

Abstract
Effective adaptive immune responses depend on activation of CD4+ T cells via the presentation of antigen peptides in the context of major histocompatibility complex (MHC) class II. The structure of an antigen strongly influences its processing within the endolysosome and potentially controls the identity of peptides that are presented to T cells. A recombinant immunotoxin, comprising exotoxin A domain III (PE-III) from Pseudomonas aeruginosa and a cancer-specific antibody fragment, has been developed to manage cancer, but its effectiveness is limited by the induction of neutralizing antibodies. Here, we observed that this immunogenicity is substantially reduced by substituting six residues within PE-III. Although these substitutions targeted T-cell epitopes, we demonstrate that reduced conformational stability and protease resistance were responsible for the reduced antibody titer. Analysis of mouse T-cell responses coupled with biophysical studies on single-substitution versions of PE-III suggested that modest but comprehensible changes in T-cell priming can dramatically perturb antibody production. The most strongly responsive PE-III epitope was well-predicted by a structure-based algorithm. In summary, single-residue substitutions can drastically alter the processing and immunogenicity of PE-III but have only modest effects on CD4+ T-cell priming in mice. Our findings highlight the importance of structure-based processing constraints for accurate epitope prediction.
Keywords: antigen processing, T-helper cells, T cell, antigen presentation, antibody, protein structure, protein stability, proteomics, proteolysis, cancer, CD4+ T cells, deimmunization, immunotoxin, vaccine
Introduction
The accurate prediction of T-cell epitopes could be used for the deimmunization of therapeutics and allergens, as well as the design of more effective vaccines and cancer therapies. Computational prediction of peptide affinity for major histocompatibility complex (MHC)2 antigen–presenting proteins has become more accurate with the accumulation of peptide-binding data and application of machine learning, but progress has not uniformly advanced to epitope prediction (1, 2). CD4+ helper T-cell epitope–containing peptides are displayed by MHC class II molecules (MHCII) and are mostly derived from exogenous antigens that simultaneously encounter proteolytic enzymes and MHCII molecules in the endosomal pathway (3, 4). CD8+ cytotoxic T-cell epitope-containing peptides are mostly derived from endogenous antigens that are processed by the cytoplasmic proteasome and subsequently loaded into MHC class I molecules (MHCI) in the endoplasmic reticulum (5). Although peptide–MHCI-binding affinity is a strong predictor for CD8+ T-cell epitopes, peptide–MHCII-binding affinity is a weak predictor for CD4+ T-cell epitopes in many antigens (1, 6–9). Previous work by our laboratory has shown that the antigen three-dimensional structure influences antigen processing in the endosomal pathway and therefore the selection of peptides that are presented by MHCII. Dominant helper T-cell epitopes were found to overlap flexible loops between elements of secondary structure and between protein domains (10–14). Flexible protein segments can more easily conform to protease active sites and are ideal sites for proteolytic cleavage. With this knowledge, we have developed an epitope-prediction algorithm that predicts immunogenicity with significant accuracy (15) without consideration of peptide–MHCII-binding affinity.
In this paper we present our studies on domain III of exotoxin A from Pseudomonas aeruginosa (PE) as a model antigen. Exotoxin A has been studied for decades as a potential cancer therapy in the form of a recombinant immunotoxin (RIT) (16, 17). In its original form, the exotoxin A RIT contained a portion of domain I and the entirety of domains II and III of exotoxin A linked to a cancer-specific antibody fragment (Fab). The recombinant protein is administered to patients intravenously, where it binds to cell-surface proteins, is internalized, and results in cell death via ADP-ribosylation of elongation factor 2 (18). Clinical trials proved challenging, as the bacterial component of the PE RIT is strongly immunogenic, resulting in the induction of neutralizing antibodies after as little as one treatment, rendering the therapy ineffective (19). CD4+ T-cell responses are required for the induction of potent antibody responses, so human CD4+ T-cell epitopes within PE were silenced by removing unnecessary segments, including most of domain II, and introducing six amino acid substitutions within domain III. The resulting immunotoxin, RIT-T18, induced a smaller CD4+ T-cell response from human cells (20) and a reduced CD4+ T-cell and serum antibody response in mice (21, 22). The mutations introduced to exotoxin A domain III (PE-III) were designed to reduce the affinity of epitope-containing peptides for the MHC class II molecule while maintaining the structure and enzymatic activity of PE-III (22). The successful “deimmunization” of PE-III provides an opportunity to use both the WT and deimmunized PE-III as model antigens to more finely probe the relationship between antigen structure and antigen processing/presentation to CD4+ T cells.
To that end, we purified the wildtype (WT) PE-III, T18 PE-III, and two single mutant variants of PE-III to study how these mutations have altered the structure, processing, and immune response. We observed that the deimmunizing mutations significantly altered PE-III stability and susceptibility to antigen-processing proteases. The mutations also modified presentation of epitope-containing peptides and CD4+ T-cell epitope immunogenicity, as well as serum antibody levels in mice. We also observed that a single mutation was sufficient to reduce serum antibody titers below that observed for the six-mutant T18 while causing only modest changes in the priming of CD4+ T cells.
Results
WT PE-III and all mutant variants used for this study were purified from Escherichia coli grown in autoinduction media (23), with a typical yield of 10–15 mg/liter of culture.
Assessing the folding stability of PE-III and mutant variants
We hypothesized that the amino acid substitutions intended to disrupt peptide–MHCII interactions have instead altered the conformational stability of PE-III. To test this, we first used homology modeling and the GROMACS96 software suite to estimate changes in the free energy of PE-III folding caused by individual epitope-silencing mutations (Fig. S1) (24). Based on the GROMACS software calculations, we predicted decreases in free energy for PE-III variants containing the R427A, R494A, and R505H mutations and an increase in free energy for PE-III L477H and L552E mutations. We selected the R494A and L552E single mutations for further study, as they were predicted to have opposing effects on PE-III stability and were not located in any previously mapped mouse epitopes (20). We also analyzed the regional stabilities of WT PE-III using the COREX/best algorithm to generate a structural thermodynamic ensemble (25). The COREX/best residue-specific stabilities were compared with results calculated for R494A and L552E single-mutant variants. We observed changes in predicted stability of protein substructures located near previously identified T-cell epitopes (data not shown), as well an overall decrease in stability for the R494A variant and an overall increase in stability for the L552E variant. Based on the GROMACS and COREX analyses, we predicted that WT, T18, and the single-mutant PE-III R494A and L552E variants would exhibit differences in folding stability. We next measured folding stability of purified PE-III variants using acid- and denaturant-induced unfolding (Fig. 1, A and C). From the acid unfolding data, EC50 values were determined for each mutant PE-III variant using a sigmoidal dose–response regression model (Fig. 1B). The single mutant variant L552E unfolded at a significantly lower pH value than WT PE-III, whereas R494A unfolded at a significantly higher pH value. T18 PE-III exhibited no difference in acid-induced unfolding when compared with WT PE-III. Free energies of unfolding were determined by fitting the denaturant-induced unfolding curves as described previously (14, 26). The measured free energy of unfolding for T18 PE-III did not differ from that for WT PE-III, whereas the single mutant variants exhibited significant changes in measured free energy compared with WT PE-III (Fig. 1D). Our findings that the single mutant variants R494A and L552E exhibited decreased and increased resistance to unfolding, respectively, matched our predictions from computational techniques.
Figure 1.

Deimmunizing mutations alter the folding stability of PE-III. A, acid-induced denaturation of PE-III reported by Bis-ANS fluorescence (n = 3). B, analysis of best-fit pH 50 values; error bars indicate standard error; asterisks indicate significance by one-way ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). C, chemical denaturation of PE-III using GdnHCl and reported by intrinsic tryptophan fluorescence (representative of n = 3). The ratio of the intensity at a given [GdnHCl] to that at zero [GdnHCl] was plotted for each concentration, and the data were fitted to equations that solve for the free energy of unfolding at zero [GdnHCl] (ΔG0′). D, analysis of ΔG0′ values determined in C. Error bars indicate standard error, and asterisks indicate significance by one-way ANOVA.
Structural characterization of deimmunizing PE-III mutations
We have demonstrated that some single-amino acid substitutions intended to deimmunize PE-III have altered the protein's stability and resistance to unfolding. We next sought to explain these changes by observing the effects of these mutations on the crystallographic structure of PE-III. To date, we have been unable to crystallize PE-III R494A or T18, but we have obtained diffraction-quality crystals of a variant of T18 lacking the L477H mutation (T18-H477L). A high-resolution structure for WT PE-III is also already available (PDB code 1XK9) (27). Using the WT PE-III structure as a search model, we solved the structure of PE-III T18-H477L by molecular replacement at a resolution of 1.47 Å. Statistics and software information are detailed in Table S1. In the WT PE-III structure, we observed arginine 494 participating in a hydrogen bond with glutamate 445 (Fig. 2A). When alanine is substituted for arginine 494 in T18-H477L, this interaction is eliminated, and glutamate 445 changes conformation to a more solvent-exposed position. Elimination of this hydrogen bond pair coincided with a large increase in the crystallographic B-factors for the loop spanning residues 480–496 (Fig. 2, B and C). The replacement of leucine 552 with glutamate in T18-H477L introduces a new hydrogen bond between glutamate 552 and arginine 513 (Fig. 2A). In WT PE-III, arginine 513 is more than 5.0 Å away from the nearest amino acid side chain or backbone carbonyl, too far to be participating in any meaningful structural interactions. The introduction of this hydrogen bond pair is not accompanied by a change in B-factors (Fig. 2B). In a structural analysis using the COREX algorithm described above, we observed contrasting effects of the R494A and L552E mutations on residue stabilities that are consistent with the structural changes introduced by these mutations (Fig. 2D). Residue stabilities were drastically decreased in the region where the R494A mutation eliminates the hydrogen bond, although they were noticeably increased in the region where the L552E mutation introduces a hydrogen bond. These data demonstrate that the R494A and L552E mutations have modified the structure of PE-III, resulting in significant changes in folding free energy and acid resistance.
Figure 2.

Deimmunizing mutations alter the structure of PE-III. A, orientation of residues at the 494 and 552 positions in the WT (green, PDB code 1XK9) and T18-H477L (cyan, PDB code 6EDG) structures. B, Comparison of the crystallographic B-factors from the WT and T18-H477L structures. Arrows indicate locations of the R494A and L552E mutations. C, approximate locations of the proteinase K cleavage sites within the WT PE-III structure. D, COREX stability constants of the WT and T18-H477L structures. Arrowheads indicate locations of the R494A and L552E mutations.
Destabilizing PE-III mutations increase proteolytic susceptibility
The presentation of antigen peptides bound to class II MHC molecules depends on proteolytic processing of those antigens. Previous work from our laboratory has demonstrated that the proteolytic processing of antigens preferentially occurs at sites within and between protein domains that are flexible, solvent-accessible, and lacking in secondary structure (11–14, 28). These regions are readily accessible for interactions with proteases and can easily conform to protease active sites, making them the initial sites of proteolytic cleavage within an antigen. Furthermore, previous studies have shown that protease-resistant antigens are more immunogenic than their protease-sensitive counterparts (29–31). Therefore, we predicted that proteolysis of PE-III initiates in flexible, unstructured regions of the protein and that mutations would alter PE-III susceptibility to proteolytic degradation according to changes in PE-III stability (32). To test these hypotheses, we first performed limited proteolysis using a single protease, proteinase K. Proteolysis experiments were conducted over a pH range from 7.6 to 4.6 in intervals of 1 pH unit to simulate the environment of the antigen-processing compartment. Degradation of WT PE-III by proteinase K at pH 7.6 is rapid and produces few fragments (Fig. 3A). As the pH decreases, larger PE-III fragments accumulate, whereas the activity of the protease remains consistent to a pH as low as 4.6, indicating that PE-III becomes more resistant to proteolysis by proteinase K at low pH (Fig. 3A). To identify the initial proteinase K cleavage sites, we isolated the two largest fragments (18 and 15 kDa, Fig. 3A) from an SDS-polyacrylamide gel, digested them with trypsin, and sequenced the peptides using LC and tandem MS (LC-MS/MS). High-scoring tryptic peptides from both fragments aligned to the C-terminal region of PE-III suggesting the cleavage sites are located within the N-terminal region (Table S2 and Fig. 3C). Sequenced tryptic peptides and molecular-weight estimations indicated that proteinase K generated the C-terminal 18-kDa fragment by cleavage within the segment of residues 456–462 and the C-terminal 15-kDa fragment by cleavage in the segment of residues 490–496 (Fig. 3C). To confirm the location of these preferential cleavage sites, we generated a variant of WT PE-III in which residues 485–492 (PE-III Δ485–492) were replaced by a histidine–glycine linker to inhibit proteolysis at this site. Although the deletion did not entirely block proteolysis, it did reduce cleavage of intact PE-III, increase the accumulation of the 18-kDa fragment, and decrease the accumulation of the 15-kDa fragment. These data show that initial proteolytic cleavage of PE-III preferentially occurs at two unstructured loops within the protein. These two regions also have the highest B-factors measured from the crystal structures of WT and T18-H477L, respectively (Fig. 2, B and C).
Figure 3.

Limited proteolysis of PE-III variants demonstrates folding stability influences proteolytic susceptibility. A, limited proteolysis of WT PE-III with proteinase K (Prot K). The indicated amount of protease was incubated with WT PE-III for 15 min at 37 °C at the indicated pH prior to analysis by SDS-PAGE and Coomassie staining. Arrowheads indicate the fragments selected for analysis by MS. B, limited proteolysis of PE-III variants with proteinase K at pH 5.6. Arrowheads indicate the bands analyzed in D (top) and E (bottom). C, limited proteolysis of WT and Δ485–492 PE-III with proteinase K at the indicated pH. Arrowheads indicate locations of proteinase K cleavage sites in either variant detected by MS. D, mean relative quantity of the intact PE-III variant after proteolysis with 0.5 μg of proteinase K as compared with the undigested lane. Error bars indicate standard deviation (n = 3). E, mean relative quantity of the major 15-kDa fragment of PE-III variant after proteolysis with 0.5 μg of proteinase K, as compared that of WT PE-III. Error bars indicate standard deviation, and asterisks indicate significance by one-way ANOVA (n = 3).
We next performed limited proteolysis of PE-III with cathepsin S, an important antigen-processing enzyme (3). As seen with proteinase K, PE-III becomes more resistant to proteolysis by cathepsin S as the pH decreases to pH 4.6, when PE-III begins to unfold (Figs. 4A and 1B). Three major fragments of 18, 15, and 13 kDa were subjected to tryptic sequencing (Table S2). The identities of tryptic peptides and molecular-weight estimations indicated that cathepsin S cleaved PE-III in the region of residues 456–462 to generate the C-terminal 18-kDa fragment, residues 514–518 to generate the N-terminal 15-kDa fragment, and residues 527–531 to generate the N-terminal 13-kDa fragment (Fig. 4A). The locations of these sites were corroborated by cathepsin S digestion of PE-III Δ485–492, in which essentially the same N-terminal fragments were observed but shifted down in molecular weight, as expected for a reduction in size corresponding to the 486–492 deletion (Fig. 4C).
Figure 4.

Destabilized PE-III variants are more susceptible to proteolysis by lysosomal proteases. A, indicated amount of cathepsin S was incubated with WT PE-III for 3 h at 37 °C at the indicated pH prior to analysis by SDS-PAGE and Coomassie staining. Arrowheads indicate fragments selected for analysis by MS. B, limited proteolysis of PE-III variants by cathepsin S at pH 5.6 for 3 h at 37 °C. C, C-terminal cleavage sites were confirmed by limited proteolysis of a PE-III variant lacking the 490s loop mentioned above. Schematic showing the locations of the cathepsin S cleavage sites within the primary structure of PE-III is placed below the gel. D, proteolysis of the PE-III variants using lysosomal extracts at different pH values analyzed by SDS-PAGE and Coomassie staining.
Proteinase K is not associated with antigen processing, and although cathepsin S is an important antigen-processing enzyme, the lysosome contains a variety of exo- and endoproteases capable of participating in antigen processing (3). Therefore, we sought to more accurately assess differences in proteolytic processing using lysosomal extracts from a mouse macrophage cell line, RAW264.7 (29). PE-III variants were incubated with lysosomal extracts in pH conditions corresponding to the early (pH 5.9), mid (pH 5.2), and late (pH 4.5) lysosomal environments (Fig. 4D) (31). At the highest pH condition, no degradation was detected for PE-III WT or L552E, whereas R494A and T18 were substantially degraded by 24 h. These differences were exaggerated at pH 5.2, as both PE-III R494A and T18 were completely degraded by 24 h, whereas little proteolysis was detected for WT PE-III and L552E. At the lowest pH condition, WT PE-III and L552E persisted for up to 12 h, while R494A and T18 were completely degraded by 6 h. Thus, we concluded that WT and L552E are much more resistant to lysosomal proteases than R494A and T18 across various pH conditions.
Proteolysis with proteinase K of the WT and three mutant PE-III variants at decreasing pH values revealed differences in the accumulation of specific fragments such as the C-terminal 15-kDa fragment mentioned above. These differences were most obvious at pH 5.6 where accumulation of the C-terminal 15-kDa fragment is greatest (Fig. 3B). In the condition with the least protease present, there was as much or more degradation of PE-III variants, compared with that of WT PE-III (Fig. 3D). However, accumulation of the C-terminal 15-kDa fragments was significantly reduced for the PE-III variants R494A and T18, suggesting that the mutant fragments were more sensitive to proteolysis than the WT or L552E fragments (Fig. 3E). A similar pattern of proteolytic susceptibility and fragment accumulation was also observed for cathepsin S proteolysis (Fig. 4B), suggesting that proteolysis by both model proteases and lysosomal proteases is directed by the structure of the substrate.
Deimmunizing mutations alter the processing and presentation of stabilized PE-III variants in antigen-presenting cells
Peptides derived from the lysosomal processing of protein antigens must be bound to the peptide-binding regions of MHC class II molecules before they are displayed to CD4+ T cells on the surface of antigen-presenting cells. Because the deimmunizing mutations have altered PE-III stability and proteolytic processing, we hypothesized that the mutations would alter the spectrum of PE-III–derived peptides displayed by antigen-presenting cells, as modeled by the macrophage-like RAW264.7 cell line. Previous studies have used immunoprecipitation to isolate solubilized peptide–MHC complexes and then identify peptide sequences using biochemical techniques, including MS (33, 34). However, immunopurification has several weaknesses that limit its usefulness for MHC class II ligand interrogation. The purification of peptide–MHCII complexes results in a large amount of sample loss and thus requires a very large (>108) number of cells; there also appears to be a bias toward the identification of high-affinity peptides that selectively remain bound to the MHCII molecule during the purification process (35). To mitigate these issues, we utilized mild-acid elution to strip peptides from the surface of antigen-presenting cells that have been incubated with PE-III or its mutant variant (36, 37). Eluted peptides were then purified and subjected to LC and tandem MS (LC-MS/MS) for identification (Fig. 5A). We considered only peptides of 12 residues or longer to eliminate the possibility that they were associated with MHCI. Typically, 1–2% of the identified peptides were derived from PE-III, and they formed nested clusters whose members differed by at most a few residues, typical of MHCII-bound peptides (Fig. 5B). Peptides detected by this method could not be positively identified as MHCII ligands, but they were derived from PE-III that had been internalized to subcellular compartments that are associated with antigen processing and normally contain MHCII molecules (Fig. S2). Peptides eluted from cells fed WT, R494A, or L552E were similar in diversity, except for a cluster of peptides centered around the site of the L552E mutation. This cluster was completely absent from samples obtained from cells fed PE-III T18. Overall, less diversity in peptides was observed from cells fed PE-III T18. The most densely populated peptide clusters were centered on, or adjacent to, regions of high-solvent accessibility (Fig. 5C). Two large clusters centered around residues 450 and 480 were located adjacent to highly solvent-accessible regions of PE-III that serve as preferred protease-cleavage sites (Fig. 5C).
Figure 5.

In vitro processing and presentation of PE-III variants generate nested clusters of peptides. A, schematic for the mild acid elution workflow. Antigen-presenting cells are incubated with PE-III protein for 6 h before peptides are eluted with acid and analyzed by LC-MS/MS. B, eluted peptides detected by MS aligned to the PE-III sequence (black lines). Numbered red lines indicate PE-III epitopes reported here. C, solvent-accessible surface area determined from the WT PE-III structure (PDB code 1XK9) variant aligned to the graph in B. Red arrowhead indicates approximate locations of proteinase K cleavage site; blue arrowheads indicate approximate locations of cathepsin S cleavage sites, and the green arrowhead indicates a shared proteinase K (Prot K) and cathepsin S (Cat S) cleavage site.
Destabilizing PE-III mutations reduce serum antibody response with minor changes to CD4+ T-cell priming
The amino acid substitutions originally made to the full-length recombinant immunotoxin were intended to disrupt peptide binding to MHC class II, and the resulting mutant immunotoxin T18 elicited significantly reduced CD4+ T-cell and serum-antibody responses in immunized mice (21). We next sought to replicate these findings using PE-III variants. We began with weekly intravenous administrations to BALB/c mice to mimic clinical treatment with RIT in the absence of any adjuvant. After 4 weeks, mice immunized with PE-III WT or L552E had elevated PE-III–specific serum IgG, whereas PE-III T18 induced a significantly reduced serum IgG response (Fig. 6A and Fig. S3). PE-III R494A immunization induced a drastically reduced serum IgG response compared with WT PE-III and PE-III T18. Next, we performed CD4+ T-cell epitope mapping with a separate group of mice, following a single immunization with PE-III variants and double-mutant heat-liable E. coli enterotoxin (R192G/L211A) as adjuvant (38). To restimulate PE-III–specific CD4+ T cells, we used a set of 21 overlapping 20-residue peptides with a 10-residue offset (Table S3). We detected responding T cells by IL-2 ELISpot and identified epitopes using the Wilcoxon signed-rank test. Peptides that passed the Wilcoxon test and generated an average of 20 spots per mouse or greater were considered epitopes. CD4+ T cells from mice immunized with WT PE-III responded strongly to peptides 2, 3, 8, 14, 15, and 16 (Fig. 6B). All groups of mice responded to peptides 3 and 14, and particular groups responded variously to peptides 2, 4, 8, 13–15, and 19 (Fig. 5B, red lines, and Table 1). All epitope-mapping observations are summarized in Table 1. Peptide sequences matching mapped epitopes were also observed in the sets of peptides eluted from antigen-presenting cells. Interestingly, the CD4+ T-cell responses to peptides 15 and 16 were absent from mice immunized with PE-III T18, and the peptide cluster containing peptides 15 and 16 was absent from the eluate of cells fed PE-III T18 (Figs. 5B and 6B). However, the appearance of a response to peptide 8 exclusively in WT-immunized mice was not consistent with elution results, as peptides overlapping peptide 8 were observed in all elution experiments, regardless of the antigen administered (Figs. 5B and 6B).
Figure 6.

Deimmunized PE-III variants induce lower antibody titers but effectively prime CD4+ T cells. A, BALB/c mice were immunized i.v. for 4 weeks, and serum samples were taken 4 days after each immunization. PE-III–specific antibodies were detected by direct ELISA. Each line indicates average titer as logEC50 determined by nonlinear regression. Error bars indicate standard error; asterisks indicate significance by one-way ANOVA (n = 8 per group). Data are representative of three independent experiments. B, BALB/c mice were immunized once intranasally with PE-III and dmLT. Spleens were isolated, and splenocytes were restimulated with peptides spanning the sequence of WT PE-III with mutant peptides substituted as necessary in duplicate. Bars show average spot-forming units (SFU) per 5 × 105 cells. Error bars indicate standard deviation; asterisks indicate significance by the Wilcoxon signed-rank test, and a mean SFU greater than 20. Dashed line indicates an SFU value 2 S.D. over background.
Table 1.
Peptides identified as epitopes in BALB/c mice
| PE-III WT | PE-III R494A | PE-III L552E | PE-III T18 |
|---|---|---|---|
| 2 | 2 | 2 | |
| 3 | 3 | 3 | 3 |
| 4 | |||
| 8 | |||
| 13 | 13 | 13 | |
| 14 | 14 | 14 | 14 |
| 15 | 15 | ||
| 16 | |||
| 19 |
Discussion
In this study we tested the following hypotheses: (i) the epitope-silencing mutations made to domain III of Pseudomonas exotoxin A (PE-III) have altered the structure and stability of the protein, and (ii) these changes are responsible for the reported reduction in CD4+ T-cell and serum antibody responses in the corresponding therapeutic immunotoxins (19–22). We also identified previously unreported CD4+ T-cell epitopes in mice. Earlier work has shown that antigens resistant to unfolding and proteolysis are much more immunogenic than antigens with the same three-dimensional structure but are less stable and more susceptible to proteolysis (29, 30). Others have also proposed models for antigen processing in which there is an ideal level of protein conformational stability for antigen processing (31). Unstable antigens are degraded too quickly to be loaded onto class II MHC molecules; and for an excessively stabilized antigen, processing occurs too slowly for peptide–MHCII complexes to be generated. Here, we have examined the effects of two single epitope-silencing mutations as well as the combination of six deimmunizing mutations on the stability, proteolytic processing, presentation, and immunogenicity of epitope-containing peptides of PE-III.
We observed that the mutations altered PE-III structure and stability and that individual mutations can reduce or enhance serum-antibody response in immunized mice. The mutations were designed to reduce the affinity of epitope-containing peptides for the MHCII molecule (20). Our results show that all PE-III variants examined here are capable of priming T cells, and thus the disruption of peptide–MHC interactions alone cannot explain the reduced antibody response to certain PE-III variants. An earlier epitope-mapping study identified domain II of the WT immunotoxin as the most immunogenic domain and showed that several mice responded weakly to peptides corresponding to peptides 4 and 5 used in this study. The deimmunized immunotoxin elicited a very weak T-cell response to a single peptide, approximately corresponding to peptide 3 used in this study (21). However, there are several important differences between the previous and current studies, such as the route and frequency of immunization, as well as the antigen dose and adjuvant used. Regardless of these differences, we have reinforced the identification of the region covered by peptides 2–4 as the most immunogenic and have identified other epitopes that had not been reported.
The overall native conformation of PE-III was not disrupted by either of the two single-residue substitutions or the combination of all six residue substitutions. All four PE-III proteins exhibited cooperative unfolding transitions when treated with acid or chemical denaturant (Fig. 1). However, the single-residue substitutions shifted conformational stability in different directions. Using X-ray crystallography of the five-residue substituted T18-H477L, we determined that the changes in stability resulting from the R494A and L552E substitutions were likely due to the removal and addition of a hydrogen bond, respectively (Fig. 2A). These changes in hydrogen bonds were also reflected in the crystallographic B-factors and stability analysis obtained from the COREX algorithm (Fig. 2, B and D), which show that the R494A substitution caused a substantial increase in flexibility in the 490s loop. The L552E mutation had no discernable effect on B-factors but did cause a modest increase in COREX residue stabilities in the region of the mutation. The structural effects of the other three mutations (R427A, F443A, and R505H) were unclear, as Arg-427 and Arg-505 interact almost exclusively with solvent, whereas the F443A mutation appears to alter hydrophobic interactions, the effects of which are difficult to analyze. However, it is possible that these additional mutations within T18-H477L also contribute to the structural differences observed between WT PE-III and T18-H477L.
Our previous work has shown that flexible regions within and between protein domains are the favored sites for proteolytic cleavage and that the terminal segments of early processing intermediates provide CD4+ T-cell epitopes (10–14, 28, 39). In this study, we observed that proteolysis of PE-III with proteinase K produces two major fragments resulting from cleavage within the 460s loop and 490s loop (Fig. 3). These two loops are characterized by a lack of secondary structure, low COREX stability factors, high-crystallographic B-factors, and high-solvent accessibility, which are all indicative of flexibility and promote proteolytic susceptibility. Interestingly, PE-III became less sensitive to proteolysis at pH 6.6 and 5.6 and then resumed sensitivity at the pH of the global unfolding transition (pH 4.6, Fig. 1B). Likewise, at reduced pH, the two major proteinase K fragments accumulated, suggesting they had become more resistant to further proteolysis. Of potential interest to PE-III immunology, these fragments appeared to be much less resistant to proteolysis in the deimmunized PE-III variants R494A and T18, suggesting that a similar processing intermediate may have become a poorer source of one or more T-cell epitopes. Indeed, the epitopes contained in peptide 16 (and to a lesser extent peptide 15) were less immunogenic in PE-III variants R494A and T18. Whether a proteinase K-like enzyme participates in antigen processing is unknown. Aside from the well-studied lysosomal enzymes, the secretory-pathway serine proteases have been implicated in antigen processing (40, 41).
We observed a similar pattern of limited proteolysis by the authentic antigen-processing enzyme, cathepsin S. As the pH is reduced to pH 5.6, intact WT PE-III becomes more resistant to proteolysis by cathepsin S, and a fragment resulting from cleavage at the 460s loop accumulates (Fig. 4A). This same fragment accumulates even further in the cathepsin S digestion of stabilized mutant L552E. In the immune response, the epitope contained in peptide 16 was dramatically reduced in both L552E and T18 (which contains the L552E mutation). Because peptides corresponding to peptide 16 were absent or reduced from the pool of peptides eluted from RAW264.7 cells, we speculate that a processing intermediate is over-stabilized in variants containing the L552E mutation and therefore is a poor source of peptide 16. Taken together, the results with proteinase K and cathepsin S are consistent with the accumulation of a processing intermediate following cleavage at the 460s loop that affects the presentation of an epitope contained in peptide 16. This phenomenon is similar to our previous observation of a correlation between the accumulation of a proteolytic fragment in vitro with the presentation of an adjacent epitope to a T-cell hybridoma in a series of engineered variants of bacteriophage T4 Hsp10 (13).
We also observed that cathepsin-S cleaves within the C-terminal region of PE-III, near the location of epitopes mapped in this study (Fig. 5C). Cathepsin S appears to be distinct from proteinase K, in that it cleaves efficiently in regions of moderate flexibility and solvent accessibility (Figs. 4B and 5C), as well as in regions of high flexibility and solvent accessibility, which is characteristic of proteinase K (Figs. 4A and 5C).
When we subjected the PE-III variants to proteolysis by macrophage lysosomal extracts, we observed that the R494A and T18 variants exhibited greater proteolytic susceptibility compared with the WT or stabilized L552E variant. These results are consistent with previous studies that correlated increased sensitivity to lysosomal extracts with decreased immunogenicity in vivo (30, 31). Large proteolytic fragments that might represent early processing intermediates were not observed in the course of digestion with lysosomal extracts, which we propose is due mainly to the presence of exopeptidase activities, such as has been described for cathepsin B (42).
The deimmunization of the PE-III recombinant immunotoxin resulted in a significant decrease in neutralizing serum antibodies in immunized mice (21). It was important that we replicate the deimmunization that was previously observed for the Fab-containing full-length RIT. The lack of domain II and the Fab could affect pharmacokinetics and the clearance rate of PE-III and thus the exposure time of the molecule to the immune system. Nevertheless, the six-residue substituted PE-III T18 induced a lower level of serum IgG in immunized mice, consistent with previous findings (Fig. 6A) (21). Surprisingly, the single-mutant variant R494A exhibited substantially lower serum IgG compared with both PE-III WT and T18. Despite the reduced antibody induced by some variants upon intravenous immunization, all PE-III variants tested in this study were capable of priming CD4+ T cells, as detected by IL-2 ELISpot with splenocytes following intranasal immunization with adjuvant (Fig. 6B). Therefore, these results suggest that the deimmunization of PE-III was not achieved by the wholesale elimination of CD4+ T-cell epitopes. Nevertheless, we did observe differences in the epitope specificity of CD4+ T cells primed by different PE-III variants.
Our observations raise the question of how the antibody response to PE-III T18 was reduced or, in the case of PE-III R494A, essentially eliminated, given the subtle changes in CD4+ T-cell priming observed here. Robust antibody responses require two antigen-processing and presentation steps, the initial priming of CD4+ T cells by dendritic cells, and later cognate peptide–MHC interactions between CD4+ T cells and antigen-specific B cells at the border between T- and B-cell zones and in the germinal center within the lymph node (43). It is possible that the deimmunizing mutations affect the phenotypes of primed T cells without diminishing their numbers. Past work has demonstrated that the abundance of peptide–MHC complexes and the avidity of the T-cell receptor influence effector T-cell fate (44). Deimmunized PE-III variants could exhibit a reduced capacity to induce T follicular helper cells that are essential for robust antibody responses (45, 46). It is also possible that the deimmunizing mutations modify processing and presentation distinctively within the B cells, such that the B cells are unable to solicit help from T follicular helper cells. These possibilities are not mutually exclusive and may be occurring simultaneously to disrupt the antibody response against the deimmunized PE-III variants.
Structure-based epitope prediction captures the most strongly immunogenic PE-III epitope but fails to accurately predict others. The PE-III epitopes contained within peptides 2–4 lie within the most stable region of the protein, located near the N terminus and near a preferred protease cleavage site within the 460s loop, and therefore it requires only a single proteolytic event to yield the free peptide–MHCII complex. Peptide 3 is strongly predicted to become an epitope by our structure-based prediction algorithm, which generates a profile of antigen processing likelihood (APL) score on the basis of multiple types of conformational stability data (Fig. 7 and Fig. S4) (15). Peptide 3 is predicted to bind well to the I-Ad (Fig. 7) and I-Ed (Fig. S5) MHC molecules, whereas peptides 2 and 4 have poor predicted MHCII affinity, which may be responsible for their greatly reduced immunogenicity compared with peptide 3.
Figure 7.
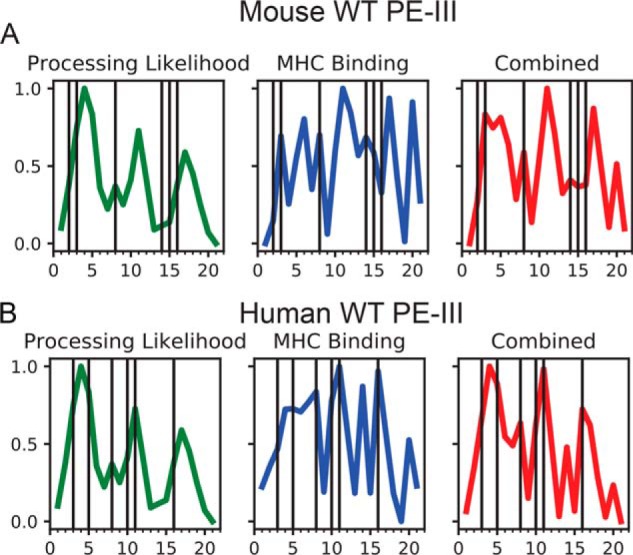
Dominant mouse and human PE-III epitopes are well-predicted by a structure-based epitope prediction algorithm. A, prediction of mouse PE-III epitopes using structure-based processing likelihood (15), MHC-binding prediction for the I-Ad MHCII molecule, and their combination. B, prediction of human PE-III epitopes using structure-based processing likelihood, MHC-binding prediction, and their combination. Vertical lines indicate positions of experimentally mapped epitopes. Bottom tic marks represent 20 residue peptides detailed in Table S3.
The epitopes within peptides 8 and 16 were the only epitopes that were sensitive to the deimmunizing mutations made to PE-III. The epitope in peptide 8 lies between the 460s and 490s loops. Although peptide 8 is readily processed and accumulates on model antigen–presenting cells (Fig. 5B), it is not strongly predicted to be an epitope by APL, due to its relatively low conformational stability (Fig. S4). In contrast, peptide 8 is predicted to bind to the I-Ad MHCII molecule with affinity above the 60th percentile. An explanation for the poor immunogenicity of peptide 8 in mutant PE-III variants is unclear and may be due to divergent conformational effects, depending on the PE-III variant used. Mice immunized with R494A may not respond to peptide 8 because the epitope is destroyed by proteolysis rather than presented, a hypothesis that is consistent with the increased proteolytic susceptibility of intact R494A and its epitope-containing fragments (Fig. 3). In contrast, mice immunized with L552E or T18 may not respond to peptide 8 because the epitope is sequestered by the stabilized structure in L552E and T18, as has been described for particular epitopes in other antigens (14, 47). Peptide 16 elicited a response from only mice immunized with WT PE-III, and this may be for the same reasons as noted for peptide 8. These two epitopes are located in different β-strands of the same β-sheet in PE-III, which could account for similar processing fates. The disruption of PE-III structure by the R494A mutation causes proteolytic destruction of both peptides 8 and 16, whereas the stabilization provided by the L477H and L552E mutations reduces their processing efficiency (Fig. 5B) (47). Peptide 16 does contain the L552E mutation; however, this mutation results in a negligible change in predicted binding affinity to both the I-Ad and I-Ed MHCII molecules (data not shown) (48).
The epitopes contained within peptides 14 and 15 correspond to a relatively unstable region of the protein flanked by two very stable regions and are therefore not well predicted by APL scores. Peptides 14 and 15 are predicted to bind to the I-Ad MHC molecule as strongly as peptides 3 and 8. The characteristics of high-affinity binding and location within a large unstable region are compatible with a “protease-independent” mechanism of presentation, in which this region of PE-III may unfold and bind to the MHCII before the epitopes can be destroyed by proteolysis (Fig. 5B) (49). We note that peptides 11 and 17 are predicted to have strongly MHCII-binding epitopes and reasonably favorable APL scores, but epitopes were not found. As yet, the poorly understood mechanisms must account for their poor immunogenicity. We are currently exploring methods to account for different processing pathways to improve the predictive ability of APL.
Interestingly, the newly mapped PE-III epitopes in mice are similar to those reported in RIT-treated human patients (peptides 8 and 16, Fig. 7B) (20), suggesting that processing of PE-III generates a set of epitope-containing peptides that are common to mice and humans and then a subset binds to the available MHCII molecules. The human PE-III epitopes are all well-predicted by APL, and the prediction was improved when combined with MHCII affinity predicted for the allele DRB1_15, the most common allele within the cohort used for the epitope mapping study (20).
In conclusion, our data demonstrate that single-residue substitutions drastically altered the structure, processing, and immunogenicity of PE-III while having modest effects on CD4+ T-cell priming in mice. The dominant mouse PE-III epitope is well predicted by APL, whereas subdominant epitopes are poorly predicted because they are protease-independent or for reasons that remain obscure. These results provide new insights into the importance of structure-based processing constraints for accurate epitope prediction and highlight the need to combine both APL and MHCII-binding predictions to produce a more holistic understanding of antigen processing and presentation through further experimental study.
Experimental procedures
Cloning and protein purification
Coding sequences for all four PE-III variants were codon-optimized for E. coli and cloned by Genscript using the NdeI and EcoRI sites of pET-22b(+) for lactose-inducible expression and a C-terminal hexahistidine tag. FASTA files containing the amino acid sequence for each variant are available in the supporting information. Plasmid DNA was used to transform BL21 E. coli cells with selection by ampicillin resistance at 50 μg/ml. Protein expression was induced by culturing the cells in autoinduction media as described in 1-liter batches at 27 °C with shaking at 250 rpm (23). Cells were harvested by centrifugation at 7300 × g for 45 min, and cell paste was used immediately or stored at −80 °C. Cell paste was resuspended in purification buffer A (50 mm Tris-HCl, pH 7.5, 25 mm NaCl), including 1 mm phenylmethylsulfonyl fluoride (PMSF), and lysed with a French pressure cell at 16,000 p.s.i. Lysate was clarified by centrifugation at 38,400 × g for 30 min and filtered through cheesecloth. His-tagged PE-III was then purified using nickel-nitrilotriacetic acid affinity chromatography. Bound protein was eluted in steps using a combination of buffer A and buffer B (buffer A plus 250 mm imidazole) and analyzed by SDS-PAGE. Fractions containing PE-III were pooled and further purified to >90% homogeneity by ion-exchange chromatography using Q-Sepharose (GE Healthcare) and a gradient of buffer A and buffer C (50 mm Tris-HCl, pH 7.5, 1 m NaCl). PE-III–containing fractions were pooled and concentrated using Millipore Amicon ultracentrifugal filters with a MWCO of 10,000 Da. Protein for crystallization was further purified by gel filtration using a Pharmacia Superose 6 10/300 GL column and buffer D (50 mm Tris-HCl, pH 7.5, 150 mm NaCl). Protein concentration was determined by absorbance at 280 nm. Purified PE-III was aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C. FITC-labeled PE-III was prepared using the FluoReporter® FITC protein-labeling kit (ThermoFisher Scientific) according to the manufacturer's instructions.
In silico stability analysis
Homology models for PE-III variants were generated using Swiss-Model (https://swissmodel.expasy.org/) and the WT PE-III structure (PDB code 1XK9) as a template. Homology models were used with Swiss PDBViewer (https://spdbv.vital-it.ch/)3 (59) to perform GROMACS96 free energy calculations.
In vitro stability analysis
For acid-induced unfolding experiments, the hydrophobic dye BIS-ANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid, Invitrogen) was used to monitor protein unfolding by fluorescence spectroscopy with an excitation wavelength of 390 nm. Emissions were scanned from 400 to 500 nm. Different pH conditions were generated using phosphate-citrate buffer, where 0.2 m dibasic sodium phosphate and 0.1 m citric acid were mixed until the desired pH was reached. Protein was mixed with dye in phosphate-citrate buffer ranging from pH 7.6 to 2.6 at a concentration of 0.1 μm protein and 1.0 μm dye. Fluorescence intensities averaged for the range 476 to 485 nm and for three replicates were analyzed by nonlinear regression using a sigmoidal dose-response variable slope regression model (Prism 6). For unfolding free energy determinations, 0.5 μm protein was mixed with guanidine-HCl in PBS in 0.25 m steps from 0 to 5 m, and intrinsic tryptophan fluorescence was measured by exciting at 285 nm and scanning emissions from 300 to 400 nm. Fluorescence intensities averaged for the range 330 to 340 nm and for three replicates were analyzed by nonlinear regression as described (14, 26). Best-fit values of pH50 and ΔG0′ were compared using ANOVA (Prism 6).
PE-III crystallography and structure determination
After ion-exchange, PE-III was further purified using a Superose 6 size-exclusion column (Pharmacia) and concentrated. Crystals were grown using the hanging-drop vapor-diffusion method. The crystallization solution was prepared by mixing on a siliconized coverslip, 2 μl of protein solution (10 mg/ml PE-III 0.5 mm PJ34 (Sigma-Millipore, CAS 344458-15-7)) with 2 μl of 60% precipitation solution (0.66 m sodium citrate, pH 7.5, 0.54 mm sodium azide, 0.3 mm DTT). After 24 h at room temperature, coverslips were transferred to fresh wells containing 100% precipitation solution (1.1 m sodium citrate, pH 7.5, 0.9 mm sodium azide, 0.5 mm DTT) for 2–3 days at room temperature until crystals appeared. Crystals were flash-frozen in liquid nitrogen in a mother liquor (1.1 m citrate, pH 7.5, 0.5 mm PJ34, and 20% w/v glycerol), and diffraction data were collected remotely at the Canadian Light Source 08ID-1 beamline. Data collection, refinement, and modeling information are detailed in Table S1. Briefly, data were processed using XDS/XSCALE (50), solved with molecular replacement using the WT PE-III structure (PDB code 1XK9) (27, 51), and iteratively refined using refmac5 (52) within the CCP4 (53) software suite and Coot (54).
Limited proteolysis
Proteolysis reactions with proteinase K and cathepsin S were performed in phosphate-citrate buffer at the indicated pH. Each 20-μl reaction contained 10 μg of protein and 0, 0.5, or 1.0 μg of proteinase K (ThermoFisher Scientific) or 0, 0.25, or 0.5 μg of recombinant human cathepsin S (Sigma-Millipore). 1 mm DTT was included for cathepsin S proteolysis reactions. Lysosomal extract degradation reactions were performed in 50 mm sodium citrate, pH 5.9, 5.2, and 4.5, plus 2 mm DTT. Each 200-μl reaction contained 0.25 μg/μl PE-III and 0.4 μg/μl lysosomal extract. Reaction mixtures were incubated at 37 °C for the indicated time and terminated by the addition of an equal volume of Bio-Rad Laemmli Sample Buffer containing 5 mm PMSF and 150 mm 2-mercaptoethanol. Samples were boiled for 5 min and then analyzed by SDS-PAGE, using a 4–20% Bio-Rad TGX gradient gel. Gels were stained with Coomassie Blue and scanned on a Bio-Rad Chemidoc MP imaging system and analyzed with Bio-Rad ImageLab software. Proteolytic cleavage sites were identified by trypsin sequencing of fragments excised from SDS-polyacrylamide gels. Briefly, each gel slice was destained using a 20-volume excess of 50 mm ammonium bicarbonate and 50% methanol for 20 min, twice. Destained gel slices were dehydrated by incubating in a 20 volume excess of 75% acetonitrile for 20 min. Dried slices were then incubated in a 5-volume excess of 20 μg/ml MS-grade trypsin dissolved in 50 mm ammonium bicarbonate at 37 °C overnight. Each sample was subjected to a 60-min chromatographic method employing a gradient from 2 to 25% acetonitrile in 0.1% formic acid (ACN/FA) over the course of 30 min, a gradient to 50% ACN/FA for an additional 10 min, a step to 90% ACN/FA for 8 min, and a re-equilibration into 2% ACN/FA. Chromatography was carried out in a trap-and-load format using a PicoChip source (New Objective, Woburn, MA), trap column C18 PepMap 100, 5 μm, 100 Å, and the separation column was PicoChip REPROSIL-Pur C18-AQ, 3 μm, 120 Å, 105 mm. The entire run was 0.3 μl/min flow rate. Survey scans were performed in the Orbitrap utilizing a resolution of 120,000 between 375 and 1600 m/z. Data-dependent MS2 scans were performed in the linear ion trap using a collision-induced dissociation of 25%. Raw data were searched using Proteome Discoverer 2.2 using SEQUEST. The Protein FASTA database was Mus musculus (TaxID = 10090) version 2017-07-05 with the PE-III sequence added. Static modifications included carbamidomethyl on cysteines (=57.021) and dynamic modification of oxidation of methionine (=15.9949). Parent ion tolerance was 10 ppm; fragment mass tolerance was 0.6 Da, and the maximum number of missed cleavages was set to 2. Only high-scoring peptides were considered utilizing a false discovery rate (FDR) of 1%.
In vitro processing and presentation
RAW264.7 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Premium, Atlanta Biologicals) and 1% penicillin/streptomycin (Gibco) at 37 °C and 5% CO2 in a humidified incubator.
For confocal microscopy, 1 × 104 cells were seeded into cover glass chambers (ThermoFisher Scientific) and incubated at 37 °C in 5% CO2 overnight. The next day, the growth media were removed and replaced with fresh media containing 25 μg/ml FITC-labeled WT PE-III and incubated for 30 min at 37 °C in 5% CO2. Endosomal compartments were labeled with 50 nm LysoTracker Deep Red (ThermoFisher Scientific) for 30 min at 37 °C in 5% CO2. Media were removed, and cells were washed three times with PBS. Fluorescent images were collected using a Nikon eclipse Ti2 confocal microscope with a Plan Fluor 40× oil differential interference contrast objective. FITC was excited at 487.7 nm, and emission was recorded at 525 nm. LysoTracker was excited at 561.4 nm, and emission was recorded at 595 nm. Images were analyzed and processed using NIS elements software (Nikon).
For mild acid elution, RAW264.7 cells were seeded into a 175-cm2 flask at 500,000 cells/ml and incubated until 80% confluent, about 10 million cells per flask, and five flasks were used for one experiment. PE-III WT or mutant variant was then added to the culture medium to a concentration of 50 μg/ml, and cells were incubated for 6 h to allow for antigen uptake, processing, and presentation. Then cells were washed three times with 10 ml of phosphate-buffered saline (PBS, 50 mm sodium phosphate, 125 mm sodium chloride) and incubated with 10 ml of phosphate-citrate buffer, pH 3.2, for 5 min (36). Phosphate-citrate buffer was prepared by mixing 0.2 m sodium phosphate dibasic with 0.1 m citric acid until the desired pH is reached (pH 3.2 is 40 mm dibasic sodium phosphate 80 mm citric acid). Eluate was collected and stored at −80 °C until purification and analysis, and cells were washed with 10 ml PBS, which was replaced with complete media. Cells were allowed to recover overnight before the same process was repeated two more times. Eluate fractions were clarified by centrifugation at 4700 × g for 15 min and pooled before being applied to a Sep-PAK (Waters) C-18 cartridge that was activated with 3 ml of acetonitrile followed by 3 ml of ddH2O. Cartridges were washed with 50 ml of ddH2O, and bound material was eluted using 400 μl of 60% acetonitrile in ddH2O. Solvent was evaporated in a heated speed-vac until <50 μl remained, and 300 μl of phosphate-citrate, pH 3.2, was added. Samples were analyzed immediately or stored at −20 °C until analysis. PE-III–specific peptides within the eluate were identified by MS. All reagents were purchased from ThermoFisher Scientific (Waltham, MA) unless otherwise stated. Each sample was aliquoted and reduced with 10 mm tris(2-carboxyethyl)phosphine for 1 h at 55 °C. Cysteines were alkylated with 20 mm iodoacetamide at room temperature for 30 min in the dark. The protein aliquots were desalted using a methanol/chloroform extraction (55) before drying to completion in a speed vacuum concentrator. The dried pellets were dissolved in 2% acetonitrile and 0.1% formic acid. LC-MS was performed using a Dionex U3000 nano flow system coupled to a ThermoFisher Scientific fusion mass spectrometer. Each sample was subjected to a 90-min chromatographic method employing a gradient from 2 to 25% acetonitrile in 0.1% formic acid (ACN/FA) over the course of 60 min, a gradient to 50% ACN/FA for an additional 10 min, a step to 90% ACN/FA for 10 min, and a re-equilibration into 2% ACN/FA. Chromatography and survey scans were as described above. Parent ion tolerance was 10 ppm; fragment mass tolerance was 0.6 Da, and the maximum number of missed cleavages was set to 12, because cleavage is nonspecific. Only high-scoring peptides were considered utilizing a FDR of 1%. Identified peptides were aligned to the whole protein sequence using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/)3 (60) and BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).3 Solvent-accessible surface area was calculated using MolMol (56). Graphs and figures were made using OriginPro 8 and Adobe Illustrator.
Immunogenicity of PE-III variants
Female BALB/c mice aged 6–8 weeks (Charles River Laboratories) were used for all animal experiments. Mice were allowed to acclimate to our facility for 1 week after arrival. For the intravenous immunization experiments mice were administered 5 μg of PE-III in 100 μl of PBS weekly for 4 weeks by retroorbital injection under 3% v/v isoflurane anesthesia and oxygen. Four days after each immunization, serum was collected by submandibular vein puncture and stored at −20 °C. PE-III–specific serum antibody titer was analyzed by ELISA. Then 96-well ELISA plates (Corning) were coated overnight with 100 μl of 2 μg/ml PE-III in PBS at 4 °C. Plates were then blocked for 1 h at room temperature with 200 μl of 5% w/v nonfat dry milk (NFDM) in PBS with 0.1% v/v Tween 20 (PBST). Serum was diluted 1:10 in PBST + 5% NFDM, then serially diluted 1:5 in each column on the plate and incubated for 2 h at room temperature. Plates were washed five times with PBST, and IgG was detected by incubating plates with a 1:2000 dilution of goat anti-mouse IgG-HRP (Invitrogen) in PBST + 5% NFDM. Plates were washed five times with PBST and developed for 5 min with 100 μl of TMB substrate (2.5 mm TMB, 3% v/v hydrogen peroxide, 20 mm sodium acetate, pH 6.0). The development reaction was stopped with 100 μl of 0.1 m phosphoric acid, and absorbance at 450 nm was read using a BioTek plate reader. Data were fit using a sigmoidal dose-response variable–slope nonlinear regression model with the logEC50 serving as the titer and compared using ANOVA and GraphPad Prism 6. For epitope mapping experiments, mice were immunized once intranasally with 5 μg of PE-III and 1 μg of double-mutant (R192G/L211A) heat-liable E. coli enterotoxin (dmLT) as adjuvant. After 10 days, mice were euthanized by CO2 asphyxiation, and spleens were aseptically removed and homogenized using gentle MACS C tubes (Miltenyi). Splenocytes were isolated by passing the homogenate through a 40-μm filter and centrifugation. Red blood cells were removed using ACK lysis buffer (Gibco), and splenocytes were plated at a density of 9 × 104 cells per well in 180 μl of RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (Gibco) on microfilter ELISpot plates (Millipore MAIPS45) precoated with anti-mouse IL-2 antibodies (eBioscience). Cells were restimulated with 0.4 μg of PE-III–specific synthetic peptides (Pepscan, Netherlands), 0.04 μg of intact PE-III or dmLT, or 10 μg of concanavalin A (Sigma) in complete media to a total volume of 200 μl for 36 h at 37 °C 5% CO2. Plates were washed five times, and spots were developed using a biotinylated secondary antibody and HRP. Spots were counted by computer-assisted image analysis (Immunospot 5.0, Cellular Technology Ltd.). Results are shown in spot forming units (SFU) per 5 × 105 cells. All mouse experiments followed institutional guidelines approved by the Tulane Animal Care and Use Committee.
Epitope prediction
Structure-based epitope prediction was performed as described with minor changes (15). The number of proteins homologous to PE-III is inadequate for sequence entropy to be reliably calculated; and thus, aggregate conformational stability was based on B-factors, solvent-accessible surface area, and COREX/Best residue stabilities. Mapped epitopes are reported here or previously (20). MHC binding prediction was performed using the NetMHCII 2.3 server (http://www.cbs.dtu.dk/services/NetMHC/)3 (57, 58).
Author contributions
D. L. M. and S. J. L. conceptualization; D. L. M., R. R. M., and S. J. L. formal analysis; R. R. M. and S. J. L. supervision; R. R. M. and S. J. L. funding acquisition; D. L. M., R. R. M., and S. J. L. investigation; D. L. M., H.-W. P., R. R. M., and S. J. L. methodology; D. L. M. writing-original draft; R. R. M. and S. J. L. project administration; D. L. M. and S. J. L. writing-review and editing; H.-W. P. resources; R. R. M. and S. J. L. software.
Supplementary Material
Acknowledgments
We thank Jessie Guidry for assisting with the proteomics work. Proteomics work was supported by National Institutes of Health Grants P20 RR018766, P20 GM103514, and P30 GM103514 and a special appropriation from the Louisiana State University School of Medicine Office of the Dean.
This work was supported by National Institutes of Health Grant R21-AI122199 (to R. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5, Tables S1–S3, and supporting Ref. 1.
The atomic coordinates and structure factors (code 6EDG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Pseudomonas exotoxin A domain III T18H477L: 6EDG–X-ray crystallography has been deposited in Worldwide Protein Data Bank.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MHC
- major histocompatibility complex
- MHCI
- MHC class I molecule
- MHCII
- MHC class II molecule
- GdnHCl
- guanidine hydrochloride
- RIT
- recombinant immunotoxin
- PE
- Pseudomonas exotoxin
- HRP
- horseradish peroxidase
- PMSF
- phenylmethylsulfonyl fluoride
- PDB
- Protein Data Bank
- ANOVA
- analysis of variance
- ACN
- acetonitrile
- FA
- formic acid
- FDR
- false discovery rate
- NFDM
- nonfat dry milk
- APL
- antigen-processing likelihood
- ddH2O
- double-distilled H2O
- TMB
- 3,3′,5,5′-tetramethylbenzidine.
References
- 1. Wang P., Sidney J., Dow C., Mothé B., Sette A., and Peters B. (2008) A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4, e1000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jensen K. K., Andreatta M., Marcatili P., Buus S., Greenbaum J. A., Yan Z., Sette A., Peters B., and Nielsen M. (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406 10.1111/imm.12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watts C. (2001) Antigen processing in the endocytic compartment. Curr. Opin. Immunol. 13, 26–31 10.1016/S0952-7915(00)00177-1 [DOI] [PubMed] [Google Scholar]
- 4. Latek R. R., and Unanue E. R. (1999) Mechanisms and consequences of peptide selection by the I-Ak class II molecule. Immunol. Rev. 172, 209–228 10.1111/j.1600-065X.1999.tb01367.x [DOI] [PubMed] [Google Scholar]
- 5. Blum J. S., Wearsch P. A., and Cresswell P. (2013) Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 10.1146/annurev-immunol-032712-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma C., Whiteley P. E., Cameron P. M., Freed D. C., Pressey A., Chen S.-L., Garni-Wagner B., Fang C., Zaller D. M., Wicker L. S., and Blum J. S. (1999) Role of APC in the selection of immunodominant T cell epitopes. J. Immunol. 163, 6413–6423 [PubMed] [Google Scholar]
- 7. Gelder C., Davenport M., Barnardo M., Bourne T., Lamb J., Askonas B., Hill A., and Welsh K. (1998) Six unrelated HLA-DR-matched adults recognize identical CD4+ T cell epitopes from influenza A haemagglutinin that are not simply peptides with high HLA-DR binding affinities. Int. Immunol. 10, 211–222 10.1093/intimm/10.2.211 [DOI] [PubMed] [Google Scholar]
- 8. Phelps R. G., Jones V. L., Coughlan M., Turner A. N., and Rees A. J. (1998) Presentation of the Goodpasture autoantigen to CD4 T cells is influenced more by processing constraints than by HLA class II peptide binding preferences. J. Biol. Chem. 273, 11440–11447 10.1074/jbc.273.19.11440 [DOI] [PubMed] [Google Scholar]
- 9. Kim J., Sette A., Rodda S., Southwood S., Sieling P. A., Mehra V., Ohmen J. D., Oliveros J., Appella E., Higashimoto Y., Rea T. H., Bloom B. R., and Modlin R. L. (1997) Determinants of T cell reactivity to the Mycobacterium leprae GroES homologue. J. Immunol. 159, 335–343 [PubMed] [Google Scholar]
- 10. Landry S. J. (1997) Local protein instability predictive of helper T-cell epitopes. Immunol. Today 18, 527–532 10.1016/S0167-5699(97)01152-3 [DOI] [PubMed] [Google Scholar]
- 11. Dai G., Carmicle S., Steede N. K., and Landry S. J. (2002) Structural basis for helper T-cell and antibody epitope immunodominance in bacteriophage T4 Hsp10. Role of disordered loops. J. Biol. Chem. 277, 161–168 10.1074/jbc.M102259200 [DOI] [PubMed] [Google Scholar]
- 12. Carmicle S., Dai G., Steede N. K., and Landry S. J. (2002) Proteolytic sensitivity and helper T-cell epitope immunodominance associated with the mobile loop in Hsp10s. J. Biol. Chem. 277, 155–160 10.1074/jbc.M107624200 [DOI] [PubMed] [Google Scholar]
- 13. Carmicle S., Steede N. K., and Landry S. J. (2007) Antigen three-dimensional structure guides the processing and presentation of helper T-cell epitopes. Mol. Immunol. 44, 1159–1168 10.1016/j.molimm.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 14. Nguyen H.-N., Steede N. K., Robinson J. E., and Landry S. J. (2015) Conformational instability governed by disulfide bonds partitions the dominant from subdominant helper T-cell responses specific for HIV-1 envelope glycoprotein gp120. Vaccine 33, 2887–2896 10.1016/j.vaccine.2015.04.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mettu R. R., Charles T., and Landry S. J. (2016) CD4+ T-cell epitope prediction using antigen processing constraints. J. Immunol. Methods 432, 72–81 10.1016/j.jim.2016.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kondo T., FitzGerald D., Chaudhary V. K., Adhya S., and Pastan I. (1988) Activity of immunotoxins constructed with modified Pseudomonas exotoxin A lacking the cell recognition domain. J. Biol. Chem. 263, 9470–9475 [PubMed] [Google Scholar]
- 17. Kreitman R. J., and Pastan I. (1997) Recombinant toxins containing human granulocyte-macrophage colony-stimulating factor and either Pseudomonas exotoxin or diphtheria toxin kill gastrointestinal cancer and leukemia cells. Blood 90, 252–259 [PubMed] [Google Scholar]
- 18. Chaudhary V. K., Mizukami T., Fuerst T. R., FitzGerald D. J., Moss B., Pastan I., and Berger E. A. (1988) Selective killing of HIV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature 335, 369–372 10.1038/335369a0 [DOI] [PubMed] [Google Scholar]
- 19. Mazor R., Onda M., and Pastan I. (2016) Immunogenicity of therapeutic recombinant immunotoxins. Immunol. Rev. 270, 152–164 10.1111/imr.12390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazor R., Vassall A. N., Eberle J. A., Beers R., Weldon J. E., Venzon D. J., Tsang K. Y., Benhar I., and Pastan I. (2012) Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl. Acad. Sci. U.S.A. 109, E3597–E3603 10.1073/pnas.1218138109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mazor R., Crown D., Addissie S., Jang Y., Kaplan G., and Pastan I. (2017) Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell. Mol. Immunol. 14, 432–442 10.1038/cmi.2015.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mazor R., Eberle J. A., Hu X., Vassall A. N., Onda M., Beers R., Lee E. C., Kreitman R. J., Lee B., Baker D., King C., Hassan R., Benhar I., and Pastan I. (2014) Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc. Natl. Acad. Sci. U.S.A. 111, 8571–8576 10.1073/pnas.1405153111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 24. Fraternali F., and Van Gunsteren W. F. (1996) An efficient mean solvation force model for use in molecular dynamics simulations of proteins in aqueous solution. J. Mol. Biol. 256, 939–948 10.1006/jmbi.1996.0139 [DOI] [PubMed] [Google Scholar]
- 25. Hilser V. J., and Whitten S. T. (2014) Using the COREX/BEST server to model the native-state ensemble. Methods Mol. Biol. 1084, 255–269 10.1007/978-1-62703-658-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manyusa S., and Whitford D. (1999) Defining folding and unfolding reactions of apocytochrome b5 using equilibrium and kinetic fluorescence measurements. Biochemistry 38, 9533–9540 10.1021/bi990550d [DOI] [PubMed] [Google Scholar]
- 27. Yates S. P., Taylor P. L., Jørgensen R., Ferraris D., Zhang J., Andersen G. R., and Merrill A. R. (2005) Structure–function analysis of water-soluble inhibitors of the catalytic domain of exotoxin A from Pseudomonas aeruginosa. Biochem. J. 385, 667–675 10.1042/BJ20041480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mirano-Bascos D., Tary-Lehmann M., and Landry S. J. (2008) Antigen structure influences helper T-cell epitope dominance in the human immune response to HIV envelope glycoprotein gp120. Eur. J. Immunol. 38, 1231–1237 10.1002/eji.200738011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delamarre L., Pack M., Chang H., Mellman I., and Trombetta E. S. (2005) Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307, 1630–1634 10.1126/science.1108003 [DOI] [PubMed] [Google Scholar]
- 30. Delamarre L., Couture R., Mellman I., and Trombetta E. S. (2006) Enhancing immunogenicity by limiting susceptibility to lysosomal proteolysis. J. Exp. Med. 203, 2049–2055 10.1084/jem.20052442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Machado Y., Freier R., Scheiblhofer S., Thalhamer T., Mayr M., Briza P., Grutsch S., Ahammer L., Fuchs J. E., Wallnoefer H. G., Isakovic A., Kohlbauer V., Hinterholzer A., Steiner M., Danzer M., et al. (2016) Fold stability during endolysosomal acidification is a key factor for allergenicity and immunogenicity of the major birch pollen allergen. J. Allergy Clin. Immunol. 137, 1525–1534 10.1016/j.jaci.2015.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park C., and Marqusee S. (2004) Probing the high energy states in proteins by proteolysis. J. Mol. Biol. 343, 1467–1476 10.1016/j.jmb.2004.08.085 [DOI] [PubMed] [Google Scholar]
- 33. Karunakaran K. P., Yu H., Jiang X., Chan Q., Goldberg M. F., Jenkins M. K., Foster L. J., and Brunham R. C. (2017) Identification of MHC-bound peptides from dendritic cells infected with Salmonella enterica strain SL1344: implications for a nontyphoidal Salmonella vaccine. J. Proteome Res. 16, 298–306 10.1021/acs.jproteome.6b00926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karunakaran K. P., Yu H., Jiang X., Chan Q., Moon K.-M., Foster L. J., and Brunham R. C. (2015) Outer membrane proteins preferentially load MHC class II peptides: implications for a Chlamydia trachomatis T cell vaccine. Vaccine 33, 2159–2166 10.1016/j.vaccine.2015.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hassan C., Kester M. G., Oudgenoeg G., de Ru A. H., Janssen G. M., Drijfhout J. W., Spaapen R. M., Jiménez C. R., Heemskerk M. H., Falkenburg J. H., and van Veelen P. A. (2014) Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. J. Proteomics 109, 240–244 10.1016/j.jprot.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 36. Storkus W. J., Zeh H. J., Salter R. D., and Lotze M. T. (1993) Identification of T-cell epitopes: rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. Emphasis Tumor Immunol. 14, 94–103 10.1097/00002371-199308000-00003 [DOI] [PubMed] [Google Scholar]
- 37. Gebreselassie D., Spiegel H., and Vukmanovic S. (2006) Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum. Immunol. 67, 894–906 10.1016/j.humimm.2006.08.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Norton E. B., Bauer D. L., Weldon W. C., Oberste M. S., Lawson L. B., and Clements J. D. (2015) The novel adjuvant dmLT promotes dose sparing, mucosal immunity and longevity of antibody responses to the inactivated polio vaccine in a murine model. Vaccine 33, 1909–1915 10.1016/j.vaccine.2015.02.069 [DOI] [PubMed] [Google Scholar]
- 39. Dai G., Steede N. K., and Landry S. J. (2001) Allocation of helper T-cell epitope immunodominance according to three-dimensional structure in the human immunodeficiency virus type I envelope glycoprotein gp120. J. Biol. Chem. 276, 41913–41920 10.1074/jbc.M106018200 [DOI] [PubMed] [Google Scholar]
- 40. Zhu H., Liu K., Cerny J., Imoto T., and Moudgil K. D. (2005) Insertion of the dibasic motif in the flanking region of a cryptic self-determinant leads to activation of the epitope-specific T cells. J. Immunol. 175, 2252–2260 10.4049/jimmunol.175.4.2252 [DOI] [PubMed] [Google Scholar]
- 41. Schneider S. C., Ohmen J., Fosdick L., Gladstone B., Guo J., Ametani A., Sercarz E. E., and Deng H. (2000) Cutting edge: introduction of an endopeptidase cleavage motif into a determinant flanking region of hen egg lysozyme results in enhanced T cell determinant display. J. Immunol. 165, 20–23 10.4049/jimmunol.165.1.20 [DOI] [PubMed] [Google Scholar]
- 42. Bond J. S., and Barrett A. J. (1980) Degradation of fructose-1,6-bisphosphate aldolase by cathepsin B. Biochem. J. 189, 17–25 10.1042/bj1890017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gitlin A. D., Shulman Z., and Nussenzweig M. C. (2014) Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 637–640 10.1038/nature13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tubo N. J., Pagán A. J., Taylor J. J., Nelson R. W., Linehan J. L., Ertelt J. M., Huseby E. S., Way S. S., and Jenkins M. K. (2013) Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796 10.1016/j.cell.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fazilleau N., McHeyzer-Williams L. J., Rosen H., and McHeyzer-Williams M. G. (2009) The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 10, 375–384 10.1038/ni.1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shulman Z., Gitlin A. D., Weinstein J. S., Lainez B., Esplugues E., Flavell R. A., Craft J. E., and Nussenzweig M. C. (2014) Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345, 1058–1062 10.1126/science.1257861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. So T., Ito H.-O., Koga T., Watanabe S., Ueda T., and Imoto T. (1997) Depression of T-cell epitope generation by stabilizing hen lysozyme. J. Biol. Chem. 272, 32136–32140 10.1074/jbc.272.51.32136 [DOI] [PubMed] [Google Scholar]
- 48. Vita R., Overton J. A., Greenbaum J. A., Ponomarenko J., Clark J. D., Cantrell J. R., Wheeler D. K., Gabbard J. L., Hix D., Sette A., and Peters B. (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412 10.1093/nar/gku938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villadangos J. A., and Ploegh H. L. (2000) Proteolysis in MHC class II antigen presentation: who's in charge? Immunity 12, 233–239 10.1016/S1074-7613(00)80176-4 [DOI] [PubMed] [Google Scholar]
- 50. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 10.1107/S0021889897006766 [DOI] [Google Scholar]
- 52. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Friedman D. B. (2007) Quantitative proteomics for two-dimensional gels using difference gel electrophoresis. Methods Mol. Biol. 367, 219–239 [DOI] [PubMed] [Google Scholar]
- 56. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 57. Nielsen M., Lundegaard C., Worning P., Lauemøller S. L., Lamberth K., Buus S., Brunak S., and Lund O. (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 10.1110/ps.0239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Andreatta M., and Nielsen M. (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 10.1093/bioinformatics/btv639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- 60. Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Park Y. M., Buso N., and Lopez R. (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 10.1093/nar/gkv279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.