B (2007) Regulation of Nox and Duox enzymatic activity and expression. Free radical biology & medicine 43, 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee CF, Qiao M, Schroder K, Zhao Q, and Asmis R (2010) Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circulation research 106, 1489–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Groemping Y, and Rittinger K (2005) Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386, 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burg ND, and Pillinger MH (2001) The neutrophil: function and regulation in innate and humoral immunity. Clin Immunol 99, 7–17 [DOI] [PubMed] [Google Scholar]
- 99.Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, and Silliman CC (2005) Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. Journal of leukocyte biology 78, 1025–1042 [DOI] [PubMed] [Google Scholar]
- 100.Dana R, Malech HL, and Levy R (1994) The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem J 297 ( Pt 1), 217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shmelzer Z, Haddad N, Admon E, Pessach I, Leto TL, Eitan-Hazan Z, Hershfinkel M, and Levy R (2003) Unique targeting of cytosolic phospholipase A2 to plasma membranes mediated by the NADPH oxidase in phagocytes. J Cell Biol 162, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Omori K, Ohira T, Uchida Y, Ayilavarapu S, Batista EL Jr., Yagi M, Iwata T, Liu H, Hasturk H, Kantarci A, and Van Dyke TE (2008) Priming of neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. Journal of leukocyte biology 84, 292–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, Iwata T, Yagi M, Hasturk H, and Van Dyke TE (2010) Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J Immunol 184, 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Henderson LM, and Chappell JB (1992) The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochem J 283 ( Pt 1), 171–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brechard S, Bueb JL, and Tschirhart EJ (2005) Interleukin-8 primes oxidative burst in neutrophil-like HL-60 through changes in cytosolic calcium. Cell Calcium 37, 531–540 [DOI] [PubMed] [Google Scholar]
- 106.Doussiere J, Bouzidi F, and Vignais PV (2002) The S100A8/A9 protein as a partner for the cytosolic factors of NADPH oxidase activation in neutrophils. European journal of biochemistry 269, 3246–3255 [DOI] [PubMed] [Google Scholar]
- 107.Lominadze G, Rane MJ, Merchant M, Cai J, Ward RA, and McLeish KR (2005) Myeloid-related protein-14 is a p38 MAPK substrate in human neutrophils. J Immunol 174, 7257–7267 [DOI] [PubMed] [Google Scholar]
- 108.Beckett CS, Pennington K, and McHowat J (2006) Activation of MAPKs in thrombin-stimulated ventricular myocytes is dependent on Ca2+-independent PLA2. American journal of physiology. Cell physiology 290, C1350–1354 [DOI] [PubMed] [Google Scholar]
- 109.Schenten V, Brechard S, Plancon S, Melchior C, Frippiat JP, and Tschirhart EJ (2010) iPLA2, a novel determinant in Ca2+- and phosphorylation-dependent S100A8/A9 regulated NOX2 activity. Biochim Biophys Acta 1803, 840–847 [DOI] [PubMed] [Google Scholar]
- 110.Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, and Asmis R (2012) NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arteriosclerosis, thrombosis, and vascular biology 32, 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tan C, Day R, Bao S, Turk J, and Zhao QD (2014) Group VIA phospholipase A2 mediates enhanced macrophage migration in diabetes mellitus by increasing expression of nicotinamide adenine dinucleotide phosphate oxidase 4. Arteriosclerosis, thrombosis, and vascular biology 34, 768–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shmelzer Z, Karter M, Eisenstein M, Leto TL, Hadad N, Ben-Menahem D, Gitler D, Banani S, Wolach B, Rotem M, and Levy R (2008) Cytosolic phospholipase A2alpha is targeted to the p47phox-PX domain of the assembled NADPH oxidase via a novel binding site in its C2 domain. J Biol Chem 283, 31898–31908 [DOI] [PubMed] [Google Scholar]
- 113.Corbalan-Garcia S, Rodriguez-Alfaro JA, and Gomez-Fernandez JC (1999) Determination of the calcium-binding sites of the C2 domain of protein kinase Calpha that are critical for its translocation to the plasma membrane. Biochem J 337 ( Pt 3), 513–521 [PMC free article] [PubMed] [Google Scholar]
- 114.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, and Bolotina VM (2004) A novel mechanism for the store-operated calcium influx pathway. Nature cell biology 6, 113–120 [DOI] [PubMed] [Google Scholar]
- 115.Moon SH, Jenkins CM, Mancuso DJ, Turk J, and Gross RW (2008) Smooth muscle cell arachidonic acid release, migration, and proliferation are markedly attenuated in mice null for calcium-independent phospholipase A2beta. J Biol Chem 283, 33975–33987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee SH, Park DW, Park SC, Park YK, Hong SY, Kim JR, Lee CH, and Baek SH (2009) Calcium-independent phospholipase A2beta-Akt signaling is involved in lipopolysaccharide-induced NADPH oxidase 1 expression and foam cell formation. J Immunol 183, 7497–7504 [DOI] [PubMed] [Google Scholar]
- 117.Lee JG, Lim EJ, Park DW, Lee SH, Kim JR, and Baek SH (2008) A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal 20, 2266–2275 [DOI] [PubMed] [Google Scholar]
- 118.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, and Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America 101, 10679–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van Velzen AG, Suzuki H, Kodama T, and van Berkel TJ (1999) The role of scavenger receptor class A in the adhesion of cells is dependent on cell type and cellular activation state. Exp Cell Res 250, 264–271 [DOI] [PubMed] [Google Scholar]
- 120.Price LS, Leng J, Schwartz MA, and Bokoch GM (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell 9, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nikolic DM, Gong MC, Turk J, and Post SR (2007) Class A scavenger receptor-mediated macrophage adhesion requires coupling of calcium-independent phospholipase A(2) and 12/15-lipoxygenase to Rac and Cdc42 activation. J Biol Chem 282, 33405–33411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sharma J, Eickhoff CS, Hoft DF, Marentette JO, Turk J, and McHowat J (2014) Absence of calcium-independent phospholipase A2 beta impairs platelet-activating factor production and inflammatory cell recruitment in Trypanosoma cruzi-infected endothelial cells. Physiol Rep 2, e00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xie Z, Gong MC, Su W, Xie D, Turk J, and Guo Z (2010) Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem 285, 8628–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Somlyo AP, and Somlyo AV (1994) Signal transduction and regulation in smooth muscle. Nature 372, 231–236 [DOI] [PubMed] [Google Scholar]
- 125.Bone RN, Gai Y, Magrioti V, Kokotou MG, Ali T, Lei X, Tse HM, Kokotos G, and Ramanadham S (2015) Inhibition of Ca2+-independent phospholipase A2beta (iPLA2beta) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 64, 541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ashley JW, Hancock WD, Nelson AJ, Bone RN, Tse HM, Wohltmann M, Turk J, and Ramanadham S (2016) Polarization of Macrophages toward M2 Phenotype Is Favored by Reduction in iPLA2beta (Group VIA Phospholipase A2). J Biol Chem 291, 23268–23281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu S, Xie Z, Zhao Q, Pang H, Turk J, Calderon L, Su W, Zhao G, Xu H, Gong MC, and Guo Z (2012) Smooth muscle-specific expression of calcium-independent phospholipase A2beta (iPLA2beta) participates in the initiation and early progression of vascular inflammation and neointima formation. J Biol Chem 287, 24739–24753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiao L, Inhoffen J, Gan-Schreier H, Tuma-Kellner S, Stremmel W, Sun Z, and Chamulitrat W (2015) Deficiency of Group VIA Phospholipase A2 (iPLA2beta) Renders Susceptibility for Chemical-Induced Colitis. Dig Dis Sci 60, 3590–3602 [DOI] [PubMed] [Google Scholar]
- 129.Jiao L, Gan-Schreier H, Tuma-Kellner S, Stremmel W, and Chamulitrat W (2015) Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim Biophys Acta 1852, 1646–1657 [DOI] [PubMed] [Google Scholar]
- 130.Jiao L, Gan-Schreier H, Zhu X, Wei W, Tuma-Kellner S, Liebisch G, Stremmel W, and Chamulitrat W (2017) Ageing sensitized by iPLA2beta deficiency induces liver fibrosis and intestinal atrophy involving suppression of homeostatic genes and alteration of intestinal lipids and bile acids. Biochim Biophys Acta 1862, 1520–1533 [DOI] [PubMed] [Google Scholar]
- 131.Sharma J, Blase JR, Hoft DF, Marentette JO, Turk J, and McHowat J (2016) Mice with genetic deletion of group VIA phospholipase A2beta exhibit impaired macrophage function and increased parasite load in trypanosoma cruzi-induced myocarditis. Infect Immun 84, 1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalyvas A, Baskakis C, Magrioti V, Constantinou-Kokotou V, Stephens D, Lopez-Vales R, Lu JQ, Yong VW, Dennis EA, Kokotos G, and David S (2009) Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain 132, 1221–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gilroy DW, Newson J, Sawmynaden P, Willoughby DA, and Croxtall JD (2004) A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J 18, 489–498 [DOI] [PubMed] [Google Scholar]
- 134.Malley KR, Koroleva O, Miller I, Sanishvili R, Jenkins CM, Gross RW, and Korolev S (2018) The structure of iPLA2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nature communications 9, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jenkins CM, Wolf MJ, Mancuso DJ, and Gross RW (2001) Identification of the calmodulin-binding domain of recombinant calcium-independent phospholipase A2beta. implications for structure and function. J Biol Chem 276, 7129–7135 [DOI] [PubMed] [Google Scholar]
- 136.Jenkins CM, Yan W, Mancuso DJ, and Gross RW (2006) Highly selective hydrolysis of fatty acyl-CoAs by calcium-independent phospholipase A2beta. Enzyme autoacylation and acylCoA-mediated reversal of calmodulin inhibition of phospholipase A2 activity. J Biol Chem 281, 15615–15624 [DOI] [PubMed] [Google Scholar]
- 137.Wolf MJ, and Gross RW (1996) The calcium-dependent association and functional coupling of calmodulin with myocardial phospholipase A2. Implications for cardiac cycle-dependent alterations in phospholipolysis. J Biol Chem 271, 20989–20992 [DOI] [PubMed] [Google Scholar]
- 138.Wang Z, Ramanadham S, Ma ZA, Bao S, Mancuso DJ, Gross RW, and Turk J (2005) Group VIA phospholipase A2 forms a signaling complex with the calcium/calmodulin-dependent protein kinase IIbeta expressed in pancreatic islet beta-cells. J Biol Chem 280, 6840–6849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mouchlis VD, Chen Y, McCammon JA, and Dennis EA (2018) Membrane allostery and unique hydrophobic sites promote enzyme substrate specificity. J Am Chem Soc 140, 3285–3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kokotou MG, Limnios D, Nikolaou A, Psarra A, and Kokotos G (2017) Inhibitors of phospholipase A2 and their therapeutic potential: an update on patents (2012–2016). Expert Opin Ther Pat 27, 217–225 [DOI] [PubMed] [Google Scholar]
- 141.Ali T, Kokotos G, Magrioti V, Bone RN, Mobley JA, Hancock W, and Ramanadham S (2013) Characterization of FKGK18 as inhibitor of group VIA Ca2+-independent phospholipase A2 (iPLA2beta): candidate drug for preventing beta-cell apoptosis and diabetes. PLoS One 8, e71748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mouchlis VD, Limnios D, Kokotou MG, Barbayianni E, Kokotos G, McCammon JA, and Dennis EA (2016) Development of potent and selective inhibitors for Group VIA calcium-independent phospholipase a2 guided by molecular dynamics and structure-activity relationships. J Med Chem 59, 4403–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mouchlis VD, Morisseau C, Hammock BD, Li S, McCammon JA, and Dennis EA (2016) Computer-aided drug design guided by hydrogen/deuterium exchange mass spectrometry: A powerful combination for the development of potent and selective inhibitors of Group VIA calcium-independent phospholipase A2. Bioorg Med Chem 24, 4801–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hirabayashi T, Anjo T, Kaneko A, Senoo Y, Shibata A, Takama H, Yokoyama K, Nishito Y, Ono T, Taya C, Muramatsu K, Fukami K, Munoz-Garcia A, Brash AR, Ikeda K, Arita M, Akiyama M, and Murakami M (2017) PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nat Commun 8, 14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ohno Y, Kamiyama N, Nakamichi S, and Kihara A (2017) PNPLA1 is a transacylase essential for the generation of the skin barrier lipid omega-O-acylceramide. Nat Commun 8, 14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, and Gimeno RE (2005) Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res 46, 2477–2487 [DOI] [PubMed] [Google Scholar]
- 147.Liu GY, Moon SH, Jenkins CM, Li M, Sims HF, Guan S, and Gross RW (2017) The phospholipase iPLA2gamma is a major mediator releasing oxidized aliphatic chains from cardiolipin, integrating mitochondrial bioenergetics and signaling. J Biol Chem 292, 10672–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mancuso DJ, Jenkins CM, Sims HF, Cohen JM, Yang J, and Gross RW (2004) Complex transcriptional and translational regulation of iPLA2gamma resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. European journal of biochemistry 271, 4709–4724 [DOI] [PubMed] [Google Scholar]
- 149.Moon SH, Jenkins CM, Liu X, Guan S, Mancuso DJ, and Gross RW (2012) Activation of mitochondrial calcium-independent phospholipase A2gamma (iPLA2gamma) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J Biol Chem 287, 14880–14895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Moon SH, Liu X, Cedars AM, Yang K, Kiebish MA, Joseph SM, Kelley J, Jenkins CM, and Gross RW (2018) Heart failure-induced activation of phospholipase iPLA2gamma generates hydroxyeicosatetraenoic acids opening the mitochondrial permeability transition pore. J Biol Chem 293, 115–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Moon SH, Mancuso DJ, Sims HF, Liu X, Nguyen AL, Yang K, Guan S, Dilthey BG, Jenkins CM, Weinheimer CJ, Kovacs A, Abendschein D, and Gross RW (2016) Cardiac Myocyte-specific Knock-out of Calcium-independent Phospholipase A2gamma (iPLA2gamma) decreases oxidized fatty acids during ischemia/reperfusion and reduces infarct size. J Biol Chem 291, 19687–19700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Su X, Mancuso DJ, Bickel PE, Jenkins CM, and Gross RW (2004) Small interfering RNA knockdown of calcium-independent phospholipases A2 beta or gamma inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. J Biol Chem 279, 21740–21748 [DOI] [PubMed] [Google Scholar]
- 153.Jacobson DA, Weber CR, Bao S, Turk J, and Philipson LH (2007) Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem 282, 7442–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Moran JM, Buller RM, McHowat J, Turk J, Wohltmann M, Gross RW, and Corbett JA (2005) Genetic and pharmacologic evidence that calcium-independent phospholipase A2beta regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J Biol Chem 280, 28162–28168 [DOI] [PubMed] [Google Scholar]
- 155.Sharma J, Turk J, and McHowat J (2010) Endothelial cell prostaglandin I2 and platelet-activating factor production are markedly attenuated in the calcium-independent phospholipase A2beta knockout mouse. Biochemistry 49, 5473–5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.