

Abstract
Approval of generic drugs by the US Food and Drug Administration (FDA) requires the product to be pharmaceutically equivalent to the reference listed drug (RLD) and demonstrate bioequivalence (BE) in effectiveness when administered to patients under the conditions in the RLD product labeling. Effectiveness is determined by drug exposure at the target sites. However, since such measurement is usually unavailable, systemic exposure is assumed to equal target site exposure and systemic BE to equal target site BE. This assumption, while it often applies to small molecule drug products that are readily dissolved in biological fluids and systemically absorbed, is unlikely to apply to nanotechnology products (NP) that exist as heterogeneous systems and are subjected to dimension- and material-dependent changes. This commentary provides an overview of the intersecting and spatial-dependent processes and variables governing the delivery and residence of oncologic NP in solid tumors. In order to provide a quantitative perspective of the collective effects of these processes, we used quantitative systems pharmacology (QSP) multi-scale modeling to capture the physicochemical and biological events on several scales (whole-body, organ/suborgan, cell/subcellular, spatial locations, time). QSP is an emerging field that entails using modeling and computation to facilitate drug development; an analogous approach (i.e., model-informed drug development) is advocated by to FDA. The QSP model-based simulations illustrated that small changes in NP attributes (e.g., size variations during manufacturing, interactions with proteins in biological milieu) could lead to disproportionately large differences in target site exposure, rending systemic BE unlikely to equal target site BE.
Keywords: FDA, nanotechnology, quantitative systems pharmacology, systemic bioequivalence, target site bioequivalence
The purpose of this commentary is to illustrate how the multiple, intersecting dynamic and spatial-dependent processes and variables affect the delivery and residence of oncologic nanotechnology products (NP) in solid tumors and how they confound the bioequivalence (BE) determination and render systemic BE unlikely to equal target site BE. Parts of this commentary were presented at an FDA Public Workshop in October 2017 (1).
In the USA, approval of generic drugs by the Food and Drug Administration (FDA) requires the generic product to be pharmaceutically equivalent to the reference listed drug (RLD) and demonstrate BE in effectiveness when administered to patients under the conditions specified in the RLD product labeling. Effectiveness is determined by the drug exposure at the intended target sites. Since target site exposure measurement is usually unavailable, systemic exposure is assumed to equal target site exposure and systemic BE to equal target site BE. This assumption, while it often applies to small-molecule drug products that are designed to be systemically absorbed and are readily dissolved in biological fluids, is unlikely to apply to NP that exist as heterogeneous systems and are subjected to dimension- and material-dependent changes.
The first part of this commentary outlines the FDA definition of nanotechnologies and the FDA guidance on BE determination. The second part provides an overview of the processes and variables governing the delivery and residence of NP in a tumor. Lastly, the kinetic processes on several scales (whole-body, organ/suborgan, cell/subcellular, spatial locations, time) were captured using quantitative systems pharmacology (QSP)–based multi-scale modeling, and simulations were performed to demonstrate why systemic BE is unlikely to equal target site BE for NP.
QSP, an emerging field that entails using modeling and computation to interpret, interrogate, and integrate drug effects spanning from molecule to whole organism, is expected to enhance the efficiency in drug development (2). FDA has, since late 2017, advocated the use of an analogous approach, model-informed drug development, in the development and approval of innovator and generic drugs. The three focus areas for generic drugs are (a) mechanistic-based absorption models, (b) model-based development and BE determination, and (c) data-based knowledge discovery for review optimization of Abbreviated New Drug Application (ANDA) (3).
FDA DEFINITION OF NANOTECHNOLOGIES AND FDA GUIDANCES ON BE DETERMINATION OF SMALL-MOLECULE AND NANOTECHNOLOGY PRODUCTS
FDA Definition of Nanotechnologies.
Nanomaterials range from simple nanocrystals, organic (e.g., liposome, polymeric NP) and inorganic nanomaterials (e.g., iron colloids), to complex-structure integrated NP (e.g., core-shell, surface-modified NP). FDA, in its 2017 draft guidance, specifically states that the agency has not established regulatory definitions of nanotechnology, nanomaterial, nanoscale, or other related terms. FDA will, as described in its 2014 nanotechnology considerations guidance, continue to consider nanotechnology products as entities engineered to have at least one external dimension or an internal or surface structure, in the nanoscale range (~1–100 nm) or entities having up to 1000-nm dimension plus dimension-dependent physical or chemical properties or biological effects (4,5).
At present, there are a total of 49 FDA-approved innovator NP including 33 parenteral and 16 non-parenteral NP (6–9). Among the 33 parenteral NP, 19 contain active pharmaceutical ingredients (API) and 10 are for oncologic indications (Table I), and the remaining 14 include 5 intravenous fat emulsions for nutrition, 8 intravenous iron-polymer complexes for iron deficiency anemia, and 1 metal-nonmetal complex of technetium sulfur colloid for imaging. Additional FDA-approved entities include 239 parenteral protein and peptide products (12) that would be within the size range and share the dimension-dependent properties of FDA-defined NP.
Table I.
FDA-approved innovator parenteral API-containing NP. Abelcet® is included in a FDA NP list (6,7), but has been reported to have an average diameter of >1000 nm (10,11). NA, not available
| Trade name | API | Formulation | Average diameter, nm | Route of administration | Approval year |
|---|---|---|---|---|---|
| Doxil® | Doxorubicin | Liposome | 85 | Intravenous | 1995 |
| DaunoXome® | Daunorubicin | Liposome | 45 | Intravenous | 1996 |
| AmBisome® | Amphotericin B | Liposome | 60–70 | Intravenous | 1997 |
| Visudyne® | Verteporfin | Liposome | 18–104 | Intravenous | 2000 |
| Marqibo® | Vincristine sulfate | Liposome | 100 | Intravenous | 2012 |
| Onivyde® | Irinotecan HCL | Liposome | 110 | Intravenous | 2015 |
| Vyxeos® | Daunorubicin/cytarabine | Liposome | 100 | Intravenous | 2017 |
| Abelcet® | Amphotericin B | Lipid complex | 1600–11,100 | Intravenous | 1995 |
| Amphotec® | Amphotericin B | Lipid complex | 150 | Intravenous | 1996 |
| Onpattro® | Patisiran | Lipid complex | < 100 | Intravenous | 2018 |
| Abraxane® | Paclitaxel | Protein complex | 130 | Intravenous | 2005 |
| Taxol® | Paclitaxel | Micelle | 13 | Intravenous | 1995 |
| Taxotere® | Docetaxel | Micelle | 11 | Intravenous | 1996 |
| Jevtana® | Cabazitaxel | Micelle | NA | Intravenous | 2010 |
| Somatuline® | Lanreotide acetate | Nanotube | 24 | Subcutaneous | 2007 |
| Invega Sustenna® | Paliperidone palmitate | Nanocrystal | NA | Intramuscular | 2009 |
| Invega Trinza® | Paliperidone palmitate | Nanocrystal | NA | Intramuscular | 2015 |
| Ryanodex® | Dantrolene sodium | Nanocrystal | NA | Intravenous | 2014 |
| Aristada Initio® | Aripiprazole lauroxil | Nanocrystal | NA | Intramuscular | 2018 |
FDA Guidance on BE of Generic Medicines.
FDA requires a generic drug to be the same as an already marketed brand-name drug in dosage form, safety, strength, route of administration, quality, performance characteristics, and intended use, so that the generic drug works in the same way and provides the same clinical benefit as its brand-name version (13). FDA defines BE as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study (14).
FDA Guidance on BE Studies.
FDA recommends several types of BE studies, in the following descending order of preference: (a) pharmacokinetic (PK) study, (b) pharmacodynamic study, (c) clinical study, (d) accepted in vitro study, and (e) any other approach deemed adequate by FDA (15).
The FDA-recommended PK studies for BE determination comprise (a) conducting a randomized, crossover trial with test and reference products in human subjects, (b) measuring the maximum concentration (Cmax) and the total drug exposure in serum (area under concentration-time curve or AUC) in the subjects, and (c) obtaining the 90% confidence interval for the test product-to-reference product ratio of the averages (population geometric means) of Cmax and AUC. Systemic BE is established if the 90% confidence interval falls within a defined range, usually 80–125% of the test product-to-reference product ratio of the averages (16–18).
The above systemic BE paradigm has been successfully applied to products comprising small-molecule drugs, typically a single API, that dissolve in biological fluids such as plasma and interstitial fluid and are intended to be absorbed and distributed via the systemic circulation. However, other products that do not have these properties present significantly greater challenges. For example, FDA acknowledges difficulties in establishing BE when the drug cannot be readily measured in the blood, when the therapeutic effect is due to localized delivery to the target site, or when the product contains a mixture of API, and recommends it may be appropriate to use other additional means to establish target site BE, e.g., clinical endpoints, pharmacodynamic endpoints, or suitably designed and validated in vitro studies, in a product-specific manner (17,19). In October 2017, FDA issued a draft guidance for ANDA; this document includes some NP as complex formulations that require additional considerations and recommends sponsors of such products to seek formal meetings to clarify regulatory expectations early in product development (20).
FDA Draft Guidance on BE Determination of NP.
FDA has published a total of 7 documents mentioning nanomaterials between June 2014 and April 2018 (4,5,20–24). In general, a generic NP must meet the same five requirements recommended for non-NP drugs: (a) safe and effective, (b) pharmaceutically equivalent to RLD, (c) bioequivalent to RLD, (d) adequately labeled, and (e) manufactured in compliance with cGMP regulations (25). The 2017 FDA draft guidance has provided instructions and nonbinding recommendations regarding the following key components: (a) potential risk factors for products containing nanomaterials; (b) chemistry, manufacturing, and controls; (c) nonclinical studies; (d) clinical development; (e) bioanalytical methods; (f) in vitro tests with human biomaterials; and (g) immunogenicity. This guidance recommends the generic NP to have formulation sameness and systemic BE as RLD (7). These recommendations are reiterated in a recent publication authored by FDA scientists outlining the paradigm for the development of generic parenteral NP (6). These FDA guidance documents also acknowledge significant challenges to the demonstration of formulation sameness or BE of NP due to the following reasons: (a) NP (including excipients) are generally heterogeneous mixtures with diverse properties (e.g., materials, drug release mechanisms, bio-distribution, particle morphology, particle size and distribution, surface property), (b) complicated manufacturing processes, and (c) more than one therapeutically active entity (e.g., free API or NP-associated API) present in both systemic circulation and target site. FDA further recognizes that NP concentrations in systemic circulation may not always reflect NP concentration at the target site, that systemic BE may not equal target site BE, and that in most cases evidence of comparable PK parameters in blood/plasma in conventional BE studies alone may not be sufficient to satisfy the requirements for generic NP approval. FDA Office of Generic Drugs plans to develop BE guidance on a product-by-product basis.
The remaining sections of this commentary provide an in-depth discussion of the complications and challenges that are the scientific basis of the above FDA considerations, plus the computer QSP-based simulations to demonstrate how small changes in two NP attributes (particle size, interactions with cells) could lead to disproportionally large differences in target site exposure for NP that have systemic BE.
DETERMINANTS AND BARRIERS OF NP TRANSPORT TO TARGET SITES
Target Sites of NP.
Potential target sites of NP are tissue interstitium (e.g., diagnostics or therapeutics), cell membrane (e.g., antibodies), or intracellular compartments (e.g., DNA, antisense, RNAi). Figure 1 shows the multiple processes involved in the NP delivery from injection site to extracellular or intracellular target. The numerous determinants and barriers affecting drug/NP transport to the intended target sites in solid tumors are summarized below. Readers are referred to earlier reviews for more details and original citations (26–36).
Fig. 1. Determinants of target site delivery and residence of NP.
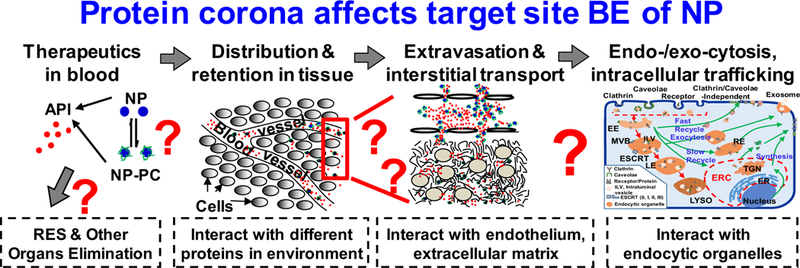
Following introduction into the systemic circulation (e.g., intravenous injection), therapeutics (small or large molecules, or their NP carriers) are distributed in blood. The concentration or amount of NP reaching a target cell is determined by the following events: (a) removal from the systemic circulation by RES cells/organs or elimination by metabolism and excretion, (b) transport to organs including tumors via the systemic circulation, (c) extravasation (transvascular transport by diffusion or convection) into tissue interstitium, and (d) interstitial transport by diffusion and convection to reach individual tumor cells. After reaching the target cell, NP is internalized by one or more of several mechanisms: (a) non-endocytic pathways: diffusion/active transport of small-molecule therapeutics across the cell membranes and fusion of NP with the cell membrane, and (b) endocytic pathways: clathrin-mediated endocytosis, caveolae-mediated endocytosis, clathrin- and caveolae-independent endocytosis and macropinocytosis. In an endocytic pathway, the cargo is presented to the early endosomes (EE) and get sorted into recycling endosomes (RE) that are either directly recycled back to the cell membrane or indirectly through the endosome recycling center (ERC), or late endosomes (LE) that fuse with lysosomes (LYSO). One aspect of the sorting results in budding of EE to form intraluminal vesicles (ILV), a component of multivesicular bodies (MVB) that either mature into LE or are fused with the cell membrane followed by releasing the ILV vesicles and their contents as exosomes. ERC, endocytic recycling compartment; ESCRT, endosomal sorting complexes required for transport; TGN, trans-Golgi network. Note the formation of NP-coating PC in blood due to NP interactions with serum proteins, and the continuous exchange of proteins on NP-PC with proteins in tissue microenvironment to form new NP-PC complex. Drawing of subcellular organelles is not to scale. See text for references. Figure and legend are adapted from Figure 1 of (26) and reprinted with permission
Transport of NP from Blood to Tumor Interstitium.
The processes governing the transport of NP from injection site to tumor interstitium include NP disposition in non-target organs (e.g., degradation in blood, hepatic metabolism, renal excretion, interactions with proteins or blood cells, surface opsonization and entrapment by phagocytic system and reticuloendothelial (RES) organs). These processes are dimension- and material-dependent. For example, renal excretion is primarily for small NP with <6 nm diameter. NP is transported via blood circulation and enters the tumor interstitium by transvascular diffusion and convection. NP cargo such as small-molecule drugs can be released within systemic blood, tissue interstitium, or intracellularly; the released cargo can interact with blood components, undergo elimination, or be transported to its intended targets.
Spatial-Dependent Delivery and Residence of NP in Tumors.
Compared with normal tissues, vasculature in tumors is more disorganized, is more tortuous, and presents greater flow resistance, but also has much larger openings (100–780 nm vs. 5–15 nm in most normal tissues) and is much leakier. Larger tumors show heterogeneous blood vessel distribution, with a central avascular or necrotic region, an intermediate semi-necrotic and poorly perfused region, and a stably perfused periphery. Tumors typically do not have functional lymphatic systems. These properties of tumor blood and lymph vasculatures affect the delivery and residence of NP, and result in spatial-dependent and uneven distribution within a tumor.
Transport of NP Within Tumor Interstitium.
Tissue interstitium comprises cells, and connective and supportive tissues. The fibrous proteins (e.g., collagen, elastin, fibronectin) and polysaccharides (e.g., hyaluronan, glycosaminoglycan) in extracellular matrix (ECM), as well as the tumor cells, present physical resistance to interstitial diffusive and convective transport. In general, densely packed tumor cells or ECM components reduce the tissue porosity and NP diffusivity, and NP binding to ECM components or cells reduce the fraction of free NP available for diffusive transport. The high interstitial fluid pressure, secondary to absence of lymphatic drainage and accumulation of macromolecules and waste, reduces the transvascular and interstitial convective transport. Because NP interactions with tumor cells/ECM are also determined by NP properties (e.g., size, surface charge or modifications, nanomaterials, shape, curvature), interstitial NP transport is jointly determined by NP properties and tumor properties.
Cell Binding, Internalization, and Intracellular Processing of NP.
The multi-step endocytosis and processing in multiple intracellular endocytic organelles are shown in Fig 1.
NP binds to cell membrane via specific binding, e.g., NP-receptor interaction, or via non-specific binding driven by electrostatic interactions (e.g., positively charged NP binding to negatively charged cell membrane components), followed by internalization. NP enters cells via multiple mechanisms, e.g., caveolae- or clathrin-mediated endocytosis (for NP of 60–80 nm and ~100–200 nm diameter, respectively), clathrin- and caveolae-independent endocytosis, or other non-receptor-related mechanisms such as fluid-phase macropinocytosis (for NP of >200 nm and larger particles of up to few micrometers). NP such as liposomes can also enter cells via fusion of NP lipids with cell membrane.
After endocytosis, the internalized vesicles fuse with each other to form a larger vesicle or early endosome. Contents in early endosomes are sorted into fast or slow recycling endosomes which return the contents, e.g., membrane receptor, to cell membrane. Early endosomes can evolve into multivesicular bodies that proceed to either the acidified late endosomes (pH 5–6) and then enzyme-rich lysosomes (pH 4.5–5) where their contents are degraded, or migrate to a pericellular location where their contents are released as exosomes.
NP-COATING PROTEIN CORONA IS ANOTHER CONFOUNDING FACTOR
NP-Coating Protein Corona.
Adsorption of proteins in biological milieu (e.g., apolipoprotein, complement protein, prothrombin, vitronectin, immunoglobulin, fibrinogen, serum albumin) on NP surface leads to protein corona (PC). This process occurs in a wide variety of NP (e.g., metallic, metal oxide, carbon based, polymer coated, polymeric, quantum dots, liposomes). While interactions between foreign particulates and endogenous proteins, e.g., opsonization, have been demonstrated for decades, their effects on the formation and evolution of PC and the consequent changes in NP properties and disposition came into focus only ~10 years ago. PC has since been proposed as a potentially important determinant of NP disposition and functionality. For example, presence of PC affects the in vivo NP bio-distribution and clearance. PC proteins, such as immunoglobulins, complement family proteins, fibrinogen, and lipoproteins, elicit opsonization and cause entrapment in RES organs, whereas apolipoprotein B and apolipoprotein E enable transport across the blood-brain barrier and albumin prolongs the circulation time. Readers are referred to several reviews for more details and original citations (28,36–48).
Compared with liposomal and polymeric NP that have been extensively studied for 50+ years, there have been much fewer studies on NP-PC. For example, a PubMed database search showed only 226 publications in 2017 contained both “nanoparticles” and “corona” as key words. Most published studies have focused on the analysis of protein composition of PC formed in vitro or in vivo and on the NP interactions with single proteins in vitro. These studies have demonstrated PC comprises multiple proteins and the binding of one protein to NP induces conformational changes and affects the subsequent binding of other proteins. Relatively little is known regarding PC formation under different physiological and pathological conditions, the pharmacological consequences of different PC, or the quantitative effects of PC on target site NP exposure. Below is a discussion on how this knowledge gap on PC poses a challenge for the evaluation of target site BE.
Time- and Environment-Dependent Formation and Evolution of PC.
PC formation, mediated by van der Waals forces and electrostatic interactions, occurs rapidly and is completed within minutes. PC formation depends on NP properties (material, surface charge and modifications, size, shape, curvature), environmental factors (ECM composition, pH, temperature, shear stress, ionic strength), and time. The first layer of high affinity proteins on the NP surface or hard corona is covered by a layer of low-affinity proteins or the soft corona. The relationship between NP materials/structures and PC protein composition is not clear, as only a small fraction of the several thousand proteins in human plasma are present in the hard corona and they are not necessarily the most abundant or the proteins with the highest binding affinity.
The formation and stability of PC, due to the reversibility of noncovalent binding, depend on the concentrations and types of proteins available in the biological milieu. Hence, the protein composition of PC evolves as NP translocates from one biological compartment to another, e.g., from blood to tumor tissues or lymph (49,50). PC composition also evolves over time, e.g., the low binding affinity albumin in the silica NP-PC formed at early times in vitro was replaced by the high affinity fibrinogen at 50 min (51). The same also occurs in vivo; about 50% of the initial proteins of pegylated liposome PC formed in vivo in mice change over time (52), and complement C3 protein in the PC preformed in human plasma are replaced by other proteins after injection to mice (53).
Effects of PC Formation and Evolution on NP Delivery and Residence at Target Site.
The formation of NP-coating PC has multiple effects, including increased effective size, change of surface charge from positive to neutral/negative, increased opsonization, increased uptake in RES organs, and pathobiological changes (e.g., hemolysis, endothelial cell death) (28,54). The change of the surface charge of silica NP from neutral to negative reduced cell binding by up to 3-fold (55). PC on Abraxane®, the albumin-bound paclitaxel NP, acts as a barrier and reduces the drug release over 8 h from 100% in the protein-free phosphate-buffered saline to <10% in human plasma and < 30% in fetal bovine serum (56). The presence of PC proteins that are ligands to membrane receptors, e.g., apolipoprotein B (ligand to low density lipoprotein receptors), enhances NP internalization (57). These various changes, in turn, affect the NP transvascular and interstitial transport, internalization, intracellular processing, and consequently the target site delivery and residence. Similarly, the evolution of PC protein compositions can be expected to cause spatial-, environment-, and time-dependent changes in NP disposition and functionality.
Effect of PC on NP Toxicity and Efficacy.
PC reduces NP toxicity, e.g., red blood cell hemolysis by bare silica NP (54), cytotoxicity of bare cationic NP in fibroblast cells (58), and release of toxic zinc ions from zinc oxide NP (secondary to the PC-mediated stabilization of NP) (59). PC also causes toxicities or reduces the NP efficacy; examples include (a) increased immunogenicity of negatively charged poly(acrylic acid)-conjugated gold NP due to denaturation and conformation change of fibrinogen (which promotes interaction with integrin receptor) and Mac-1 (which causes the release of inflammatory cytokines and inflammatory response) (60), (b) activation of immune reactions and inflammation caused by the presence of complement proteins in PC, and (c) reduced access of gold NP surface to amyloid beta proteins resulting in lowered NP inhibition of protein fibrillation in the treatments of Parkinsonism and Alzheimer’s disease (61).
Species Differences and Subject-Specific Factors: Potential Impact on Clinical Translation of Nanotechnologies and Regulatory Considerations.
Evaluation of efficacy and toxicity in animals is a main staple in drug development. For example, immunodeficient mice lacking T and/or B cell immunity are routinely used to evaluate the therapeutic efficacy in human xenograft tumors, whereas other animals such as immunocompetent rodents and dogs are used to evaluate the host toxicities. Inter-species differences in PC compositions have been shown to affect the interactions of positively charged, neutral and negatively charged polystyrene NP with macrophages (62). However, the potential impact of inter-species differences in PC on the preclinical-to-clinical NP product development remains largely unknown.
The formation and evolution of PC also represent a challenge on BE determination. It is well-established that serum proteins in humans vary with age, ethnicity, and physiological or disease states (63,64), and differences in NP-PC among cancer and non-cancer patients have been reported (65). Inter-subject variabilities in the PC-forming proteins can be expected to affect NP disposition and play a role in determining the target site exposure. This, in turn, raises questions regarding the selection of appropriate subject population for BE studies. For example, should the subjects be selected based on their demographics or their plasma/target site protein profiles, and, since PC formation and evolution is a time-dependent process, when are the appropriate time points for BE determination?
QSP-BASED SIMULATIONS TO DEMONSTRATE SYSTEMIC BE IS UNLIKELY TO EQUAL TARGET SITE BE FOR ONCOLOGIC NP
QSP-Based Multi-scale Models.
As discussed above, the delivery and residence of NP at the target site are determined by a wide range of physical, chemical, and biological factors that are highly dependent on where the NP resides (i.e., whole-body, organ/suborgan, or cell/subcellular), which, in turn, is dependent on the time scale. Accordingly, a computational approach to determine BE would require models that can account for the scale-dependent processes. One approach is QSP-based multi-scale models to capture and integrate the different processes on individual scales. For example, we developed a multi-scale model that successfully predicted the spatiotemporal changes of drug concentrations in tumors located on the omentum during and after intraperitoneal chemotherapy; this model was validated in mice (66,67). A similar approach was used here to demonstrate how PC formation and evolution, by changing NP properties and NP-tumor cell interactions, affect the target site NP exposure. These in silico studies provided a quantitative perspective of how these PC-related changes can cause substantial differences of NP delivery and residence at the intended intracellular or extracellular targets.
Briefly, we used QSP-based multi-scale models to capture some of the above-described environment-, spatial-, time-, and concentration-dependent processes of NP. In these models, a tumor is viewed as a unit where the intratumoral heterogeneities are represented in mathematical terms (e.g., high blood flow for a highly perfused tumor vs. low blood flow to the necrotic region, large volume for a primary tumor vs. small volume for a micro-metastasis). The models account for the transvascular convective and diffusive NP transport (Eq. 1), the convective and diffusive NP transport in the interstitium (Eq. 2), NP binding to cells (Eq. 3), and NP internalization in cells (Eq. 4). In these equations, Cblood and Cint are the respective NP concentrations in blood and interstitial fluid, Cbound is cell surface– bound NP, Cintracell is intracellular NP, u is velocity of interstitial fluid flow, and Pe is Peclet number (ratio of convection to diffusion). These equations capture the spatiotemporal-dependent fluid and mass transport, and were solved numerically using computational fluid dynamics and finite element method. Please see our earlier publications for the mathematic basis of these equations (66,67) and Table IIA for the definitions of all other terms.
Table II.
Definitions of model parameter and their values. Some values were obtained from the literature (references in brackets). Dint was computed using the stokes-einstein equation with corrections for interstitial fluid viscosity and volume fraction as previously described (6,8). Sv/ V is spatial-dependent; we used the literature value for highly vascularized tumors (i.e., 200 cm−1) and assigned lower values for the tumor center (40 or 180 cm−1) to reflect the heterogeneity in tumor vessels (lower number and size in center vs. periphery (69–7,2)). σNP was calculated as previously described (73) and was set to 1 in normal tissues. While direct measures of the diffusive permeability Pd are not available, measurements of the effective permeability (Peff), which represents a combination of convective and diffusive transport, are available. Due to the negligible transvascular convection secondary to elevated interstitial fluid pressure, Peff is assumed to approximate Pd (see (74)). The reported values of Peff in tumors range from between 2 × 10−8 to 5 × 10−8 cm/s for NP of ~100 nm diameter (75,76) and 5.7 × 10−7 cm/s for dextran (~8 nm) (77). These values were used to determine the Pd values for the smaller NP (20 and 80 nm diameters) by linear interpolation. For the larger NP, we used a constant value of 3.5 × 10−8 cm/s. For NP-cell interaction parameters (kon/koff/kin/Bmax), we used the previously reported values for HSPC-liposomes (68). rtumor, rel is the normalized radial coordinate for tumor, 0 refers to tumor center, and 1 refers to tumor border
| Tumor | Normal | |
|---|---|---|
| A. Organ/suborgan scale model: NP transport in tumor and normal tissues | ||
| NP diffusion coefficient in interstitium (Dint), cm2/s | Calculated | Calculated |
| Vessel surface per unit tissue volume (Sv/V), cm−1 (78) | 40 or 180 at rtumor, rel = 0; 200 at rtumor, rel =1 | 70 |
| Volume fractions of blood vessel φv, interstitium φi, and tumor cell φc (79) | 0.05, 0.025, and 0.925 at rtumor, rel = 0; 0.225, 0.06, and 0.715 at rtumor, rel =1 | Same as rtumor, rel =1 |
| NP vascular diffusive permeability (Pd), cm/s | Calculated | 0 |
| Interstitial hydraulic conductivity (K), cm2/mmHg/s (80) | 2.5 × 10−7 | 2.5 × 10−7 |
| Hydraulic permeability of vessel walls (Lpv), cm/mmHg/s (80) | 1.86 × 10−6 | 3.6 × 10−8 |
| Lymphatic vessel pressure (Pl), mmHg | No lymphatics | 0 |
| Lymphatic flow per unit tissue volume (Lpl ⋅ Sl/V), (mmHg·s)−1 (81) | 0 | 2.22 × 10−4 |
| Blood vessel pressure (Pv), mmHg (79,80) | 11.7 | 17 |
| Vascular oncotic pressure (πv), mmHg (80) | 19.8 | 19.8 |
| Interstitial oncotic pressure (πi), mmHg (80) | 17.3 | 10 |
| NP reflection coefficient across vascular endothelium for extravasation (σNP) | Calculated | Calculated |
| Protein reflection coefficient across vascular endothelium (σp) (80,82,83) | 8.7 × 10−5 | 0.91 |
| B. Cellular/subcellular scale model: HSPC-liposome binding to cells and internalization (68) | ||
| Particle size, nm (mean ± SD) | 131 ± 1.9 | |
| Polydispersity index (mean ± SD) | 0.029 ± 0.015 | |
| Zeta potential, mV (mean ± SD) | −9.7 ± 0.5 | |
| Total liposome binding sites per unit tumor volume (Bmax), M | 1.3 × 10−7 | |
| Rate constant of NP binding to cell membrane (kon), M−1 s−1 | 5.5 × 104 | |
| Rate constant of NP dissociation from membrane binding sites (koff), s−1 | 3.4 × 10−4 | |
| (1) |
| (2) |
| (3) |
| (4) |
Equations 1–4 were used with the Darcy’s Law and Transport of Diluted Species Modules in COMSOL
Multiphysics 4.4 (COMSOL, Los Angeles, CA) to simulate the NP delivery and residence in a solid tumor, in order to evaluate the effects of three parameters (NP size, NP binding to cells, and internalization) on target site exposure. Note that all three parameters are affected by PC formation and evolution.
The model assumptions were (a) a spherical tumor (1 cm diameter) surrounded by a layer of normal tissue (0.5 cm thick) with NP input via extravasation from blood vasculature (i.e., blood/plasma concentration-time (C-T) profile of NP is the drug source); (b) intratumoral spatial heterogeneities with respect to blood perfusion, vessel density, interstitial pressure, and interstitial/vessel/cell volume fractions; (c) lower blood perfusion to tumor center relative to tumor periphery; (d) vessel surface area per unit volume (Sv/V) in tumor declines linearly with increasing distance from tumor periphery; (e) NP extravasates from blood via transvascular convection and diffusion; (f) transport of NP in tumor and normal tissue interstitium is by diffusion and convection in a porous matrix; (g) normal interstitial fluid pressure in normal tissue and higher pressure in tumor; (h) tumor blood vessel pore size of 200 nm; (i) NP in the tissue interstitial fluid binds and enters cells; (j) lymphatic vessels are present in normal tissue but absent in tumor tissue and hence negligible NP removal from tumor via the lymphatics (i.e., the last term in Eq. 2 equals zero in the case of tumor); (k) no NP binding to ECM; (l) no NP degradation and no changes in NP size in tumor interstitium; (m) internalized NP remains in cells (i.e., no exocytosis); (n) in the tumor-to-normal tissue interface, the values of transport model parameters were changed from those in tumor to those in normal tissue using linear interpolation over an arbitrary layer with thickness equaled to 2% of tumor radius; (o) no changes in tumor properties or structures over the duration-of-interest; and (p) no changes in NP-cell interaction parameters.
With respect to model parameter values, the values of NP transport parameters were taken from the literature or calculated using established equations (e.g., Stoke-Einstein equation), and were adjusted for the particle size (e.g., the two parameters that determine the transvascular transport, σNP and Pd); the model parameters are summarized in Table IIA. The simulations used a randomly selected biphasic plasma C-T profile (from 0 to 60 h) typical of intravenously injected NP as the input for the tumor. Note the systemic C-T profile would have accounted for the effects of NP properties on their distribution and clearance in other organs, e.g., effect of particle size on the clearance by different RES organs due to differences in their inter-endothelial vascular fenestration, i.e., 50–100 nm in the liver and 200–500 nm in the spleen (84). With respect to the spatial-dependent vessel distribution, due to the absence of quantitative data in the literature, we evaluated two situations where the Sv/V declined linearly with distance from tumor periphery (total reduction of 10% or 80% relative to the value in the periphery). The simulated results indicate (a) >99% of the extravasated NP occurred via convection in tumor periphery where there was a substantial pressure gradient in the tumor-normal tissue interface, (b) negligible extravasation in tumor center due to elevated interstitial fluid pressure, and (c) diffusive transvascular transport was >1000 times lower compared with convective extravasation due to the low Pd/Peff values. The insignificant transvascular transport in tumor center relative to the transport in tumor periphery indicates NP delivery to tumor was not sensitive to changes in Sv/V in tumor center resulting in <1% difference in the total NP in the tumor when Sv/V was reduced by 10% or 80%. Subsequent simulations used the 10% lower Sv/V value in tumor center relative to tumor periphery.
With respect to changes in NP size, we evaluated several small sizes (20, 80, and 140 nm diameter) that are within the range of some approved NP (e.g., Taxol® micelles with average size of 13 nm, liposomal Doxil® of 85 nm, Abraxane® of 139 nm), and several larger sizes (160, 180, 200, and 220 nm). The larger sizes were selected because they are in the proximity of the vessel pore size of 200 nm and because PC formation increased NP particle size, e.g., a 2014 study demonstrated interactions of Abraxane® with proteins in fetal bovine serum and human plasma led to PC formation and, based on the electron microscopy data, increased the diameter by ~33% from 139 nm (85) to 185 nm (56,85).
With respect to NP-cell interaction parameters (i.e., rate constants for NP association, dissociation, and internalization, or kon, koff, and kin, respectively), there are no data for, e.g., Abraxane®, in the literature. Hence, the simulations used our previously published data for HSPC-liposomes, which are of comparable size (131 nm average diameter) and are negatively charged (zeta potential of − 9.7 mV in water), as the baseline values (see Table IIB, (68)). Note the surface charge of Abraxane® is not provided in the drug product patents listed in the FDA Orange Book (86–88), its package insert (89), or its sponsor’s Citizen’s Petition (90), but, in view of albumin properties, is expected to be negatively charged.
The simulations yielded the C-T profiles of three NP entities (interstitial fluid, bound to cell surface, intracellular) and their sum (i.e., total in whole tumor), at different spatial locations within the tumor (e.g., tumor periphery or center). Adding up the concentrations of each entity at all locations followed by division with the tumor volume yielded their respective tumor-averaged concentrations.
Figure 2 shows the simulated C-T profiles in the whole tumor for NP with different sizes (20–220 nm diameter) but have systemic BE (i.e., same C-T in plasma). The simulation results show the particle size significantly affected the profile in several ways. First, the two smaller NP (20 and 80 nm) showed a rapidly peaking concentration at 2 h followed by a decline, whereas the three largest NP (180, 200, and 220 nm) showed a continuously increasing concentration that peaked at the last time point of 60 h. The remaining two NP with intermediate sizes (140 and 160 nm) displayed an early peak at 2 h, followed by a slight decline and then reversed to a continuous increase to reach the second peak (for 140-nm NP) or a higher peak (for 160-nm NP) at 60 h. Second, increasing the particle size drastically reduced Cmax and AUC, with the greatest changes when the NP diameter approached the blood vessel pore size (200 nm). For example, Cmax and AUC were reduced by ~50% when the diameter increased from 160 to 180 nm and by an additional >90% at 200 nm diameter, whereas none of the larger 220-nm NP entered the tumor. This data indicates the particle size determined the rate of NP delivery and residence in a tumor, and the formation of PC, by increasing the hydrodynamic diameter of NP such as Abraxane® to a value near the typical tumor vessel pore size, significantly reduces the tumor-averaged NP concentration.
Fig. 2. Effect of increasing NP diameter on target site exposure.
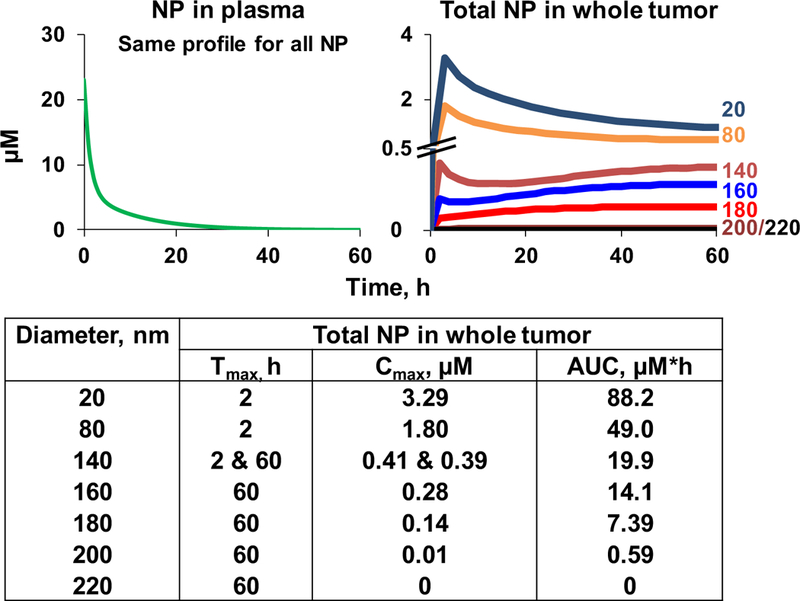
The simulations used the transport parameters in Table IIA and the HSPC-liposome-cell interaction parameters in Table IIB, with the assumption of negligible changes in the interaction parameters with the incremental diameter increases from 20 to 220 nm. The biphasic plasma C-T profile of NP was randomly selected and is typical of profiles after intravenous administration. All simulation concentrations accounted for the spatiotemporal changes and represented the total NP concentration in the whole tumor. Tmax is time of maximal concentration. Note the different scales in y-axis and the nearly overlapping curves of the 200 and 220 nm diameters
Figure 3 compares the effects of halving and doubling the baseline values of kon, koff, and kin. These rate constant changes, which correspond to lowering or increasing the NP association/dissociation or internalization, affected Cmax and AUC in the whole tumor and the extracellular and intracellular sites; the changes from baseline were between − 38 and + 53%. In general, lowering the association/dissociation and internalization reduced the Cmax and reduced the AUC in whole tumor and inside the cells, but increased the AUC of NP in interstitial fluid and cell surface–bound NP. Conversely, doubling the association/dissociation and internalization yielded the opposite results. This data indicates changes in NP-cell interactions alter the NP exposure in tumor interstitial fluid, and extracellular and intracellular locations. This data also suggests the possibility of tailoring the NP properties to optimize its interactions with biological components and its exposure at the intended target sites, e.g., antibodies for cell membrane proteins or receptors, or therapeutics acting on intracellular molecules.
Fig. 3. Effects of changes in cell association/dissociation and internalization of NP on extracellular and intracellular target site exposure to NP.
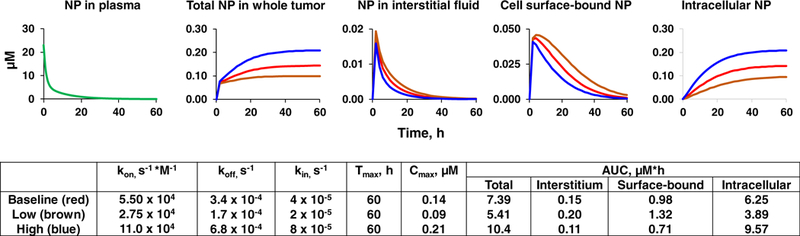
The simulations were for NP with a diameter of 180 nm. The simulations were performed using the transport parameters in Table IIA and the HSPC-liposome-cell interaction parameters in Table IIB, with the assumption of negligible changes in the liposome-cell interaction parameters due to diameter changes. The biphasic plasma C-T profile of NP was randomly selected and is typical of profiles after intravenous administration. The simulations evaluated the effects of different rate constants of cell association, dissociation, and internalization on the concentrations of NP in the whole tumor, interstitium, bound to cell surface and inside the cells; the three simulations used their baseline values, one half of baseline values (low) and twice the baseline values (high). All simulations accounted for the spatiotemporal changes and represented the tumor-averaged NP concentration. Note the different scales in y-axis of individual plots. Tmax is time of maximal concentration. The absence of exocytosis resulted in accumulation of the intracellular NP over time
The purpose of the above simulations is to provide a quantitative perspective that seemingly small changes in NP properties such as those caused by PC formation or evolution can lead to substantial differences in the delivery (rate and extent) and residence of NP in a tumor. It is noted the current model used a number of simplifying assumptions, including some that may not apply to all NP. For example, exocytosis of NP from cells may diminish the NP accumulation and residence. The model used the average NP size and did not account for the size distribution (e.g., high vs. low polydispersity index). The model further assumed no changes in NP properties or tumor properties/structures over the duration-of-interest, which may not apply as some NPs are designed to undergo chemical changes at specified conditions (e.g., acidic pH environment) and as it is known that cytotoxic treatments, by inducing apoptosis, transiently increase the tumor porosity (90). In addition, the model used the same values of NP-cell interaction parameters, whereas changes in NP size or charges are likely to alter the parameter values. We advocate further development of QSP-based multi-scale models to capture relevant physical, chemical, and biological events such that these models can be used to predict the effects of changes in NP and tumor properties, individually and collectively, on the target site exposure to NP.
CONCLUSIONS
In summary, there are many intersecting kinetic processes that determine the target site exposure of oncologic NP and cargo, including NP disposition and clearance in whole organism, transvascular and interstitial transport of NP in tumor interstitium, NP interactions with proteins in blood/ serum, NP interactions with cells/extracellular matrix components/subcellular organelles, NP internalization in cells, and release of cargo in each of these sites. Adding to the complexity is the formation of NP-coating corona in biological milieu, the continuous exchanges of corona proteins, the involvement of proteins that change with the race, age, gender, and disease states of the host, and the ability of protein corona to affect the NP transport and disposition. The large number of determinants and their inherent variabilities render systemic BE of NP unlikely to equal target site BE.
Theoretically, target site BE is achievable if the generic NP fulfills the formulation sameness requirement in the 2017 FDA draft guidance (5), i.e., having identical properties as RLD. However, in practice, formulation sameness is difficult to achieve due to batch-to-batch variations or variations in manufacturing processes. An example for the former is the different average diameters of Abraxane® reported by its sponsor in different documents (~130 nm cited in its package insert (89) vs.139 nm cited in its Citizen Petition (85)). An example for the latter is the finding that variations in manufacturing conditions (pressure or number of homogenizations, concentrations of albumin, paclitaxel, chloroform or ethanol) can cause substantial changes in the properties of paclitaxel-containing albumin complex (e.g., 2.4-fold for diameter, 6.3-fold for polydispersity index, change from being negatively charged to neutral-to-slightly positively charged) (91). Such changes are expected to significantly affect the target site exposure, as we have shown in the QSP-based simulations that a seemingly small 10% difference in the diameter of some NP can result in >90% difference in the tumor exposure.
The 2017 FDA draft guidance on NP highlights the importance of defining the critical quality attributes (CQA) with regard to their function and potential impact on product performance, and provides a detailed list of attributes focusing on quality-related issues (i.e., chemistry manufacturing, and controls) (5). This guidance discusses how the interactions of nanomaterials with multiple plasma proteins result in the formation of protein corona, which may endow NP with new biological properties, e.g., enhanced rate of dissolution and improved bioavailability, altered distribution and residence time of API, altered exposure-response profile, altered NP uptake by endocytosis, and elimination through phagocytosis by macrophages of the mononuclear phagocyte system. The same guidance further recommends the determination of protein corona formation and the percentage of corona-coated NP in human blood/plasma. A later 2018 FDA guidance on liposomal products states that interactions between liposome surface and blood proteins may affect the drug release and pharmacological properties of a liposome drug product in vivo, and that such interactions can have safety implications because of dose dumping (24). On the other hand, FDA has not addressed the potential complications of PC evolution that may lead to location- or environment-dependent changes in the NP properties or the biological consequences.
A review of the literature revealed limited investigations on determining the target site BE of NP. We propose the following considerations, in addition to the above FDA recommendations, regarding the contribution of NP-cell-biomatrix interactions and environment-dependent corona compositions to target site exposure of NP. These efforts would be assisted by acquiring additional qualitative and quantitative data, including (a) effects of extracellular matrix components and protein corona on the transport, binding, residence, and internalization of NP; (b) exchange of corona proteins in different biological matrices, as functions of time, NP concentration, and concentrations of proteins-of-interest; (c)effects of NP properties on protein corona formation and stability; (d) species differences in extracellular matrix components and protein corona formation and evolution; and (e) effects of age, gender, ethnicity, and physiological and disease states on protein corona formation and evolution in human subjects. Such data are needed to enable the in vitro-to-in vivo and the preclinical-to-clinical translation. Generation of these missing data is in alignment with one of the three FDA focus areas in the development and approval of generic drugs, i.e., data-based knowledge discovery for review optimization of ANDA.
As the approval of generic drugs by FDA requires the generic product to be therapeutically equivalent to the RLD, the inability to demonstrate target site BE based on systemic BE translates to a need of demonstrating equivalent efficacy in patients. We advocate the combined use of in silico studies with relatively limited experimental studies (e.g., in vitro and preclinical in vivo experiments) as a potentially more cost-effective path for generic NP development, as follows: (a) develop computational systems-based multi-scale models to capture the different biological and physicochemical events occurring on the whole-body, organ/suborgan, and cellular/ subcellular levels that affect the ability of NP reaching its intended target sites; (b) measure the product-specific model parameters in vitro (e.g., NP-cell-biomatrix interactions, release of API); (c) use the models and the product-specific parameters to compute the target site exposure of NP/API and use sensitivity analysis to identify the CQA; and (d) experimentally validate the CQA in vivo in preclinical models with appropriate target tissue properties (e.g., blood vessel pore size). This approach aligns with another FDA focus areas, i.e., model-based generic drug development and BE determination.
Acknowledgments
FUNDING INFORMATION
This work was supported in part by research grants R01GM100487 from the National Institute of General Medical Sciences and R01EB015253 from the National Institute of Biomedical Imaging and Bioengineering, NIH, DHHS, the Mosier Endowed Chair in Pharmaceutical Sciences at University of Oklahoma Health Sciences Center, and the Chair in Systems Pharmacology at Taipei Medical University.
Abbreviations:
- ANDA
Abbreviated New Drug Application
- API
Active pharmaceutical ingredient
- AUC
Area under concentration-time-curve
- BE
Bioequivalence
- Cmax
Maximum concentration
- CQA
Critical quality attribute
- C-T
Concentration-time
- ECM
Extracellular matrix
- FDA
US Food and Drug Administration
- kon, koff, and kin
Respective rate constants of NP binding and release from cell membrane, and internalization
- NP
Nanotechnology products
- PC
Protein corona
- PK
Pharmacokinetic
- QSP
Quantitative systems pharmacology
- RES
Reticuloendothelial system
- RLD
Reference listed drug
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
Conflict of Interest JA, GW and ZL have ownership interests in Optimum Therapeutics LLC, which is involved in developing cancer nanotechnologies.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES
- 1.Fang L, Kim MJ, Li Z, Wang Y, DiLiberti CE, Au J, et al. Model-informed drug development and review for generic products: summary of FDA Public Workshop. Clin Pharmacol Ther 2018;104:27–30. [DOI] [PubMed] [Google Scholar]
- 2.Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, Califano A, et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. An NIH White Paper by the QSP Workshop. Group 2011.
- 3.Lionberger R Using quantitative methods and modeling to transform generic drug development and review https://www.fda.gov/downloads/Drugs/NewsEvents/UCM582148.pdf 2017. Accessed 31 Oct 2018.
- 4.USFDA. Guidance for industry: considering whether an FDA-regulated product involves the application of nanotechnology https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM401695.pdf 2014. Accessed 31 Oct 2018.
- 5.USFDA. Guidance for industry: drug products, including biological products, that contain nanomaterials https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM588857.pdf 2017. Accessed 31 Oct 2018. [DOI] [PMC free article] [PubMed]
- 6.Zheng N, Sun DD, Zou P, Jiang W. Scientific and regulatory considerations for generic complex drug products containing nanomaterials. AAPS J 2017;19:619–31. [DOI] [PubMed] [Google Scholar]
- 7.Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res 2016;33:2373–87. [DOI] [PubMed] [Google Scholar]
- 8.D’Mello SR, Cruz CN, Chen ML, Kapoor M, Lee SL, Tyner KM. The evolving landscape of drug products containing nanomaterials in the United States. Nat Nanotechnol 2017;12:523–9. [DOI] [PubMed] [Google Scholar]
- 9.USFDA. Drugs@FDA database https://www.fda.gov/Drugs/InformationOnDrugs/ucm135821.htm 2018. Accessed 10 Dec 2018.
- 10.Johnson EM, Ojwang JO, Szekely A, Wallace TL, Warnock DW. Comparison of in vitro antifungal activities of free and liposome-encapsulated nystatin with those of four amphotericin B formulations. Antimicrob Agents Chemother 1998;42:1412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JM, Whitney RR, Olsen SJ, George RJ, Swerdel MR, Kunselman L, et al. Amphotericin B lipid complex therapy of experimental fungal infections in mice. Antimicrob Agents Chemother 1991;35:615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usmani SS, Bedi G, Samuel JS, Singh S, Kalra S, Kumar P, et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS One 2017;12:e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.USFDA. Generic drugs: questions & answers https://www.fda.gov/drugs/resourcesforyou/consumers/questionsanswers/ucm100100.htm 2018. Accessed 31 Oct 2018.
- 14.Code of Federal Regulations. Applications for FDA approval to market a new drug - definations, 21 CFR 314.3 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=314.3 2018. Accessed 31 Oct 2018.
- 15.Code of Federal Regulations. Types of evidence to measure bioavailability or establish bioequivalence, 21 CFR 320.24 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=320.24 2018. Accessed 31 Oct 2018.
- 16.USFDA. Guidance for industry: submission of summary bioequivalence data for ANDAs https://www.fda.gov/downloads/Drugs/…/Guidances/UCM134846.pdf 2011. Accessed 31 Oct 2018.
- 17.USFDA. Guidance for industry: bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA https://www.fda.gov/downloads/drugs/guidances/ucm377465.pdf 2013. Accessed 31 Oct 2018.
- 18.USFDA. Guidance for industry: statistical approaches to establishing bioequivalence https://www.fda.gov/downloads/drugs/guidances/ucm070244.pdf 2001. Accessed 31 Oct 2018.
- 19.USFDA. Reducing the hurdles for complex generic drug development https://www.fda.gov/NewsEvents/Newsroom/FDAVoices/ucm612010.htm 2017. Accessed 31 Oct 2018.
- 20.USFDA. Guidance for Industry: formal meetings between FDA and ANDA applicants of complex products under GD UFA https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm578366.pdf 2017. Accessed 31 Oct 2018.
- 21.USFDA. Guidance for industry: use of nanomaterials in food for animals https://www.fda.gov/downloads/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/UCM401508.pdf 2015. Accessed 31 Oct 2018.
- 22.USFDA. Guidance for industry: assessing the effects of significant manufacturing process changes, including emerging technologies, on the safety and regulatory status of food ingredients and food contact substances, including food ingredients that are color additives https://www.fda.gov/downloads/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/UCM616225.pdf 2014. Accessed 31 Oct 2018.
- 23.USFDA. Guidance for industry: safety of nanomatrials in comestic products https://www.fda.gov/downloads/Cosmetics/GuidanceRegulation/GuidanceDocuments/UCM300932.pdf 2014. Accessed 31 Oct 2018.
- 24.USFDA. Guidance for industry: liposome drug products chemistry, manufacturing, and controls; human pharmacokinetics and bioavailability; and labeling documentation https://www.fda.gov/downloads/drugs/guidances/ucm070570.pdf 2018. Accessed 31 Oct 2018.
- 25.USFDA. Orange Book preface https://www.fda.gov/drugs/developmentapprovalprocess/ucm079068.htm 2018. Accessed 31 Oct 2018.
- 26.Li Y, Wang J, Wientjes MG, Au JL. Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv Drug Deliv Rev 2012;64:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Au JL, Jang SH, Wientjes MG. Clinical aspects of drug delivery to tumors. J Control Release 2002;78:81–95. [DOI] [PubMed] [Google Scholar]
- 28.Au JL, Yeung BZ, Wientjes MG, Lu Z, Wientjes MG. Delivery of cancer therapeutics to extracellular and intracellular targets: determinants, barriers, challenges and opportunities. Adv Drug Deliv Rev 2016;97:280–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Au JL, Jang SH, Zheng J, Chen CT, Song S, Hu L, et al. Determinants of drug delivery and transport to solid tumors. J Control Release 2001;74:31–46. [DOI] [PubMed] [Google Scholar]
- 30.Au JL, Lu Z, Wientjes MG. Versatility of particulate carriers: development of pharmacodynamically optimized drug-loaded microparticles for treatment of peritoneal cancer. AAPS J 2015;17:1065–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res 2003;20:1337–50. [DOI] [PubMed] [Google Scholar]
- 32.Lu Z, Wientjes MG, Au JL. Development of drug-loaded particles for intraperitoneal therapy. In: Ceelen Wim P., Levine Edward, editors. Intraperitoneal cancer therapy: principles and practice CRC Press; 2015. p. 331–344. [Google Scholar]
- 33.Lu Z, Wang J, Wientjes MG, Au JL. Intraperitoneal therapy for peritoneal cancer. Future Oncol 2010;6:1625–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J 2010;12:492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Lu Z, Gao Y, Wientjes MG, Au JL. Improving delivery and efficacy of nanomedicines in solid tumors: role of tumor priming. Nanomedicine (Lond) 2011;6:1605–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Au JL, Abbiati RA, Wientjes MG, Lu Z. Target site delivery and residence of nano-medicines: application of quantitative systems pharmacology. Pharmacol.Rev. (Accepted) [DOI] [PMC free article] [PubMed]
- 37.Nguyen VH, Lee BJ. Protein corona: a new approach for nanomedicine design. Int J Nanomedicine 2017;12:3137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strojan K, Leonardi A, Bregar VB, Krizaj I, Svete J, Pavlin M. Dispersion of nanoparticles in different media importantly determines the composition of their protein corona. PLoS One 2017;12:e0169552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corbo C, Molinaro R, Tabatabaei M, Farokhzad OC, Mahmoudi M. Personalized protein corona on nanoparticles and its clinical implications. Biomater Sci 2017;5:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbero F, Russo L, Vitali M, Piella J, Salvo I, Borrajo ML, et al. Formation of the protein corona: the interface between nanoparticles and the immune system. Semin Immunol 2017;34:52–60. [DOI] [PubMed] [Google Scholar]
- 41.Lai ZW, Yan Y, Caruso F, Nice EC. Emerging techniques in proteomics for probing nano-bio interactions. ACS Nano 2012;6:10438–48. [DOI] [PubMed] [Google Scholar]
- 42.Ahsan SM, Rao CM, Ahmad MF. Nanoparticle-protein interaction: the significance and role of protein corona. Adv Exp Med Biol 2018;1048:175–98. [DOI] [PubMed] [Google Scholar]
- 43.Treuel L, Nienhaus GU. Toward a molecular understanding of nanoparticle-protein interactions. Biophys Rev 2012;4:137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walkey CD, Chan WC. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem Soc Rev 2012;41:2780–99. [DOI] [PubMed] [Google Scholar]
- 45.Setyawati MI, Tay CY, Docter D, Stauber RH, Leong DT. Understanding and exploiting nanoparticles’ intimacy with the blood vessel and blood. Chem Soc Rev 2015;44:8174–99. [DOI] [PubMed] [Google Scholar]
- 46.Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol 2012;7:779–86. [DOI] [PubMed] [Google Scholar]
- 47.Yang ST, Liu Y, Wang YW, Cao A. Biosafety and bioapplication of nanomaterials by designing protein-nanoparticle interactions. Small 2013;9:1635–53. [DOI] [PubMed] [Google Scholar]
- 48.Mariam J, Sivakami S, Dongre PM. Albumin corona on nanoparticles - a strategic approach in drug delivery. Drug Deliv 2015:1–9. [DOI] [PubMed]
- 49.Monteiro-Riviere NA, Samberg ME, Oldenburg SJ, Riviere JE. Protein binding modulates the cellular uptake of silver nano- particles into human cells: implications for in vitro to in vivo extrapolations? Toxicol Lett 2013;220:286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonvin D, Aschauer U, Alexander DTL, Chiappe D, Moniatte M, Hofmann H, et al. Protein corona: impact of lymph versus blood in a complex in vitro environment. Small 2017;13. [DOI] [PubMed] [Google Scholar]
- 51.Vilanova O, Mittag JJ, Kelly PM, Milani S, Dawson KA, Radler JO, et al. Understanding the kinetics of protein-nanoparticle corona formation. ACS Nano 2016;10:10842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadjidemetriou M, Al-Ahmady Z, Kostarelos K. Time- evolution of in vivo protein corona onto blood-circulating PEGylated liposomal doxorubicin (DOXIL) nanoparticles. Nanoscale 2016;8:6948–57. [DOI] [PubMed] [Google Scholar]
- 53.Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, et al. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat Nanotechnol 2017;12:387–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol 2013;8:772–81. [DOI] [PubMed] [Google Scholar]
- 55.Kurtz-Chalot A, Klein J, Pourchez J, Boudars D, Bin V, Alcantara GB, et al. Adsorption at cell surface and cellular uptake of silica nanoparticles with different surface chemical functionalizations: impact on cytotoxicity. J NanoparticleRes 2014;16:2738. [Google Scholar]
- 56.Behzadi S, Serpooshan V, Sakhtianchi R, Muller B, Landfester K, Crespy D, et al. Protein corona change the drug release profile of nanocarriers: the Boverlooked^ factor at the nanobio interface. Colloids Surf B Biointerfaces 2014;123:143–9. [DOI] [PubMed] [Google Scholar]
- 57.Lara S, Alnasser F, Polo E, Garry D, Lo Giudice MC, Hristov DR, et al. Identification of receptor binding to the biomolecular corona of nanoparticles. ACS Nano 2017;11:1884–93. [DOI] [PubMed] [Google Scholar]
- 58.McConnell KI, Shamsudeen S, Meraz IM, Mahadevan TS, Ziemys A, Rees P, et al. Reduced cationic nanoparticle cytotoxicity based on serum masking of surface potential. J Biomed Nanotechnol 2016;12:154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin H, Chen R, Casey P, Ke PC, Davis T, Chen C. Reducing the cytotoxicity of ZnO nanoparticles by a pre-formed protein corona in a supplemented cell culture medium. RSC Adv 2015;5:73963–73. [Google Scholar]
- 60.Deng ZJ, Liang M, Monteiro M, Toth I, Minchin RF. Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat Nanotechnol 2011;6:39–44. [DOI] [PubMed] [Google Scholar]
- 61.Mirsadeghi S, Dinarvand R, Ghahremani MH, Hormozi- Nezhad MR, Mahmoudi Z, Hajipour MJ, et al. Protein corona composition of gold nanoparticles/nanorods affects amyloid beta fibrillation process. Nanoscale 2015;7:5004–13. [DOI] [PubMed] [Google Scholar]
- 62.Muller LK, Simon J, Rosenauer C, Mailander V, Morsbach S, Landfester K. The transferability from animal models to humans: challenges regarding aggregation and protein corona formation of nanoparticles. Biomacromolecules 2018;19:374–85. [DOI] [PubMed] [Google Scholar]
- 63.Hanash S Disease proteomics. Nature 2003;422:226–32. [DOI] [PubMed] [Google Scholar]
- 64.Conrads TP, Fusaro VA, Ross S, Johann D, Rajapakse V, Hitt BA, et al. High-resolution serum proteomic features for ovarian cancer detection. Endocr Relat Cancer 2004;11:163–78. [DOI] [PubMed] [Google Scholar]
- 65.Caputo D, Papi M, Coppola R, Palchetti S, Digiacomo L, Caracciolo G, et al. A protein corona-enabled blood test for early cancer detection. Nanoscale 2017;9:349–54. [DOI] [PubMed] [Google Scholar]
- 66.Au JL, Guo P, Gao Y, Lu Z, Wientjes MG, Tsai M, et al. Multiscale tumor spatiokinetic model for intraperitoneal therapy. AAPS J 2014;16:424–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abbiati RA, Au JL. Quantitative systems pharmacology on cancer drug delivery to target sites: application of chemical engineering tools. In: Manca D, editor. Quantitative systems pharmacology: models and model-based systems with applications Elsevier; 2018. p. 239–270. [Google Scholar]
- 68.Gao Y, Li M, Chen B, Shen Z, Guo P, Wientjes MG, et al. Predictive models of diffusive nanoparticle transport in 3-dimensional tumor cell spheroids. AAPS J 2013;15:816–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia 1999;1:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nitta N, Takakusagi Y, Kokuryo D, Shibata S, Tomita A, Higashi T, et al. Intratumoral evaluation of 3D microvasculature and nanoparticle distribution using a gadolinium-dendron modified nano-liposomal contrast agent with magnetic resonance micro-imaging. Nanomedicine 2018;14:1315–24. [DOI] [PubMed] [Google Scholar]
- 71.Searle EJ, Telfer BA, Mukherjee D, Forster DM, Davies BR, Williams KJ, et al. Akt inhibition improves long-term tumour control following radiotherapy by altering the microenvironment. EMBO Mol Med 2017;9:1646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stapleton S, Milosevic M, Allen C, Zheng J, Dunne M, Yeung I, et al. A mathematical model of the enhanced permeability and retention effect for liposome transport in solid tumors. PLoS One 2013;8:e81157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deen WM. Hindered transport of large molecules in liquid-filled pores. AICHE J 1987;33:1409–25. [Google Scholar]
- 74.Meijer EF, Baish JW, Padera TP, Fukumura D. Measuring vascular permeability in vivo. Methods Mol Biol 2016;1458:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Q, Krol A, Wright A, Needham D, Dewhirst MW, Yuan F. Tumor microvascular permeability is a key determinant for antivascular effects of doxorubicin encapsulated in a temperature sensitive liposome. Int J Hyperth 2008;24:475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuan F, Leunig M, Huang SK, Berk DA, Papahakjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res 1994;54:3352–6. [PubMed] [Google Scholar]
- 77.Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res 1986;31:288–305. [DOI] [PubMed] [Google Scholar]
- 78.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res 1989;37:77–104. [DOI] [PubMed] [Google Scholar]
- 79.Stapleton S, Milosevic M, Tannock IF, Allen C, Jaffray DA. The intra-tumoral relationship between microcirculation, interstitial fluid pressure and liposome accumulation. J Control Release 2015;211:163–70. [DOI] [PubMed] [Google Scholar]
- 80.Jain RK, Tong RT, Munn LL. Effect of vascular normalization, by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res 2007;67:2729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. II. Role of heterogeneous perfusion and lymphatics. Microvasc Res 1990;40:246–63. [DOI] [PubMed] [Google Scholar]
- 82.Reed RK, Townsley MI, Taylor AE. Estimation of capillary reflection coefficients and unique PS products in dog paw. Am J Phys 1989;257:H1037–41. [DOI] [PubMed] [Google Scholar]
- 83.Ballard K, Perl W. Osmotic reflection coefficients of canine subcutaneous adipose tissue endothelium. Microvasc Res 1978;16:224–36. [DOI] [PubMed] [Google Scholar]
- 84.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 2015;33:941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Celgene Corporation and Abraxis BioScience LLC Citizen Petition. https://www.regulations.gov/contentStreamer?documentId=FDA-2015-P-0732-0001&attachmentNumber=1&contentType=pdf 2015. Accessed 31 Oct 2018.
- 86.Desai NP, Soon-Shiong P, and Trieu V Compositions and methods of delivery of pharmacological agents U.S. Patent 7,820,788 2010.
- 87.Desai NP, Soon-Shiong P, and Trieu V Compositions and methods of delivery of pharmacological agents U.S. Patent 8,138,229 2012.
- 88.Desai NP and Soon-Shiong P Formulations of pharmacological agents, methods for the preparation thereof and methods for the use thereof U.S. Patent 8,853,260 2014.
- 89.Abraxane® Package Insert https://media.celgene.com/content/uploads/abraxane-pi.pdf 2018. Accessed 31 Oct 2018.
- 90.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther 2007;322:80–8. [DOI] [PubMed] [Google Scholar]
- 91.Lomis N, Westfall S, Farahdel L, Malhotra M, Shum-Tim D, Prakash S. Human serum albumin nanoparticles for use in cancer drug delivery: process optimization and in vitro charac- terization. Nanomaterials (Basel) 2016;6:116. [DOI] [PMC free article] [PubMed] [Google Scholar]