

Abstract
We recently uncovered a regulatory pathway of the muscle isoform of glycogen phosphorylase (PYGM) that plays an important role in regulating immune function in T cells. Here, using various enzymatic, pulldown, and immunoprecipitation assays, we describe signaling cross-talk between the small GTPases RAS and RAP1A, member of RAS oncogene family (RAP1) in human Kit 225 lymphoid cells, which, in turn, is regulated by the epidermal growth factor receptor (EGFR). We found that this communication bridge is essential for glycogen phosphorylase (PYG) activation through the canonical pathway because this enzyme is inactive in the absence of adenylyl cyclase type 6 (ADCY6). PYG activation required stimulation of both exchange protein directly activated by cAMP 2 (EPAC2) and RAP1 via RAS and ADCY6 phosphorylation, with the latter being mediated by Raf-1 proto-oncogene, Ser/Thr kinase (RAF1). Consistent with this model, PYG activation was EGFR-dependent and may be initiated by the constitutively active form of RAS. Consequently, PYG activation in Kit 225 T cells could be blocked with specific inhibitors of RAS, EPAC, RAP1, RAF1, ADCY6, and cAMP-dependent protein kinase. Our results establish a new paradigm for the mechanism of PYG activation, which depends on the type of receptor involved.
Keywords: epidermal growth factor receptor (EGFR), RAS protein, phosphorylase, adenylate cyclase (adenylyl cyclase), Raf kinase, glycogen phosphorylase, adaptive immunity, GTPase, cAMP-regulated guanine nucleotide exchange factor II (EPAC2), cell signaling.
Introduction
Glycogen phosphorylase (PYG,6 EC 2.4.1.1) catalyzes and regulates the breakdown of glycogen (glycogenolysis) from its intracellular stores to release glucose 1-phosphate. In mammals, PYG exists in three different isoforms, each encoded by a different gene: brain (PYGB), liver (PYGL), and muscle (PYGM) (1). Recently, we and others reported that PYGM is also expressed in T lymphocytes (2, 3), where it plays a crucial role in the control of interleukin (IL)-2–stimulated T-cell migration and proliferation (3–5). PYG activation occurs in an isoform-dependent manner; activation can be regulated by reversible serine 14 phosphorylation (PGYL), or by both serine 14 phosphorylation and allosteric modification (PYGB and PYGM) (6–9). Both mechanisms are mediated through the control of adenylyl cyclase activity (7, 9), converting ATP to cAMP, a key second messenger in eukaryotic intracellular signaling (6, 10).
cAMP mediates myriad cellular functions by acting on effector molecules, such as EPAC, a guanine nucleotide exchange factor (GEF) (11), and cAMP-dependent protein kinases (PKAs) (11, 12), which leads to the phosphorylation of PYG at serine 14 (7). In addition, hydrolysis of ATP generates AMP (13), which can mediate PYG activation by allosteric modification (8, 10). Against this background, we have previously reported that the transition of PYGM from the inactive to the active state in IL-2–stimulated T lymphocytes occurs by allosteric modifications that are dependent on the small GTPase RAC1 (RAS-related C3 botulinum toxin substrate 1) and independent from the adenylyl cyclase (ADCY)/cAMP pathway. Mechanistically, RAC1 binds to the RAC-binding domain of PYGM (amino acids 191–270), which is adjacent to the AMP-binding site (3).
Small GTPases of the RAS superfamily are key players in the complex signaling networks that control cellular physiology (14–17). They are positively regulated by signals from a large number of membrane receptors, including G protein-coupled receptors (16, 17), T-cell antigen receptor (18, 19), B-cell receptor (20, 21), IL-2 receptor (IL-2R) (3), and platelet-derived growth factor or epidermal growth factor receptor (EGFR) (16, 17, 22, 23). Small GTPases function as molecular switches that cycle between an inactive and an active GTP-bound state, with the transition between each state regulated by GEFs. In their active configuration, GTPases interact with downstream effector molecules to promote numerous biological responses (16, 24). There are a great variety of GTPase effector molecules, such as kinases (including RAF-1 prot-oncogene) (22, 23), PYGM (3), and also GEFs such as RAS-guanyl-releasing protein 1 (19, 21), T-cell lymphoma invasion and metastasis-inducing protein 1, 2 (TIAM1, TIAM2) (25, 26) or cAMP-regulated GEFII (EPAC2) (27).
Regarding EGFR, it has been well-established that it is expressed on many different hematopoietic cell types, including macrophages (28, 29), monocytes (30), and plasma cells (31), and also in both T-helper-2 (32) and CD4 (33) cells. Of special relevance is its expression in T-helper-2 cells, because EGFR participates in the control of innate immune response at the site of inflammation (32). In this context, using Kit 225 T cells, a human T cell line established from a patient with T cell chronic lymphocytic leukemia (34), we show here that subsequent to EGFR stimulation, RAS cooperates with RAP1 to activate PYG. By combining pharmacological and genetic approaches, we identify EPAC2 as the bridge between RAS and RAP1. We also show that cross-talk between these two small GTPases regulates PYG activity through ADCY6. Overall, these results reveal an unanticipated relationship between small GTPases of the RAS family (RAS and RAP1) and glycogen metabolism through PYG. Interestingly, we demonstrate that, depending on the stimulus (IL-2 or EGF), lymphoid cells will use one signaling pathway over the other to direct different cellular responses, but both lead to activation of PYG.
Results
EPAC–RAP1 mediates PYG activation
To examine whether EPAC could mediate PYG activation, T cells were deprived of IL-2 for 48 h and were then stimulated or not for 10 min with 10 μm 8-pCPT-2′-O-Me-cAMP, a potent and selective activator of EPAC (35). Cells were then lysed, and PYG activity was determined as described under “Experimental procedures.” The EPAC activator induced robust PYG activity in T cells, which was similar to that induced by IL-2 (Fig. 1A). To test whether engagement of EPAC stimulated RAP1 activation, we repeated EPAC activation in T-cells and measured RAP1 activation using a pulldown assay. A substantial increase in the active form of RAP1 (RAP1-GTP) was observed following 10 min of 8-pCPT-2′-O-Me-cAMP treatment (Fig. 1B).
Figure 1.

EPAC–RAP1 proteins stimulate glycogen phosphorylase activation in Kit 225 T cells. Cells deprived of IL-2 for 48 h were stimulated (+) or not (−) (A and B) with 500 units ml−1 IL-2 (A) or 10 μm 8-pCPT-2′-O-Me-cAMP (A and B) for 10 min and lysed. Cell lysates were used to measure glycogen phosphorylase activity (A) and RAP1 activation (B) by an affinity precipitation assay using the GST-RBD of RalGDS. Precipitated active RAP1 (RAP1-GTP) and total RAP1 from cell lysates were analyzed by Western blotting using a specific anti-RAP1 antibody. Results are representative of five independent experiments. C, cells deprived of IL-2 for 24 h were transfected with plasmids coding for pMT2-HA (mock control), pMT2-HA-RAP1, and pMT2-HA-RAP1Q63E. At 24 h post-transfection, cells were stimulated or not with 500 units ml−1 IL-2 for 10 min and lysed. Cell extracts were used to measure glycogen phosphorylase activity. Western blot analysis of protein expression levels was carried out using a specific anti-HA antibody. D, cells deprived of IL-2 for 48 h were treated with an adenylyl cyclase inhibitor (10 μm MDL12330A) or vehicle (0.1% DMSO) for 1 h and then treated or not with 10 μm forskolin or 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min and lysed. Cell extracts were used to measure glycogen phosphorylase activity. Results presented as histograms show the mean ± S.D. (error bars) of four (A), five (C), and three (D) independent experiments performed in triplicates. ***, p < 0.001. B, box plots; each box includes values from five different samples. All values are expressed in arbitrary units. Differences between unstimulated and stimulated states were p < 0.0062 (##) in all cases (Mann–Whitney test).
To examine the putative association between RAP1 and PYG activation, T-cells were transiently transfected with either an empty vector (mock control) or cDNAs encoding WT RAP1 or the constitutively active form of RAP1 (RAP1Q63E) for 24 h, and PYG activity was examined as before. Transfection of RAP1Q63E, but not WT RAP1, activated PYG to a level comparable with that obtained with IL-2 stimulation (Fig. 1C), suggesting that the potent stimulating effect of RAP1Q63E was specific to its active configuration. Western blotting showed that WT RAP1 and RAP1Q63E were equally expressed in T cells (Fig. 1C).
We next used an ADCY inhibitor, MDL12330A (36), to determine whether some ADCY activity is needed for the EPAC–RAP1 signaling complex to control PYG pathway activation. Accordingly, T cells were pretreated or not with 10 μm MDL12330A, followed by stimulation with 10 μm forskolin or 8-pCPT-2′-O-Me-cAMP for 10 min before measurement of PYG activity. Both forskolin and 8-pCPT-2′-O-Me-cAMP induced robust PYG activity, which depended on ADCY (Fig. 1D). The result obtained with forskolin in the presence of the ADCY inhibitor was expected, because there is a direct relationship between forskolin and ADCY activation. It was, however, surprising that the ADCY inhibitor blocked EPAC action on PYG, suggesting that ADCY is situated between EPAC and PYG.
To test the aforementioned hypothesis, IL-2–deprived T cells were transfected as before with WT or RAP1Q63E. After 24 h, cells were pretreated with either 10 μm MDL12330A or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 μm forskolin for 10 min, and PYG activity was subsequently measured. As shown in Fig. 2A, RAP1Q63E induced PYG activity to a level comparable with that induced by forskolin. As expected, MDL12330A efficiently inhibited forskolin-stimulated PYG activity. MDL12330A also completely blocked PYG activity induced by RAP1Q63E (Fig. 2A). Western blotting confirmed that RAP1Q63E expression was equivalent under both conditions. Overall, these results suggest that the EPAC–RAP1 signaling axis regulates PYG activation via ADCY.
Figure 2.

RAP1 mediates glycogen phosphorylase activation via adenylyl cyclase. T cells deprived of IL-2 for 48 h were pretreated with an adenylyl cyclase inhibitor (10 μm MDL12330A) (+) (A), a RAF1 inhibitor (10 μm GW5074) (+) (B), or vehicle (0.1% DMSO) (−) (A and B) for 1 h and stimulated (+) or not (−) with 10 μm forskolin (A) or 10 μm 8-pCPT-2′-O-Me-cAMP (B) for 10 min and lysed. Cell extracts were used to measure glycogen phosphorylase activity. The results (A and B) show the mean ± S.D. (error bars) of three independent experiments performed in triplicates. ***, p < 0.001.
Beazely et al. (37) have previously reported that RAF1 kinase mediates phosphorylation and activation of ADCY6. To test the hypothesis that EPAC and RAP1 mediate PYG activation via the RAF1 kinase/ADCY6 pathway, we transfected IL-2–deprived T cells with RAP1Q63E or a control vector for 24 h, after which cells were pretreated with either 10 μm GW5074 (a RAF1 kinase inhibitor) (38) or vehicle (0.1% DMSO) for 1 h. Cells were then stimulated or not with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min, and PYG activity was measured. Both 8-pCPT-2′-O-Me-cAMP and RAP1Q63E induced PYG activity to a comparable level (Fig. 2B). GW5074 efficiently blocked PYG activity stimulated by 8-pCPT-2′-O-Me-cAMP and RAP1Q63E overexpression (Fig. 2B). Western blotting confirmed that RAP1Q63E expression was equivalent under both conditions.
To monitor the status of the EPAC–RAP1 pathway in this signaling cascade, we analyzed the levels of phosphorylated RAF1 and ERK1/2 during EPAC stimulation by Western blotting using phospho-specific antibodies to RAF1 and ERK1/2. As shown in Fig. 3A, T cells responded to EPAC stimulation by phosphorylating RAF1 and ERK1/2 upon 8-pCPT-2′-O-Me-cAMP treatment. These results indicate that the EPAC–RAP pathway regulates RAF1/ERK1/2 phosphorylation in T cells.
Figure 3.

RAF1 connects the GTPase RAP1 and adenylyl cyclase 6. A, T cells deprived of IL-2 for 48 h were stimulated (+) or not (−) with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min and lysed. Cell lysates were used to measure the levels of activated RAF1 and ERK1/2 and total loaded protein using anti-phospho-RAF1, anti-RAF1, anti-phospho-ERK1/2, and anti-ERK1/2 by Western blotting. Results are representative of four independent experiments. B, T cells deprived of IL-2 for 24 h were transfected with the pMT2-FLAG-ADCY6 vector. At 24 h post-transfection, cells were pretreated with the RAF1 inhibitor (10 μm) or vehicle (0.1% DMSO) for 1 h. Subsequently, the cells were stimulated or not with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min and lysed. Cell lysates were immunoprecipitated with an anti-FLAG antibody, and the immunoreactive bands were visualized using anti-phosphoserine (pS) and anti-FLAG antibodies, as indicated. The results are representative of five independent experiments. In box plot graphs, each box includes values from five different samples. All values are expressed in arbitrary units. Differences between unphosphorylated and phosphorylated proteins were as follows. A, ##, p < 0.0079 for RAF1; ###, p < 0.0079 for ERK1/2 and ADCY6. B, ##, p < 0.0060 (Mann–Whitney test). C, cells deprived of IL-2 for 48 h were pretreated with a RAF1 inhibitor (10 μm GW5074) or vehicle (0.1% DMSO) (−) for 1 h and stimulated (+) or not (−) with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min and lysed. Cell lysates were used to measure cAMP accumulation. The results show the mean ± S.D. (error bars) of three independent experiments performed in duplicates. ***, p < 0.001.
This result prompted us to test whether ADCY6 could link PYG to RAF1 in the EPAC–RAP1 signaling pathway. To this end, T cells overexpressing FLAG-ADCY6 were pretreated or not with 10 μm GW5074 for 1 h, after which they were stimulated with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min. Cells were then lysed, and cell lysates were immunoprecipitated as described under “Experimental procedures.” Results showed that 8-pCPT-2′-O-Me-cAMP induced ADCY6 serine phosphorylation; however, this did not occur in the presence of GW5074 (Fig. 3B, top, lane 4). Equal levels of ADCY6 expression in T-transfected cells and γ-tubulin were verified by Western blotting (Fig. 3B, second and third panels).
Next, to verify whether the absence of ADCY6 serine phosphorylation results in a blockade of cAMP generation, T cells were pretreated or not with 10 μm GW5074 for 1 h and thereafter stimulated with 10 μm 8-pCPT-2′-O-Me-cAMP for 10 min before measurement of cAMP generation. Cells were then lysed, and cAMP was determined as described under “Experimental procedures.” Results showed that 8-pCPT-2′-O-Me-cAMP induced cAMP generation; however, GW5074 prevented cAMP accumulation induced by 8-pCPT-2′-O-Me-cAMP (Fig. 3C).
Epidermal growth factor receptor mediates PYG activation in Kit 225 T cells
We previously showed that IL-2–mediated PYGM activation in T cells depends on RAC1 and is independent of PKA (3), whereas EGF-mediated stimulation of RAF1 potentiates ADCY6 activation in intact cells (37). Thus, we examined the effect of different growth factors on PYG activation in T cells. Cells were deprived of IL-2 for 48 h and were then stimulated or not with 10 ng ml−1 EGF, 50 μg ml−1 fibroblast growth factor (FGF), 20 ng ml−1 PDGF, or 10 ng ml−1 vascular endothelial growth factor (VEGF) for 10 min. PYG activity was then measured in cell lysates. As shown in Fig. 4A, only EGF stimulated PYG activity in T cells.
Figure 4.

Epidermal growth factor receptor activation requires RAF1 and adenylyl cyclase to induce glycogen phosphorylase activation. A, cells deprived of IL-2 for 48 h were unstimulated (−) or stimulated (+) with 10 ng ml−1 EGF, 50 μg ml−1 FGF, 20 ng ml−1 PDGF, or 10 ng ml−1 VEGF at 37 °C for 10 min and lysed. Cell lysates were used to measure glycogen phosphorylase (PYG) activity. The results presented as histograms show the mean ± S.D. (error bars) of three independent experiments performed in triplicates ***, p < 0.001. B, cells deprived of IL-2 for 48 h were pretreated with an adenylyl cyclase inhibitor (10 μm MDL12330A), RAF1 inhibitor (10 μm GW5074), or vehicle (0.1% DMSO) (−) for 1 h and stimulated (+) or not (−) with 10 ng ml−1 EGF and 500 units ml−1 IL-2 for 10 min and lysed. Cell lysates were used to measure PYG activity. The results show the mean ± S.D. of three independent experiments performed in triplicates. ****, p < 0.0001. C, cells deprived of IL-2 for 48 h were pretreated with 10 μm GW2974 (EGFR inhibitor), 10 μm H-89, 10 μm Rp-cAMPS (PKA inhibitors), or vehicle (0.1% DMSO) (−) and stimulated (+) or not with 10 ng ml−1 EGF at 37 °C for 10 min and lysed. Cell lysates were used to measure PYG activity. The results show the mean ± S.D. of three independent experiments performed in triplicates. ***, p < 0.001.
Given this result, we investigated the potential role of EGFR in the regulation of PYG activity via the RAF1/ADCY6 signaling pathway. T cells were deprived of IL-2 for 48 h and were then pretreated with 10 μm GW5074, 10 μm MDL12330A, or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 ng ml−1 EGF or 500 units ml−1 IL-2 for 10 min before measurement of PYG activity. As expected, neither ADCY inhibition (MDL12330A) nor RAF1 inhibition (GW5074) blocked IL-2–mediated PYG activation (Fig. 4B, gray bars). By contrast, both inhibitors strongly blocked EGFR-mediated PYG activation (Fig. 4B, closed bars). Taken together, these results suggest that EGFR-mediated PYG activation requires the involvement of RAF1 and ADCY6.
To verify the involvement of EGFR and also to examine the potential role of PKA in this signaling pathway, T cells were deprived of IL-2 for 48 h and were then pretreated with an EGFR inhibitor (GW2974, 10 μm) (39), PKA inhibitors (H-89 (10 μm) and Rp-cAMPS (10 μm)), or vehicle (0.1% DMSO) for 1 h, followed by stimulation with 10 ng ml−1 EGF for 10 min and measurement of PYG activity. EGFR tyrosine kinase inhibition with GW2974 strongly blocked EGFR-mediated PYG activity in T cells, and a similar result was found in cells pretreated either with H-89 or Rp-cAMPS (Fig. 4C). To test whether RAP1 activation depends on EGFR, IL-2–deprived T cells were pretreated with 10 μm GW2974 or vehicle (0.1% DMSO), followed or not by stimulation with 10 ng ml−1 EGF for 10 min, and RAP1 activation was subsequently measured by a pulldown assay. Control stimulated cells exhibited both robust RAP1 activation (Fig. 5A, first panel, lane 2) and RAF1 and ERK1/2 phosphorylation (Fig. 5A, third and fourth panels). By contrast, GW2974 efficiently blocked EGFR-mediated RAP1 activation and also RAF1 and ERK1/2 phosphorylation (Fig. 5A, lanes 3 and 4 of the first, third, and fourth panels). Equal levels of RAP1 and γ-tubulin expression were verified by Western blotting (Fig. 5A, second and fifth panels). Overall, the results indicate that EGF-stimulated cells control RAP1, RAF1, and ERK1/2 activation, and EGFR is required for ADCY6 and PKA to mediate PYG activation.
Figure 5.

Epidermal growth factor receptor controls glycogen phosphorylase activity through RAP1–RAF1–adenylyl cyclase 6 signaling pathway. A, cells deprived of IL-2 for 48 h were pretreated with 10 μm GW2974 or vehicle (0.1% DMSO) (−) and stimulated or not with 10 ng ml−1 EGF for 10 min and lysed. Cell lysates were used to perform a RAP1 pulldown assay. Precipitated active RAP1 (RAP1-GTP) and total RAP1, phospho-RAF1, phospho-ERK1/2, and γ-tubulin from cell lysates were analyzed by Western blotting using the indicated specific antibodies. Results are representative of five independent experiments. B, T cells deprived of IL-2 for 24 h were transfected with EGFP (esiRNA) or ADCY6 (esiRNA). At 24 h post-transfection, the cells were stimulated (+) or not (−) with 10 ng ml−1 EGF for 10 min and lysed. Cell lysates were used to perform a RAP1 pulldown assay. Precipitated active RAP1 (RAP1-GTP) and total RAP1 were analyzed by Western blotting using a specific anti-RAP1 antibody. The expression levels of endogenous ADCY6 were visualized using an anti-ADCY6 antibody and protein load was analyzed with an anti-γ-tubulin antibody, as indicated. The results shown are representative of five (A) and three (B) independent experiments. In box plot graphs, each box includes values from five (A) or three (B) different samples. All values are expressed in arbitrary units. Differences between different treatments were as follows. ##, p < 0.0001 (RAP1); ###, p < 0.05 (phospho-RAF1); ###, p < 0.0079 (phospho-ERK1/2); ###, p < 0.0022 (B); Mann–Whitney test.
To determine whether RAP1 is located, up- or downstream of ADCY6 in the signaling pathway originating from EGFR for PYG activation, we used endoribonuclease-prepared siRNA (esiRNA) and transient transfection to knock down ADCY6 expression in T cells. Thus, cells were transfected with an esiRNA targeting human ADCY6 or an esiRNA targeting enhanced GFP (EGFP), as a negative control. At 24 h post-transfection, the active endogenous RAP1 was measured by a pulldown assay in T cells stimulated or not with EGF for 10 min. As shown in Fig. 5B, EGF stimulated robust RAP1 activation in EGFP (esiRNA)-transfected T cells. Conversely, RAP1 activation was unaffected in ADCY6-knockdown cells stimulated with EGF for 10 min. Western blotting confirmed the loss of expression of ADCY6 expression after esiRNA transfection (Fig. 5B). These results indicate that RAP1 is located upstream of ADCY6.
To confirm that ADCY6 functions as a PYG-activating molecule in T cells, we repeated ADCY6 silencing and measured EGF-stimulated PYG activity. The results showed that EGF was unable to stimulate PYG activation in the absence of ADCY6 expression (Fig. 6A). In addition, the absence of ADCY6 expression prevented EGF-mediated cAMP generation (Fig. 6B).
Figure 6.

Epidermal growth factor receptor requires adenylyl cyclase 6 expression to induce glycogen phosphorylase activation. A and B, T cells deprived of IL-2 for 24 h were transfected with EGFP (esiRNA) or ADCY6 (esiRNA). At 24 h post-transfection, the cells were stimulated (+) or not (−) with 10 ng ml−1 EGF for 10 min and lysed. Cell extracts were used to measure glycogen phosphorylase activity (A) and cAMP generation (B). Protein expression levels were analyzed by Western blotting using specific anti-ADCY6 and anti-γ-tubulin antibodies. The results show the mean ± S.D. (error bars) of three and independent experiments performed in triplicates (A) and duplicates (B). ***, p < 0.001.
Epidermal growth factor receptor is linked to glycogen phosphorylase via the RAS–EPAC2–RAP1 signaling pathway
To investigate whether EPAC participates in EGF-stimulated PYG activation, T cells were deprived of IL-2 for 48 h and were then pretreated with either 10 μm ESI-09, a specific and potent inhibitor of EPAC (40, 41), or vehicle (0.1% DMSO) for 1 h and then stimulated or not with 10 μm 8-pCPT-2′-O-Me-cAMP or 10 ng ml−1 EGF for 10 min, followed by measurement of PYG activity. As expected, both EGF and 8-pCPT-2′-O-Me-cAMP induced strong PYG activity, and this was completely blocked with ESI-09 (Fig. 7A). Because our data indicate that EPAC participates in the EGFR-regulated PYG enzymatic activity, we investigated the roles of RAS and RAP1 in this signaling pathway. T cells were deprived of IL-2 for 48 h and were then pretreated with inhibitors of EPAC (10 μm ESI-09), RAP1 (10 μm GGTI 298), or RAS (50 μm salirasib) (40–46) or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 ng ml−1 EGFR for 10 min and measurement of PYG activity. As expected, ESI-09 efficiently prevented EGF-induced PYG activation relative to vehicle (Fig. 7B). In addition, in the absence of RAP1 and RAS activity (by inhibition with GGTI 298 and salirasib, respectively), EGF was unable to mediate PYG activation (Fig. 7B). Taken together, our results indicate that EGFR requires EPAC, RAP, and RAS for PYG signaling pathway activation.
Figure 7.
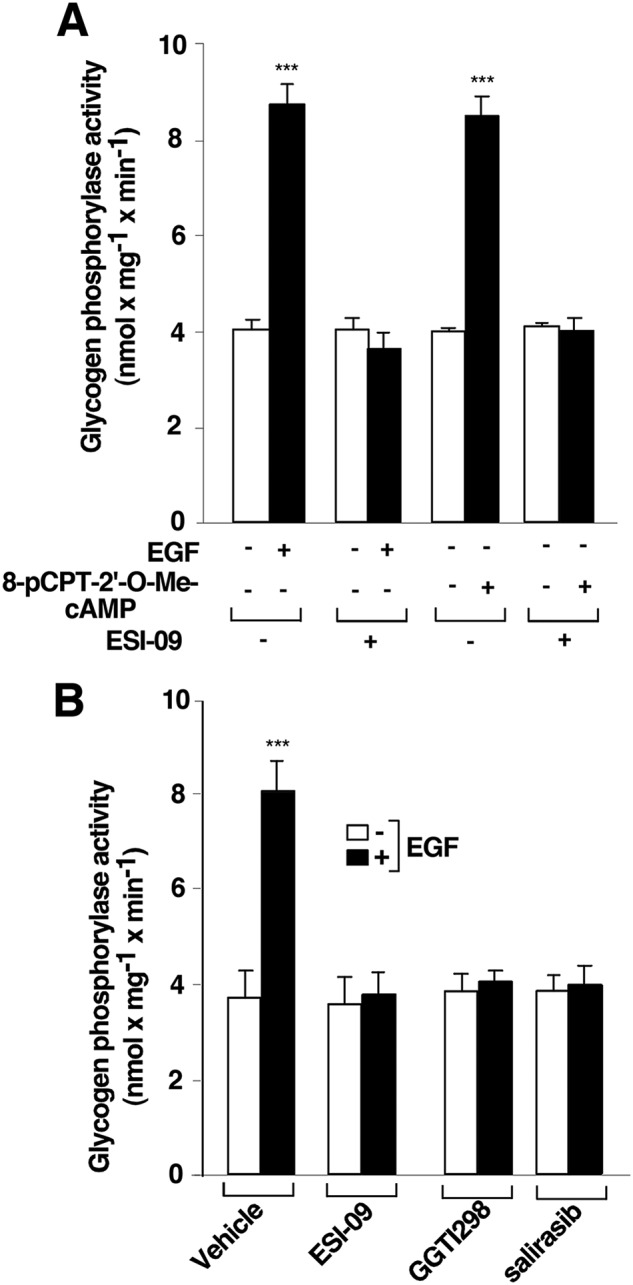
Epidermal growth factor receptor requires exchange proteins activated by cAMP (EPAC) and RAS to induce glycogen phosphorylase activation. T cells deprived of IL-2 for 48 h were pretreated with 10 μm ESI-09 (+) or vehicle (0.1% DMSO) (−) (A), with 10 μm ESI-09, 10 μm GGTI298, 50 μm salirasib (+), or vehicle (0.1% DMSO) (−) (B) for 1 h and stimulated (+) or not (−) with 10 μm 8-pCPT-2′-O-Me-cAMP and 10 ng/ml−1 EGF for 10 min and lysed. Cell extracts were used to measure glycogen phosphorylase activity. The results show the mean ± S.D. (error bars) of three (A) and seven (B) independent experiments performed in triplicates. ***, p < 0.001.
The aforementioned results led us to investigate the EGFR-established hierarchy between the GTPases RAP1 and RAS with regard to PYG activation. Thus, IL-2–deprived T cells were pretreated or not with 10 μm ESI-09, 10 μm GGTI 298, 50 μm salirasib, or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 ng ml−1 EGF for 10 min, and RAS activation was then measured by a pulldown assay. Control-stimulated cells exhibited robust RAS activation (Fig. 8A, first panel, lane 2), and this was completely blocked by RAS inhibition (Fig. 8A, first panel, lane 8). However, the EPAC and RAP inhibitors (ESI-09 and GGTI 298, respectively) did not obstruct EGFR-mediated RAS activation (Fig. 8A, first panel, lanes 4 and 6). The detection of RAS in the whole-cell lysates validated equal protein loading in all lanes (Fig. 8A, second panel).
Figure 8.

Epidermal growth factor receptor requires RAS active form to mediate RAP1 activation. T cells deprived of IL-2 for 48 h were pretreated with 10 μm ESI-09 (+) or 10 μm GGTI298 (A) or with 50 μm salirasib or vehicle (0.1% DMSO) (−) (A and B) for 1 h and stimulated (+) or not (−) with 10 ng/ml−1 EGF for 10 min and lysed. Cell extracts were used to measure RAS (A) and RAP1 (B) activation assayed by a pulldown assay. The precipitated active RAS and RAP1 (RAS-GTP and RAP1-GTP) and the total RAS and RAP1 from the cell lysates were analyzed by Western blotting using specific anti-RAS and anti-RAP1 antibodies. Immunoblots are representative of four independent experiments. In box plot graphs, each box includes values from four different samples. All values are expressed in arbitrary units. Differences between different treatments were p < 0.0001 in all cases (###) (Mann–Whitney test).
To verify that RAS is upstream of RAP1 in EGFR-stimulated T cells, IL-2–deprived cells were pretreated with 50 μm salirasib or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 ng ml−1 EGF for 10 min, and RAP1 activation was then measured by a pulldown assay. Control-stimulated cells exhibited robust RAP1 activation (Fig. 8B, top panel, lane 2), which was completely blocked by salirasib (Fig. 8B, top panel, lane 4). The detection of RAP1 in the whole-cell lysates validated equal protein loading in all lanes (Fig. 8B, second panel). Overall, our results indicate that RAS contributes to regulate EGFR-induced PYG activation via RAP1.
RAS signaling has been reported to be essential for EPAC2 to activate RAP1 in a temporally and spatially controlled manner (27). To elucidate the RAS-dependent pathway that regulates RAP1 through EPAC, we used a combination of pharmacological and genetic approaches to establish the possible functional interaction between the RAS and EPAC–RAP1 pathways. We speculated that if this interaction plays an important role in cell signaling, blockade of EPAC activity would have a direct impact on the activation of the RAP1 pathway. Thus, T cells were deprived of IL-2 for 24 h and were then transfected with a constitutively active form of H-RAS (pSG5-HA-H-RASG12V). After 24 h, cells were pretreated with 10 μm ESI-09 or vehicle (0.1% DMSO) for 1 h, followed by stimulation or not with 10 ng ml−1 EGF for 10 min, and RAP1 activation was subsequently measured by a pulldown assay. H-RASG12V overexpression induced substantial RAP1 activation, which was not further increased by EGF stimulation (Fig. 9A, top panel, lanes 1 and 2). By contrast, overexpression of H-RASG12V in the presence of the EPAC inhibitor ESI-09 blocked RAP1 activation, even in EGF-stimulated cells (Fig. 9A, top panel, lanes 3 and 4). Western blotting of RAP1 and pSG5-HA-H-RASG12V in whole-cell lysates validated equal loading of proteins (Fig. 9A, second and third panels).
Figure 9.

Epidermal growth factor receptor signaling involves RAS–EPAC–RAP pathway to activate glycogen phosphorylase. T cells deprived of IL-2 for 24 h were transfected with pSG5-HA-H-RASG12V (A) or pSG5-HA-H-RASG12V and pSG5-HA (B). At 24 h post-transfection, the cells were pretreated with 10 μm ESI-09 or vehicle (0.1% DMSO) (−) and stimulated (+) or not (−) with 10 ng ml−1 EGF for 10 min and lysed. A, cell lysates were used to measure RAP1 activation by a pulldown assay. The precipitated active RAP1 (RAP1-GTP) and the total RAP1 from the cell lysates were analyzed by Western blotting using a specific anti-RAP1 antibody. The expression levels of HA-H-RASG12V and tubulin were visualized using anti-HA and anti-γ-tubulin antibodies, as indicated. The results shown are representative of four independent experiments. In the box plot graph, each box includes values from four different samples. All values are expressed in arbitrary units. Differences between different treatments were p < 0.0001 in all cases (##) (Mann–Whitney test). B, cell extracts were used to measure glycogen phosphorylase activity. Protein expression levels were analyzed by Western blotting using specific anti-HA and anti-γ-tubulin antibodies. The results show the mean ± S.D. (error bars) of four independent experiments performed in triplicates. ***, p < 0.001.
Similar results were obtained when PYG activity was measured, with EGF maximally stimulating PYG activity in both empty vector– and H-RASG12V–transfected cells (Fig. 9B). H-RASG12V overexpression without EGF stimulation also induced maximal PYG activity, equivalent to that stimulated by EGF. By contrast, PYG activity was completely blocked in the presence of the EPAC inhibitor, both in the control-stimulated cells and in the H-RASG12V-overexpressed unstimulated or stimulated cells (Fig. 9B).
Discussion
Our study reveals a novel functional cross-talk between the small GTPase RAS and PYG guided by EGFR. This cross-talk is grounded on the involvement of the EPAC–RAP proteins situated between RAS and PYG in the signaling cascade. To the best of our knowledge, this is the first demonstration of PYG canonical activation produced in a G protein–coupled receptor–independent manner. This novel signal transduction pathway provides a molecular hierarchy in which RAS governs RAP activation, with an unusual relationship existing between these two members of the same family of small GTPases, the RAS family.
Human Kit 225 T cells express IL-2R constitutively and rely exclusively on IL-2 for cellular proliferation (34). These cells are efficiently synchronized in G0/G1 via IL-2 deprivation for 48 h, a feature that is advantageous for IL-2–mediated signaling studies. We previously showed that IL-2 stimulates activation of RAC1 and, in its active configuration, this small GTPase binds to PYGM and modulates PYGM enzymatic activity, leading to T-cell migration and proliferation in an ADCY-independent manner (3–5). This molecular mechanism is completely different from that described for the canonical cAMP–PKA–phosphorylase kinase–PYG pathway (3). Indeed, T cells can also activate PYG by the canonical pathway, as previously shown by us using forskolin, a classical ADCY activator (3). Kit 225 T cells thus provide an opportunity to decipher the involvement of other signaling pathways controlled by ADCY that involve canonical PYG pathway activation. In agreement with the results reported by Beazely et al. (37), in which RAF1 was shown to phosphorylate ADCY6, we show here that ADCY6 serine phosphorylation and PYG activation are EPAC–RAF1-dependent, a result that was further strengthened with experiments using pharmacological treatment and ADCY6 knockdown.
The functional specificity of intracellular signaling cascades depends largely on the cellular system, intracellular localization of the transducer molecules, and, ultimately, on the type of stimulus. Regarding the lymphoid system, it is well-established that tyrosine kinase–linked receptors, such as T-cell antigen receptor (18, 19), B-cell receptor (20, 21), and IL-2R (3), actively regulate immune response. Conversely, a receptor with intrinsic tyrosine kinase activity, EGFR, which is ubiquitously expressed, was traditionally considered to be absent from the hematopoietic cell lineage. However, it was recently shown that EGFR is expressed in both T helper-2 (32) and CD4 (33) cells. In this context, the Kit 225 T cells used here also respond strongly to EGFR activation, leading to PYG activation.
The EGFR signaling pathway induces a complex cascade of phosphorylation and activation events that govern several biological processes in mammalian cells, such as growth regulation, survival, proliferation, and differentiation (47–49). Active EGFR recruits the GRB2–SOS1 complex to the plasma membrane, where it drives rapid and transient activation of RAS. In its active configuration, RAS interacts with its downstream effector molecules through RAS-binding domains that can be found in the majority of RAS effectors. The first and best-characterized effector molecule of RAS is the serine threonine kinase RAF1 (23, 49). The RAS-RAF1 activated complex is by far the most important regulator of the mitogen-activated protein kinase (MAPK) pathway, a key signaling cascade in mediating the biological response of EGFR (47–49). This canonical signaling network regulated by RAS is not, however, unique. In fact, another small GTPase of the RAS family, RAP1, can also regulate the MAPK pathway through RAF1 (50–52). This scenario raises the possibility of an EGFR-mediated ADCY6/PYG pathway activation through RAS–RAF1 and/or RAP1–RAF1 complexes. Our findings not only reveal that the signaling through RAF1 to activate PYG originates exclusively from RAP, but also corroborate that RAS controls upstream RAP1 signaling.
Whereas RAF1 is the best characterized effector molecule linked to RAS, there are other RAS effector molecules involved in cell signaling, such as the GEFs TIAM1 (25), TIAM2, (26), and EPAC2 (27). Although they are all considered RAS effector molecules, TIAM1 and TIAM2 differ from EPAC2 in their interaction with RAC1 (25), with the latter interacting with RAP1 (27). Moreover, the active RAS form (RAS-GTP) only interacts specifically with EPAC2 (not with EPAC1) to activate RAP1 (27). The findings presented here point in the same direction, as pharmacological inhibition of EPAC in constitutively active H-RAS (RASG12V)-overexpressing T cells not only blocked EGFR- and RAS-mediated RAP1 activation, but also inhibited PYG activation.
In conclusion, our results provide insight into the molecular mechanisms through which EGFR stimulates RAP1 activation in T cells. We have identified EPAC2 as the GEF that specifically activates RAP1 and consequently regulates ADCY6/PYG activation, as well as the molecular mechanism through which EPAC2 activates the RAP1/PYG pathway. Regulation of this novel metabolic pathway requires the involvement of the GTPase RAS upstream of EPAC2/RAP1 and of RAP1-dependent RAF1 activity (Fig. 10). The specific role of this new unexpected signaling pathway in T-cell physiology is still unknown.
Figure 10.

Proposed model of glycogen phosphorylase regulation by epidermal growth factor receptor in human Kit 225 T cells. EGFR activation leads to the activation of the canonical RAS MAPK pathway through the GRB2–SOS complex. The results presented here document an additional signaling pathway also stimulated by EGF, which is RAS-dependent and MAPK-independent. This new signaling cascade outlines a novel regulation of the ADCY6/PKA/PK/PYG canonical pathway by a receptor with intrinsic tyrosine kinase activity (EGFR), which involves the RAS–EPAC2–RAP1–RAF1 pathway.
There is controversy over the role of ADCY and its downstream signaling pathways in lymphocyte biology. Beckner and Farrar (53, 54) have suggested that an inverse relationship exists between ADCY activity and lymphocyte proliferation. In this regard, our findings indicate that, depending on the stimulus (i.e. IL-2 or EGF), PYG can be activated in an ADCY-dependent or -independent manner. Furthermore, EGFR signaling through the ADCY6/PYG pathway could be a mechanism to deactivate IL-2 receptors in T cells and return them to their inactive state. Future studies should address the functional importance of this signaling cascade in the control of an efficient immune response.
Experimental procedures
Reagents
The following reagents were from Sigma-Aldrich: ADCY activator (forskolin), DMSO, EPAC activator (8-pCPT-2′-O-Me-cAMP), PKA inhibitors (H-89 dihydrochloride and Rp-cAMPS triethylammonium salt), RAS inhibitor (S-farnesylthiosalicylic acid, salirasib), RAP inhibitor (GGTI 298 trifluoroacetate salt hydrate), EPAC inhibitor (3-[5-(tert-butyl)isoxazol-3-yl]-2-[2-(3-chlorophenyl) hydrazono]-3-oxopropanenitrile, ESI-09), RAF1 kinase inhibitor (3-(3,5-dibromo-4-hydroxy-benzylidene)-5-iodo-1,3-dihydro-indol-2-one, GW5074), EGFR inhibitor (N4-(1-benzyl-1H-indazol-5-yl)-N6,N6-dimethyl-pyrido[3,4-d]pyrimidine-4,6-diamine, GW2974), ADCY inhibitor (MDL12330A hydrochloride), MISSION® esiRNA targeting EGFP (reference EHUEGFP) (EGFP was used as a negative transfection control because it was lacking in our cellular system), MISSION® esiRNA targeting human ADCY6 (reference EHU08099-1), and cAMP BiotrakTM EIA (GERPN2251). Mouse monoclonal anti-HA and anti-FLAG antibodies were obtained from Covance (Princeton, NJ), and mouse monoclonal anti-phosphoserine clone PSR-45, polyclonal anti-ADCY6, and anti-γ-tubulin antibodies and mouse monoclonal anti-RAS clone RAS10 antibody were obtained from Millipore (Burlington, MA). Anti-RAP1 sc:65 antibody was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX), and rabbit monoclonal anti-ERK1/2, anti-phospho-ERK1/2, anti-phospho-RAF-1, and anti-RAF-1 were obtained from Cell Signaling Technology (Danvers, MA). The enhanced chemiluminescence (ECL) reagent was obtained from GE Healthcare (Little Chalfont, UK). Recombinant human EGF, FGF, PDGF, and VEGF were obtained from Biovision (Milpitas, CA). IL-2 was provided by the National Institutes of Health AIDS Reagent Program (Germantown, MD).
Cell culture and DNA/esiRNA transfections
Kit 225 T cells were cultured with 16 units ml−1 of recombinant human IL-2 (34). For transient transfections, cells were cultured in complete Roswell Park Memorial Institute (RPMI) 1640 medium in the absence of IL-2 for 24 h. Thereafter, 2 × 107 cells were washed, resuspended in 200 μl of serum-free medium, and placed in an electroporation cuvette (0.4 mm; Sigma-Aldrich) containing 10–20 μg of different plasmids or 15 ng of esiRNAs, as indicated in the figures. Electroporation was performed in a Gene Pulser Xcell Electroporator (Bio-Rad) at 260 V and 950 microfarads (3). The cuvette contents were then transferred into 10 ml of complete RPMI 1640 medium and cultured without IL-2 for an additional 24 h.
Agonists and inhibitors
T cells were maintained without IL-2 for 48 h and subsequently treated with different stimuli: 500 units ml−1 IL-2, 10 μm forskolin, 10 μm 8-pCPT-2′-O-Me-cAMP, 10 ng ml−1 VEGF, 50 μg ml−1 FGF, 10 ng ml−1 EGF, or 20 ng ml−1 PDGF at 37 °C. In some experiments, cells were pretreated with the following inhibitors for 1 h prior to the stimulation: 10 μm ESI-09, 10 μm GGTI 298, 10 μm GW2974, 10 μm GW5074, 10 μm H-89, 10 μm Rp-cAPMS, 10 μm MDL 123300A, or 50 μm salirasib (Table 1).
Table 1.
List of inhibitors used in this study and their targets
| Name/abbreviation | Chemical name | Concentration | Targets | References |
|---|---|---|---|---|
| μm | ||||
| ESI-09 | α-[2-(3-Chlorophenyl)hydrazinylidene]-5-(1,1-dimethylethyl)-b-oxo-3-isoxazolepropanenitrile | 10 | EPAC proteins | 40, 41 |
| GGTI 298 | N-[[4-(Imidazol-4-yl)methylamino]-2-(1-naphthyl)benzoyl]leucine trifluoroacetate salt | 10 | RAP proteins | 40, 41 |
| GW 2974 | N4-(1-Benzyl-1H-indazol-5-yl)-N6,N6-dimethyl-pyrido-[3,4-d]-pyrimidine-4,6-diamine | 10 | EGFR | 39 |
| GW 5074 | 3-(3,5-Dibromo-4-hydroxybenzylidene)-5-iodo-1,3-dihydro-indol-2-one | 10 | RAF1 kinase | 38 |
| H-89 | N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride | 10 | PKA | 3 |
| RP-CAMPS | (R)-Adenosine, cyclic-3′,5′-(hydrogen phosphorothioate) triethylammonium salt | 10 | PKA | 57 |
| MDL 123300A | cis-N-(2-Phenylcyclopentyl)-azacyclotridec-1-en-2-amine hydrochloride | 10 | ADCY | 36 |
| Salirasib | 2-(((2E,6E)-3,7,11-Trimethyl-2,6,10-dodecatrienyl)sulfanyl)benzoic acid, FTS, farnesylthiosalicylic acid, S-farnesylthiosalicylic acid | 50 | RAS proteins | 44 |
Measurement of cAMP generation
Intracellular cAMP accumulation was quantified using an enzyme immunoassay system (cAMP BiotrakTM EIA) from Sigma-Aldrich. Briefly, using a centrifugal microplate adapter, cells were centrifuged at 1000–1500 × g for 3 min, and the pellet was resuspended in 200 μl of lysis buffer (0.05 m acetate buffer, pH 5.8, containing 0.02% BSA and 0.25% dodecyltrimethylammonium bromide). Lysates were used in the EIA following the manufacturer's recommendations. Finally, cAMP was quantified at 450 nm in a spectrophotometer SynergyTM HT from Bio-Tek. The data were collected using KC4 data reduction software).
Measurement of glycogen phosphorylase activity
PYG activity was assayed as described previously (55, 56), with some modifications. Briefly, cells were washed twice with cold PBS and resuspended in 500 μl of TES buffer (20 mm Tris, pH 7.4, 1 mm EDTA, 225 mm sucrose, 2.5 mm DTT, 0.1 mm PMSF, 1 μg ml−1 leupeptin, and 1 μg ml−1 aprotinin). Samples were sonicated and centrifuged at 13,500 rpm for 10 min at 4 °C. To measure PYG activity, 100 μg of total protein (1 μg/μl) was used in 200 μl of assay buffer (50 mm K2H2PO4, pH 7.5, 10 mm MgCl2, 5 mm EDTA, pH 8, 0.5 mm NADP+, 1.5 units ml−1 glucose-6-phosphate dehydrogenase, 1 unit ml−1 phosphoglucomutase, and 0.1 mg ml−1 glycogen (all from Sigma-Aldrich). Assay buffer (100 μl) containing 50 μl of TES without NADP+, glycogen, phosphoglucomutase, and glucose-6-phosphate dehydrogenase was added to 100 μg of total protein (1 μg/μl) as a blank control. A metabolic activity assay was carried out by incubating the mixture at 37 °C for 20 min. The reaction was stopped by placing the samples on ice. Sample absorbance was detected at 340 nm with a spectrophotometer (Ultrospec 3100 pro, Amersham Biosciences). The amount of NADPH formed was determined against a standard curve of known NADPH concentrations (Sigma-Aldrich).
RAP1 and RAS activation assay
RAP1 and RAS pulldown assays were performed using a GST fusion recombinant protein containing the RAP1-binding domain of Ral guanine nucleotide dissociation stimulator (RalGDS) (57). Cells maintained in the absence of IL-2 for 48 h were transfected with the indicated constructs or pretreated with specific inhibitors and stimulated with EGF as indicated and finally lysed in 600 μl of lysis buffer (10 mm Tris, pH 7.6, 150 mm NaCl, 1% IGEPAL-360, 10 mm MgCl2, 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin) (58). Cell lysates were centrifuged at 13,500 rpm for 10 min at 4 °C, and 500 μg of protein (1 μg/μl) from the soluble fraction was incubated for 1 h at 4 °C with 50 μg of GST-RBD of RalGDS fusion protein. Precipitated proteins were eluted from beads using 2× loading buffer (12 mm Tris, pH 6.8, 5% glycerol, 0.4% SDS, 140 mm 2-mercaptoethanol, and 0.02% bromphenol blue), separated by SDS-PAGE, and analyzed by Western blotting with specific monoclonal antibodies. Immunoreactive bands were visualized using the ECL reagent. Band intensities were imaged using the Molecular Imager® ChemiDocTM XRS System+Imaging Systems with Image LabTM software from Bio-Rad and analyzed with Fiji software.
Immunoprecipitation assay
T cells were transfected with pMT2-FLAG-ADCY6. Transfected cells were pretreated or not with GW5074 for 1 h, stimulated or not with 8-pCPT-2′-O-Me-cAMP for 10 min, and lysed in radioimmune precipitation assay buffer (50 mm Tris, pH 7,4, 150 mm NaCl, 1% IGEPAL-360, 0.25% sodium deoxycholate, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, 1 μg ml−1 aprotinin, and 1 μg ml−1 leupeptin). Ectopic FLAG-ADCY6 was immunoprecipitated at 4 °C for 2 h using an anti-FLAG antibody. Immune complexes were recovered using Gamma Bind Plus Sepharose beads (GE Healthcare), washed, eluted from beads, resolved electrophoretically by SDS-PAGE, and analyzed by Western blotting with anti-phosphoserine or anti-FLAG antibodies. Immunoreactive bands were visualized as above.
Statistical analysis
Student's t test for paired data was used for all comparisons with the level of significance set at p < 0.001 (***), with the exception of Fig. 4B, in which experimental groups were compared using one-way ANOVA (grouped analysis) (****, p < 0.0001). The Mann–Whitney nonparametric test for densitometric Western blotting was used for all of the box plot analyses with the level of significance set at p < 0.05. Box plots represent mean and 25th to 75th percentile, and whiskers represent minimum and maximum values. The presentation of the data is a combination of box plots and dot plots.
Author contributions
F. L. conceived and designed the work, analyzed and interpreted data, and drafted the work; A. A. S., M. L. M., and D. F. M. performed experiments and analyzed results; H. M. L. and L. A. P. analyzed results and reviewed the manuscript. A. L. conceived and designed the work, analyzed and interpreted data, and drafted the work; and J. L. Z. conceived and designed the work, analyzed and interpreted data, and drafted the work. All authors qualify for authorship, approved the final version of the manuscript, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Acknowledgments
We thank Dr. Yijuang Chern (Academia Sinica, Institute of Biomedical Sciences and Institute of Neuroscience and National Yang-Ming University Taipei, Taiwan) for the generous gift of plasmid encoding for human ADCY6. We also thank Dr. Kenneth McCreath for editorial assistance during manuscript preparation.
The authors declare that they have no conflicts of interest with the contents of this article.
- PYG
- glycogen phosphorylase
- PYGM
- glycogen phosphorylase muscle isoform
- RBD
- RAC-binding domain
- EGFP
- enhanced green fluorescent protein
- ADCY
- adenylyl cyclase
- IL
- interleukin
- GEF
- guanine nucleotide exchange factor
- PKA
- cAMP-dependent protein kinase
- FGF
- fibroblast growth factor
- PDGF
- platelet-derived growth factor
- esiRNA
- endoribonuclease-prepared siRNA
- EGFR
- epidermal growth factor receptor
- MAPK
- mitogen-activated protein kinase
- PMSF
- phenylmethylsulfonyl fluoride
- RPMI
- Roswell Park Memorial Institute.
References
- 1. David E. S., and Crerar M. M. (1986) Quantitation of muscle glycogen phosphorylase mRNA and enzyme amounts in adult rat tissues. Biochim. Biophys. Acta 880, 78–90 10.1016/0304-4165(86)90122-4 [DOI] [PubMed] [Google Scholar]
- 2. de Luna N., Brull A., Lucia A., Santalla A., Garatachea N., Martí R., Andreu A. L., and Pinós T. (2014) PYGM expression analysis in white blood cells: a complementary tool for diagnosing McArdle disease? Neuromuscul. Disord. 24, 1079–1086 10.1016/j.nmd.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 3. Arrizabalaga O., Lacerda H. M., Zubiaga A. M., and Zugaza J. L. (2012) Rac1 protein regulates glycogen phosphorylase activation and controls interleukin (IL)-2-dependent T cell proliferation. J. Biol. Chem. 287, 11878–11890 10.1074/jbc.M111.297804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Llavero F., Urzelai B., Osinalde N., Gálvez P., Lacerda H. M., Parada L. A., and Zugaza J. L. (2015) Guanine nucleotide exchange factor αPIX leads to activation of the Rac 1 GTPase/glycogen phosphorylase pathway in interleukin (IL)-2-stimulated T cells. J. Biol. Chem. 290, 9171–9182 10.1074/jbc.M114.608414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llavero F., Artaso A., Lacerda H. M., Parada L. A., and Zugaza J. L. (2016) Lck/PLCγ control migration and proliferation of interleukin (IL)-2-stimulated T cells via the Rac1 GTPase/glycogen phosphorylase pathway. Cell. Signal. 28, 1713–1724 10.1016/j.cellsig.2016.07.014 [DOI] [PubMed] [Google Scholar]
- 6. Wang J. H., and Black W. J. (1968) Relationship between structural change and allosteric transitions of glycogen phosphorylase a. J. Biol. Chem. 243, 4641–4649 [PubMed] [Google Scholar]
- 7. Keppens S., and de Wulf H. (1975) The activation of liver glycogen phosphorylase by vasopressin. FEBS Lett. 51, 29–32 10.1016/0014-5793(75)80848-9 [DOI] [PubMed] [Google Scholar]
- 8. Madsen N. B., Kasvinsky P. J., and Fletterick R. J. (1978) Allosteric transitions of phosphorylase a and the regulation of glycogen metabolism. J. Biol. Chem. 253, 9097–9101 [PubMed] [Google Scholar]
- 9. Krebs E. G. (1981) Phosphorylation and dephosphorylation of glycogen phosphorylase: a prototype for reversible covalent enzyme modification. Curr. Top. Cell. Regul. 18, 401–419 10.1016/B978-0-12-152818-8.50030-X [DOI] [PubMed] [Google Scholar]
- 10. Black W. J., and Hsueh-Ching Wang J. (1970) Studies on the allosteric activation of glycogen phosphorylase b by nucleotides. II. Nucleotide structure in relation to mechanism of activation. Biochim. Biophys. Acta 212, 257–268 10.1016/0005-2744(70)90206-8 [DOI] [PubMed] [Google Scholar]
- 11. de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., and Bos J. L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 10.1038/24884 [DOI] [PubMed] [Google Scholar]
- 12. Fuld S., Borland G., and Yarwood S. J. (2005) Elevation of cyclic AMP in Jurkat T-cells provokes distinct transcriptional responses through the protein kinase A (PKA) and exchange protein activated by cyclic AMP (EPAC) pathways. Exp. Cell Res. 309, 161–173 10.1016/j.yexcr.2005.05.016 [DOI] [PubMed] [Google Scholar]
- 13. Müller M. S., Pedersen S. E., Walls A. B., Waagepetersen H. S., and Bak L. K. (2015) Isoform-selective regulation of glycogen phosphorylase by energy deprivation and phosphorylation in astrocytes. Glia 63, 154–162 10.1002/glia.22741 [DOI] [PubMed] [Google Scholar]
- 14. Bar-Sagi D., and Hall A. (2000) Ras and Rho GTPases: a family reunion. Cell 103, 227–238 10.1016/S0092-8674(00)00115-X [DOI] [PubMed] [Google Scholar]
- 15. Mitin N., Rossman K. L., and Der C. J. (2005) Signaling interplay in Ras superfamily function. Curr. Biol. 15, R563–574 10.1016/j.cub.2005.07.010 [DOI] [PubMed] [Google Scholar]
- 16. Simanshu D. K., Nissley D. V., and McCormick F. (2017) RAS proteins and their regulators in human disease. Cell 170, 17–33 10.1016/j.cell.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wennerberg K., Rossman K. L., and Der C. J. (2005) The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 10.1242/jcs.01660 [DOI] [PubMed] [Google Scholar]
- 18. Lan R. Y., Selmi C., and Gershwin M. E. (2008) The regulatory, inflammatory, and T cell programming roles of interleukin-2 (IL-2). J. Autoimmun. 31, 7–12 10.1016/j.jaut.2008.03.002 [DOI] [PubMed] [Google Scholar]
- 19. Zugaza J. L., Caloca M. J., and Bustelo X. R. (2004) Inverted signaling hierarchy between RAS and RAC in T-lymphocytes. Oncogene 23, 5823–5833 10.1038/sj.onc.1207768 [DOI] [PubMed] [Google Scholar]
- 20. Caloca M. J., Zugaza J. L., and Bustelo X. R. (2008) Mechanistic analysis of the amplification and diversification events induced by Vav proteins in B-lymphocytes. J. Biol. Chem. 283, 36454–36464 10.1074/jbc.M803814200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caloca M. J., Zugaza J. L., Matallanas D., Crespo P., and Bustelo X. R. (2003) Vav mediates Ras stimulation by direct activation of the GDP/GTP exchange factor Ras GRP1. EMBO J. 22, 3326–3336 10.1093/emboj/cdg316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imperial R., Toor O. M., Hussain A., Subramanian J., and Masood A. (2017) Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: its clinical implications. Semin. Cancer Biol. 10.1016/j.semcancer.2017.11.016 10.1016/j.semcancer.2017.11.016 [DOI] [PubMed] [Google Scholar]
- 23. Martinelli E., Morgillo F., Troiani T., and Ciardiello F. (2017) Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat. Rev. 53, 61–69 10.1016/j.ctrv.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 24. Slack C. (2017) Ras signaling in aging and metabolic regulation. Nutr. Healthy Aging 4, 195–205 10.3233/NHA-160021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lambert J. M., Lambert Q. T., Reuther G. W., Malliri A., Siderovski D. P., Sondek J., Collard J. G., and Der C. J. (2002) Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 4, 621–625 10.1038/ncb833 [DOI] [PubMed] [Google Scholar]
- 26. Zaldua N., Gastineau M., Hoshino M., Lezoualc'h F., and Zugaza J. L. (2007) Epac signaling pathway involves STEF, a guanine nucleotide exchange factor for Rac, to regulate APP processing. FEBS Lett. 581, 5814–5818 10.1016/j.febslet.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 27. Liu C., Takahashi M., Li Y., Song S., Dillon T. J., Shinde U., and Stork P. J. (2008) Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell Biol. 28, 7109–7125 10.1128/MCB.01060-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lanaya H., Natarajan A., Komposch K., Li L., Amberg N., Chen L., Wculek S. K., Hammer M., Zenz R., Peck-Radosavljevic M., Sieghart W., Trauner M., Wang H., and Sibilia M. (2014) EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 16, 972–977 10.1038/ncb3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scholes A. G., Hagan S., Hiscott P., Damato B. E., and Grierson I. (2001) Overexpression of epidermal growth factor receptor restricted to macrophages in uveal melanoma. Arch. Ophthalmol. 119, 373–377 10.1001/archopht.119.3.373 [DOI] [PubMed] [Google Scholar]
- 30. Chan G., Nogalski M. T., and Yurochko A. D. (2009) Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. U.S.A. 106, 22369–22374 10.1073/pnas.0908787106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mahtouk K., Hose D., Rème T., De Vos J., Jourdan M., Moreaux J., Fiol G., Raab M., Jourdan E., Grau V., Moos M., Goldschmidt H., Baudard M., Rossi J. F., Cremer F. W., and Klein B. (2005) Expression of EGF-family receptors and amphiregulin in multiple myeloma: amphiregulin is a growth factor for myeloma cells. Oncogene 24, 3512–3524 10.1038/sj.onc.1208536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minutti C. M., Drube S., Blair N., Schwartz C., McCrae J. C., McKenzie A. N., Kamradt T., Mokry M., Coffer P. J., Sibilia M., Sijts A. J., Fallon P. G., Maizels R. M., and Zaiss D. M. (2017) Epidermal growth factor receptor expression licenses type-2 helper T cells to function in a T cell receptor-independent fashion. Immunity 47, 710–722.e6 10.1016/j.immuni.2017.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaiss D. M., van Loosdregt J., Gorlani A., Bekker C. P., Gröne A., Sibilia M., van Bergen en Henegouwen P. M., Roovers R. C., Coffer P. J., and Sijts A. J. (2013) Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 38, 275–284 10.1016/j.immuni.2012.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hori T., Uchiyama T., Tsudo M., Umadome H., Ohno H., Fukuhara S., Kita K., and Uchino H. (1987) Establishment of an interleukin 2-dependent human T cell line from a patient with T cell chronic lymphocytic leukemia who is not infected with human T cell leukemia/lymphoma virus. Blood 70, 1069–1072 [PubMed] [Google Scholar]
- 35. Enserink J. M., Christensen A. E., de Rooij J., van Triest M., Schwede F., Genieser H. G., Døskeland S. O., Blank J. L., and Bos J. L. (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906 10.1038/ncb874 [DOI] [PubMed] [Google Scholar]
- 36. Lim S. C., and Han S. I. (2017) MDL-12330A potentiates TRAIL-induced apoptosis in gastric cancer cells through CHOP-mediated DR5 upregulation. Korean J. Physiol. Pharmacol. 21, 397–405 10.4196/kjpp.2017.21.4.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beazely M. A., Alan J. K., and Watts V. J. (2005) Protein kinase C and epidermal growth factor stimulation of Raf1 potentiates adenylyl cyclase type 6 activation in intact cells. Mol. Pharmacol. 67, 250–259 10.1124/mol.104.001370 [DOI] [PubMed] [Google Scholar]
- 38. Chiluiza D., Krishna S., Schumacher V. A., and Schlöndorff J. (2013) Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular-signal-regulated kinases Erk1/2. J. Biol. Chem. 288, 18407–18420 10.1074/jbc.M113.463059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kesanakurti D., Chetty C., Rajasekhar Maddirela D., Gujrati M., and Rao J. S. (2012) Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 3, e445 10.1038/cddis.2012.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu Y., Chen H., Boulton S., Mei F., Ye N., Melacini G., Zhou J., and Cheng X. (2015) Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci. Rep. 5, 9344 10.1038/srep09344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen H., Ding C., Wild C., Liu H., Wang T., White M. A., Cheng X., and Zhou J. (2013) Efficient synthesis of ESI-09, a novel non-cyclic nucleotide EPAC antagonist. Tetrahedron Lett. 54, 1546–1549 10.1016/j.tetlet.2013.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miquel K., Pradines A., Sun J., Qian Y., Hamilton A. D., Sebti S. M., and Favre G. (1997) GGTI-298 induces G0-G1 block and apoptosis whereas FTI-277 causes G2-M enrichment in A549 cells. Cancer Res. 57, 1846–1850 [PubMed] [Google Scholar]
- 43. Vogt A., Sun J., Qian Y., Hamilton A. D., and Sebti S. M. (1997) The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21(WAF1/CIP1/SDI1) in a p53-independent manner. J. Biol. Chem. 272, 27224–27229 10.1074/jbc.272.43.27224 [DOI] [PubMed] [Google Scholar]
- 44. Zundelevich A., Elad-Sfadia G., Haklai R., and Kloog Y. (2007) Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Mol. Cancer Ther. 6, 1765–1773 10.1158/1535-7163.MCT-06-0706 [DOI] [PubMed] [Google Scholar]
- 45. Goldberg L., Haklai R., Bauer V., Heiss A., and Kloog Y. (2009) New derivatives of farnesylthiosalicylic acid (salirasib) for cancer treatment: farnesylthiosalicylamide inhibits tumor growth in nude mice models. J. Med. Chem. 52, 197–205 10.1021/jm801165r [DOI] [PubMed] [Google Scholar]
- 46. Riely G. J., Johnson M. L., Medina C., Rizvi N. A., Miller V. A., Kris M. G., Pietanza M. C., Azzoli C. G., Krug L. M., Pao W., and Ginsberg M. S. (2011) A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 6, 1435–1437 10.1097/JTO.0b013e318223c099 [DOI] [PubMed] [Google Scholar]
- 47. Avraham R., and Yarden Y. (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 10.1038/nrm3048 [DOI] [PubMed] [Google Scholar]
- 48. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 10.1016/S0092-8674(00)00114-8 [DOI] [PubMed] [Google Scholar]
- 49. Yarden Y. (2001) The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 37, Suppl. 4, S3–S8 [DOI] [PubMed] [Google Scholar]
- 50. Hariharan I. K. (2005) Ras and Rap: are former enemies now friends? Dev. Cell 8, 303–304 10.1016/j.devcel.2005.02.006 [DOI] [PubMed] [Google Scholar]
- 51. Stork P. J., and Dillon T. J. (2005) Multiple roles of Rap1 in hematopoietic cells: complementary versus antagonistic functions. Blood 106, 2952–2961 10.1182/blood-2005-03-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stork P. J., and Schmitt J. M. (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 12, 258–266 10.1016/S0962-8924(02)02294-8 [DOI] [PubMed] [Google Scholar]
- 53. Beckner S. K., and Farrar W. L. (1986) Interleukin 2 modulation of adenylate cyclase: potential role of protein kinase C. J. Biol. Chem. 261, 3043–3047 [PubMed] [Google Scholar]
- 54. Beckner S. K., and Farrar W. L. (1988) Potentiation of lymphokine- activated killer cell differentiation and lymphocyte proliferation by stimulation of protein kinase C or inhibition of adenylate cyclase. J. Immunol. 140, 208–214 [PubMed] [Google Scholar]
- 55. Andersen B., and Westergaard N. (2002) The effect of glucose on the potency of two distinct glycogen phosphorylase inhibitors. Biochem. J. 367, 443–450 10.1042/bj20020153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McInerney M., Serrano Rodriguez G., Pawlina W., Hurt C. B., Fletcher B. S., Laipis P. J., and Frost S. C. (2002) Glycogen phosphorylase is activated in response to glucose deprivation but is not responsible for enhanced glucose transport activity in 3T3-L1 adipocytes. Biochim. Biophys. Acta 1570, 53–62 10.1016/S0304-4165(02)00154-X [DOI] [PubMed] [Google Scholar]
- 57. Maillet M., Robert S. J., Cacquevel M., Gastineau M., Vivien D., Bertoglio J., Zugaza J. L., Fischmeister R., and Lezoualc'h F. (2003) Crosstalk between Rap1 and Rac regulates secretion of sAPPα. Nat. Cell Biol. 5, 633–639 10.1038/ncb1007 [DOI] [PubMed] [Google Scholar]
- 58. Wery-Zennaro S., Zugaza J. L., Letourneur M., Bertoglio J., and Pierre J. (2000) IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 19, 1596–1604 10.1038/sj.onc.1203458 [DOI] [PubMed] [Google Scholar]