

SUMMARY
Transcriptional and translational feedback loops in fungi and animals drive circadian rhythms in transcript levels that provide output from the clock, but post-transcriptional mechanisms also contribute. To determine the extent and underlying source of this regulation, we applied newly developed analytical tools to a long-duration, deeply sampled, circadian proteomics time course comprising half of the proteome. We found a quarter of expressed proteins are clock regulated, but >40% of these do not arise from clock-regulated transcripts, and our analysis predicts that these protein rhythms arise from oscillations in translational rates. Our data highlighted the impact of the clock on metabolic regulation, with central carbon metabolism reflecting both transcriptional and post-transcriptional control and opposing metabolic pathways showing peak activities at different times of day. The transcription factor CSP-1 plays a role in this metabolic regulation, contributing to the rhythmicity and phase of clock-regulated proteins.
In Brief
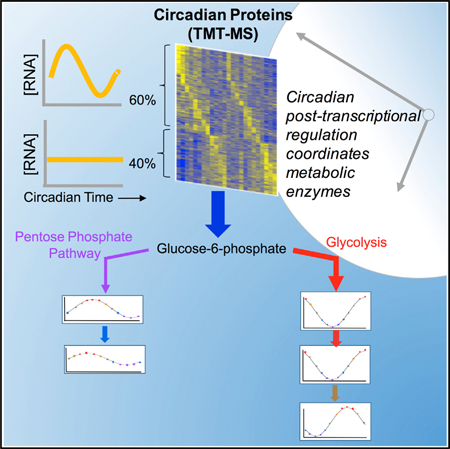
Hurley et al. used new informatic tools and deep sampling to systematically investigate the proteomic and transcriptomic output of the circadian clock, uncovering broad posttranscriptional control of output, especially of metabolic pathways. The largest portion of circadian posttranscriptional regulation appears to be imparted at the level of translation, not degradation as previously believed, highlighting a generally unrecognized importance of translational control in the regulation of circadian output.
Graphical Abstract
INTRODUCTION
Many organisms that experience recurring day and night cycles have evolved a circadian clock to organize behavior and cellular physiology such that appropriate activities occur at biologically advantageous times, regulating everything from sleep to cellular metabolism (Eckel-Mahan and Sassone-Corsi, 2013). The regulatory architecture of the clock is conserved between fungi and animals; at its core is a transcription-translation negative feedback loop (TTFL) involving a heterodimeric transcription-factor complex that functions as the positive arm and a distinct protein complex, the negative arm, whose function is to inhibit the transcriptional activity of the positive arm (Figure 1A; Dunlap and Loros, 2018; Hurley et al., 2016).
Figure 1. Neurospora Circadian Proteomic Analysis Demonstrates Rhythmic Proteins Are Enriched in Metabolic Functions and Mainly Peak after Subjective Dusk.

(A) Diagram of the positive (WC-1/WC-2) and negative (FRQ/FRH) arms of the core clock in Neurospora, showing transcriptional activation of downstream ccgs. One ancillary loop includes CSP-1, which represses WC-1 under high glucose conditions.
(B) Heatmap of rhythmic proteins showing normalized relative protein levels by circadian time of day (CT), with time points of low (blue) and high (yellow) protein levels, and proteins ordered by peak phase. By convention CT0 = subjective dawn and CT12 = subjective dusk.
(C) Histogram depicting the number of rhythmic proteins that peak in their oscillation at a given CT.
(D and E) Breakdown of FunCat terms from (D) all WT proteins detected and imputed in our analysis or (E) those proteins that are designated rhythmic in the WT proteomic dataset. Refer to legend for FunCat gene categories. WC-1/WC-2, WHITE COLLAR 1/2; FRQ, FREQUENCY; FRH, FREQUENCY-INTERACTING RNA HELICASE; CSP-1, CONIDIAL SEPARATION 1; WT, wild-type; CT, Circadian Time. *, p < 0.05; **, p ≤ 0.001.
Related to Figure S2 and Tables S1 and S2.
In addition to the activation of the negative arm genes within the core feedback loop, the positive arm influences the expression of a host of genes that have no role in the core timekeeping loop. These genes have been dubbed clock-controlled genes (ccgss; Figure 1A; Loros et al., 1989), and large-scale screens for rhythmic mRNAs and their drivers are viewed to explain clock control over cellular behavior (Bell-Pedersen et al., 2005; Partch et al., 2014). However, recent evidence has revealed additional levels of circadian regulation, including potential metabolic feedback on the positive arm through CSP-1 (Figure 1A; Sancar et al., 2012). Also, previous circadian proteomic studies detected oscillating proteins without corresponding oscillating mRNAs, although technical and analytical limitations restricted this work to a small fraction of the proteome (Chiang et al., 2014; Reddy et al., 2006; Robles et al., 2014; Wang et al., 2017a). Discordance between expressed mRNAs and translated protein levels is not a new observation. Previous research comparing mRNA and protein levels have identified correlations of 0.4 to 0.74, depending on growth conditions and the organism sampled (Vogel and Marcotte, 2012; Zhou et al., 2016). This leaves much of the variation in protein levels to be explained by other mechanisms, including post-transcriptional, translational, or post-translational regulation, such as changes in production or degradation rates. The evidence therefore suggests that solely tracking changes in mRNA levels over the circadian day is not a viable proxy for understanding what the clock controls in the cell at the functional level; one must also measure protein levels directlys.
In this work, we utilized Neurospora crassa, a well-established circadian model system (Dunlap and Loros, 2018; Hurley et al., 2016), to quantify relative protein levels every 2 hr over 48 hr, in triplicate time series, using tandem mass tag mass spectrometry (TMT-MS) (Dayon and Sanchez, 2012; Fuller et al., 2014) in two genetic backgrounds (wild-type [WT] and a csp-1 knockout), to determine the impact of post-transcriptional regulation on the clock. Our sampling density, combined with computational tools developed to address issues of missing data and batch effects (Crowell et al., 2018), has allowed the identification of a significantly larger portion of the circadian proteome than previously achieved. Overall, the rhythmic proteome was highly enriched in metabolic functions, and rhythmically expressed enzymes seem to be coordinated, resulting in the generation of metabolic pathway “phases.” We found the correlation between rhythmic mRNA and rhythmic protein was only 60%, highlighting extensive post-transcriptional regulation. Our data support the importance of translation elongation factor eEF-2 activity, rather than rhythmic protein degradation, in the circadian regulation that results in an overall peak of rhythmic proteins at circadian dusk (Caster et al., 2016; Lück et al., 2014). Finally, we found that while the gene csp-1 played an important role in circadian metabolic regulation, especially within lipid, fatty acid, and nucleotide metabolism pathways, energy availability and translational regulation may be more important determinants of the circadian proteome.
RESULTS
Neurospora Circadian Proteomic Analysis Demonstrates Rhythmic Proteins Are Enriched in Metabolic Functions and Mainly Peak after Subjective Dusks
From triplicate time course samples, normal operation of the clock was confirmed by western blot analysis of the core clock protein FREQUENCY (FRQ) (Figures S1A and S1B). Total protein lysates were subsequently extracted from fungal mats and trypsin digested for TMT-MS analysis. Each set of peptide fragments from a given time point was labeled with a unique isobaric mass tag; these labeled fragments were combined with taglabeled fragments from 7 other time point samples. As a control, tag-labeled fragments from two pooled control samples consisting of all 25 samples mixed together were also combined into the mix, creating a “ten-plex” sample. These “ten-plex” sample sets were then subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS), whereupon the second fragmentation allowed the relative quantification of identical peptides from different samples, via the differences in their isobaric mass tags (Figure S1C; also see STAR Methods).
Using TMT-MS analysis, a total of 52,251 peptides, representing 4,742 proteins (45% of the potential Neurospora proteome), were detected in >70% of all samples. This represented 3,776 proteins that had peptides at every time point, and 976 proteins for which levels of some peptides missing <30% of time points were imputed using the K-nearest neighbors (KNN) algorithm within LIMBR (Crowell et al., 2018; see STAR Methods). The data were then subjected to modeling and removal of batch effects using a time-series-specific algorithm within LIMBR that improves rhythm recognition in the presence of mass spectrometric batch effects (Crowell et al., 2018; see STAR Methods). In brief, LIMBR modeled the replicate and time series correlations to produce a matrix of residuals containing the batch effects, which were then modeled to produce linearly independent bias trends. The bias trends that met our significance threshold were then removed.
We used eJTK_cycle, a non-parametric algorithm for detecting rhythmic components in genome-scale datasets, to determine which proteins were under circadian regulation (Hughes et al., 2010; Hutchison et al., 2018; Hutchison et al., 2015). Roughly 27% of the identified proteome (1,273 proteins) significantly cycled (p < 0.05) with a circadian period, hereafter referred to as “rhythmic” (Figure S2A). All the core clock genes were detected at both the RNA and protein levels, with the exception of FRQ, likely due to low expression (Figure S2B). To analyze how peak phases of rhythmic proteins changed over the circadian day, we created a heatmap of gene products, using the normalized proteomic values (Figure 1B) and a histogram of the number of rhythmic proteins reaching peak expression at a given time of day (Figure 1C). A very small peak in rhythmic protein expression occurs in the circadian time (CT) morning (CT22–CT6), whereas the predominant peak phase in rhythmic protein expression happens in the subjective afternoon and evening (CT7–CT21), strongly centered at “dusk” (CT12–CT14).
Compared to all the detected and imputed proteins, Functional Catalogue (FunCat) (Priebe et al., 2011) analysis identified enriched gene categories (p < 0.05) involved in metabolism and the regulation of metabolism for the subset of proteins considered rhythmic (Figures 1D and 1E; Table S1), especially for those that reach peak expression during the subjective afternoon and evening (CT7–CT21) (Table S2). Similarly, phase set enrichment analysis (PSEA) identified KEGG gene sets with significant temporal coordination involved with metabolic processes, clustered near CT12 (Figure S2C; Table S3) (Zhang et al., 2016).
Comparison of WT Circadian Transcriptome and Proteome Suggests Widespread Post-transcriptional Circadian Regulation of Protein Levels
Not all potential Neurospora proteins were identified in every sample by TMT-MS. Therefore, to determine the relationship between the circadianly regulated proteome and transcriptome, we used a restricted gene list limited only to those genes identified in both the RNA sequencing (RNA-seq) and TMT-MS datasets. Using the same SVA-based tool and eJTK_cycle analysis, we found that 40% of the transcriptome (3,858 transcripts) was significantly rhythmic, an approximately 2-fold increase from prior work (Figure S3A; Hurley et al., 2014). Roughly two-thirds (2,414/3,858) of the rhythmically expressed genes corresponded to proteins also identified by TMT-MS. This subset of rhythmic transcripts (n = 2,414) appears to be representative of the full set of rhythmic transcripts identified from RNA-seq (n = 3,858), when compared by both heatmap (Figure S3B versus Figure S3C) and FunCat analyses (Figure S3D versus Figure S3E; Table S4).
Comparing the eJTK p values describing protein versus transcript rhythmicity, we found that 752 genes (59% of rhythmic proteins; Figures 2A, S4A, and S4B) were rhythmic both at the transcript and protein levels (p < 0.05). For this subset of genes rhythmic at both the transcript and protein levels (n = 752), phase delays between peak transcript and peak protein level ranged from 0 hr (77 or 10% of genes) to 22 hr, with a large average phase delay of 10.3 hr (Figure 2B).
Figure 2. Comparison of WT Circadian Transcriptome and Proteome Suggests Widespread Post-Transcriptional Circadian Regulation of Protein Levels.

(A) For all genes detected at the mRNA and protein level, mRNA rhythmicity p values (x axis) are plotted against protein rhythmicity p values (y axis).
(B) For genes that are rhythmic at both the mRNA and protein levels, a histogram depicting the number of genes with a given phase delay (in hours) between their peak mRNA and peak protein levels.
(C) Clock-style densitometry graphs of peak phase timing (CT) of rhythmic mRNA and/or rhythmic protein, for the following gene categories: “Rhythmic mRNA only” genes (n = 1,662); “Rhythmic mRNA and protein” genes (n = 752); and “Rhythmic protein only” genes (n = 521). In all cases, mRNA is in orange, while protein is in blue. WT, wild-type; CT, circadian time.
Related to Figures S3 and S4 and Table S4.
We found many significantly rhythmic transcripts without associated significantly rhythmic proteins (1,662 or 69% of the rhythmic transcriptome identified in the TMT-MS analysis; Figure 2A, area highlighted only in orange; and Figures S4A and S4B). These are easily explained if the proteins are stable. Conversely, a large proportion of significantly rhythmic proteins did not arise from significantly rhythmic mRNAs (521 proteins or 41% of the rhythmic proteome; Figure 2A, area highlighted only in blue; Figures S4A and S4B). Importantly, while comparing the residuals of the fits of the two analyses, we noted that the proteome dataset does contain more noise than the transcriptome (Figure S4C), which could account for some of the discrepancies. However, for genes with protein rhythmicity p < 0.05, 222 genes had mRNA values of 0.05 ≤ p ≤ 0.2, but another 298 genes had mRNA p > 0.2. To further show that there were indeed differences between mRNA and protein rhythmicity that were not due to the chosen p value cutoff, 1,140 genes had p < 0.1 at both the mRNA and protein level (66% of all proteins with p < 0.1 and 40% of all mRNA with p < 0.1), and 1,731 genes had p < 0.2 at both the mRNA and protein level (73% of all proteins with p < 0.2 and 51% of all mRNA with p < 0.2).
To look at overall phase distributions for these different groupings of genes (mRNA rhythmic only, rhythmic mRNA and protein, and protein rhythmic only), we plotted the peak phases of significantly rhythmic mRNA and/or significantly rhythmic protein on clock-style densitometry graphs (Figure 2C). In general, mRNA peak phases were bi-phasic, peaking at CT16 and CT22, compared to protein peak phases, which peaked near CT14. For the genes whose mRNA and protein were both significantly rhythmic, the main mRNA peak phase in the “morning” (CT22) preceded the main peak phase in protein levels in the “evening” (CT14) by at least 12 hr, which is reflected in the long phase delays that occurred in our dataset (Figure 2B).
Rhythmic Protein Degradation Is Unlikely to Be the Primary Mechanism Underlying Circadian Posttranscriptional Regulation in Neurospora
Using previously published proteomics data from mouse livers (Mauvois in et al., 2014; Reddy et al., 2006; Robles et al., 2014), Lück et al. (2014) developed an elegant model in which protein turnover elicited by rhythmic E3 ubiquitin ligase activity (inferred from transcript cycles) could drive protein rhythms, resulting in the overall peak observed in the mouse circadian proteome. As our deeper sampling and more powerful analytical techniques allowed identification of >7-fold more significantly rhythmic proteins than available previously, we used our dataset to ask whether this turnover model could also explain the timing of the overall peak in our rhythmic Neurospora proteome.
As in Lück et al. (2014), we focused on E3 ubiquitin ligases as a main mode of protein degradation and found that 10 of the 27 identified E3 ubiquitin ligase genes (37%) in our data were significantly rhythmic at the transcript level (Table S5). When we averaged the normalized mRNA levels of the 10 significantly rhythmic E3 ligase transcripts, there was no overall peak time (Figure 3A). At the protein level, only 17 E3 ubiquitin ligases were detected, and just three of those were significantly rhythmic (UPL-1 or NCU07544, NCU09866, and NCU07996). Importantly, these three significantly rhythmic proteins did not arise from any of the 10 significantly rhythmic E3 ubiquitin ligase transcripts (Table S5). Again, the p value cutoff does not explain the lack of rhythmic E3 ubiquitin ligases, as only two of the other E3 ubiquitin ligases had 0.05 ≤ p ≤ 0.2 (CUL-4 and FWD-1; Table S5). When we averaged the relative protein levels for the three rhythmic E3 ubiquitin ligases (p < 0.05; Figure 3B), there was a small potential peak around CT18, closely lagging the peak phase of significantly rhythmic proteins without corresponding significantly rhythmic mRNA (CT14) (Figure 2C) and near the peak phase in enrichment for degradation in the PSEA (Figure S2C). While this does not exclude the possibility that degradation contributes to some rhythmic protein timing, a potential peak in degradation at CT18 does not explain the circadian dusk peak timing of our significantly rhythmic proteins (Figure 2C) (Lück et al., 2014).
Figure 3. Rhythmic Protein Degradation Is Unlikely to Be the Primary Mechanism Underlying Circadian Post-transcriptional Regulation in Neurospora.

(A) Average (SEM) expression levels of rhythmic E3 ubiquitin ligase transcripts from the RNA-seq data.
(B) Average (SEM) of protein levels of rhythmic E3 ubiquitin ligases from the TMT-MS data.
(C) Average (SEM) phase delays, by time of peak mRNA phase, for genes rhythmic at both levels.
(D) The percent of genes that are rhythmic at both the mRNA and protein levels at a given time point that have a 0–2 hr phase delay, shown by time of day at which the mRNA peaks in its oscillation. Note that percentages are calculated separately for each time point. WT, wild-type; CT, circadian time; SEM, standard error of the mean; TMT-MS, tandem mass tag mass spectrometry.
See also Table S5.
Global Rhythms in Biosynthesis May Play a Major Role in Circadian Post-transcriptional Regulation
Figures 2B and 2C document the broad distribution of phase delays between peak mRNA and peak protein amounts for rhythmic proteins translated from rhythmic transcripts. As previously seen by Robles et al. (2014), the average phase delay changed in a systematic fashion across the circadian day, ranging from as short as 7.5 hr at CT10 near circadian “dusk,” to as long as 13.9 hr at CT20 near “dawn” (Figure 3C). Underlying this pattern, we found that relative to peak mRNA time, the percentage of rhythmic mRNAs that produced rhythmic proteins with little or no (i.e., 0–2 hr) phase delay varied sharply with the time of day. Approaching subjective dusk (CT12–CT14), nearly half of transcripts were translated with little or no phase delay (47% of rhythmic mRNA that peaked at CT12 and 39% of rhythmic mRNA that peaked at CT14), whereas, at CT4 and CT20, nearly all transcripts showed significant delays between the peak phase of each mRNA and protein (Figure 3D).
We posited that changes in translational activity could yield changes in phase delays. Recent work by Caster et al. (2016) reported that the circadian clock affected translation rates by regulating the phosphorylation of eukaryotic elongation factor eEF-2 (NCU07700). We noted the peak time for “protein-rhythmic only” proteins (CT12–CT14; Figure 2C) coincided with the time at which most transcripts were translated with little or no phase delay (Figure 3D), both corresponding to the peak of translational activity (Caster et al., 2016). Similarly, the phase when the lowest proportion (6%) of rhythmic mRNAs were translated with little to no delay (CT4; Figure 3D) corresponds to the predicted slowest rate of translation elongation (Caster et al., 2016).
We identified additional rhythmic proteins involved in translation, including the initiation factors eIF-1 (NCU01981), eIF-3d (NCU07380), eIF-3f (NCU01021), and eIF-3g (NCU08046). Also, at CT14, a pre-mRNA splicing factor (PRP43 [NCU01612]), an RNA helicase involved in mRNA export from the nucleus (DBP-5 [NCU01160]), and a poly(A) binding protein (PAB-1 [NCU04799]) reached peak rhythmic protein levels (Tseng et al., 1998). In addition, the delta subunit of eIF-2b (NCU01468), important in the formation of active eIF-2b that supports eIF2 activity, was significantly rhythmic at the protein level and peaked at CT14 (Jennings and Pavitt, 2014; Wortham et al., 2014). Finally, cpc-3 (NCU01187), a functional homolog of yeast GCN2 (an eIF2 alpha kinase), which plays a role in the overall repression of translation in response to amino acid starvation, was rhythmic at the protein level, peaking ~CT16 (Harding et al. 2003; Sattlegger et al., 1998; Wek et al., 2006; Wortham et al., 2014).
Metabolic Pathway Mapping Demonstrates Extensive Circadian Regulation of Metabolism at the Protein Level
Our extensive FunCat analysis demonstrated a high level of coordinated post-transcriptional circadian regulation, including 156 proteins rhythmic at just the protein level that are specifically enriched in amino acid metabolism and respiration, i.e., proteins that are directly or indirectly involved in secondary metabolism (Table S6). To visualize the extent of circadian regulation within metabolic pathways, we used the Pathway Tools software within the BioCyc Pathway Genome Database (PGDB); in BioCyc, every step in metabolism is associated with the specific proteins required for its execution, providing a straightforward means for visualizing genome-wide changes in omics data (Caspi et al., 2016). We modeled the expression of all significantly rhythmic proteins (p < 0.05) as a standard cosine wave to eliminate biological noise (see STAR Methods) and then mapped peak expression times to each protein’s respective enzyme on the metabolic map. An overall map showing the peak time of each enzyme’s oscillation was created (Figure S5), highlighting extensive circadian regulation of metabolism. Rhythmic proteins were found in the majority of BioCyc metabolic pathways, including central metabolic pathways such as glycolysis and the TCA cycle. Moreover, there appeared to be intra-pathway coordination. For example, eJTK best-fit phase and amplitudes for the rhythmic proteins within the pentose-phosphate pathway all peaked in the circadian morning. Conversely, in glycolysis and the TCA cycle, the rhythmic proteins peaked in the circadian evening (see Figure 4; Video S1).
Figure 4. Proteins that Are Rhythmic in the Pentose-Phosphate Pathway Peak in Anti-phase to Those in Glycolysis.

Enzymes and products within the pentose-phosphate pathway are shown along the left side, while enzymes and products within glycolysis are shown along the right side. Bolded and colored arrows and squares denote enzymes considered rhythmic in our analysis (p < 0.05), and the color denotes how many hours away it is from peak abundance or phase. For example, warm colors show that an enzyme is at a relatively high level at CT12, while cold colors show that an enzyme is at a low level near CT12. Inset panels show the eJTK rhythmic protein fit, with colored dots showing the time from peak protein level, by time of day (CT; see STAR Methods). CT, circadian time.
With our observation of rhythmic protein expression levels coordinated within and between metabolic pathways over the circadian day, we modeled the expression of rhythmic transcripts and then calculated the average level of rhythmic transcripts or proteins within each BioCyc pathway over the day (Figure 5). Comparing the modeled rhythmic transcriptome and proteome, peak timing of overall rhythmic mRNA averages was not well aligned with overall rhythmic protein averages in many metabolic pathways (Figures 5 and S6A–S6D). Additionally, when looking specifically at the proteome, proteins involved in other energy pathways (fermentation and aerobic respiration) also oscillate over the circadian day and peak near circadian “dusk,” in phase with glycolysis and the TCA cycle, but in antiphase to the rhythmic proteins involved in the pentose-phosphate pathway (Figure 5A). Beyond energy, some catabolic and anabolic pathway pairs peaked anti-phase relative to each other, such as fatty acid and lipid degradation and biosynthesis (Figure 5B versus Figure 5C). Likewise, fatty acid and lipid biosynthesis peaked in phase with carbohydrate and carboxylate degradation, a logical arrangement as fatty acids and lipids are produced from the by-products of glycolytic pathways (Figure 5B versus Figure 5C) (Voet et al., 2013).
Figure 5. Average Modeled Rhythmic mRNA and Protein Levels within BioCyc Categories Suggest Post-transcriptional Regulation of Metabolic Pathways.

(A–C) A graphic representation of the average of modeled rhythmic mRNA or protein levels in a given BioCyc category over the circadian day (CT). Large dots and lines are the average of all modeled rhythmic levels within a BioCyc category, and shaded areas are the standard deviations around those averages. BioCyc pathways presented are (A) energy pathways, including the pentose phosphate pathway, glycolysis, the tricarboxylic acid cycle, fermentation, and aerobic respiration; (B) degradation pathways, including fatty acid and lipid degradation and carbohydrate and carboxylate degradation; and (C) biosynthesis pathways, including fatty acid and lipid biosynthesis. In all cases, mRNA is orange, and protein is blue; and the number of proteins and mRNAs that fall into each category are displayed on the graph.
See also Figure S6.
Circadian Post-transcriptional Regulation Impacts Xylose Metabolism
The ability of filamentous fungi to deconstruct and metabolize plant cell walls is important for environmental carbon and other nutrient cycling, as well as numerous biotechnology applications such as the production of biofuels and bioproducts, like the production of ethanol from xylose derived from lignocellulosic substrates (Chen et al., 2010; Li et al., 2014). The xylan-to-ethanol pathway showed clock regulation at each stage, but the first intracellular reaction, catalyzed by xylose reductase (XR; NCU08384), was regulated at only the proteomic level (p = 6.12E–05 for protein versus p = 0.302 for mRNA; Figures 6A, S6E, and S6F). To track in vivo XR protein levels, and validate our in vitro data, we fused a codon-optimized luciferase (LUC) gene (Gooch et al., 2008) to the 3′ end of the XR coding sequence and transformed this cassette into the native xr locus of wild-type Neurospora (FGSC2489). Using a CCD camera, we followed replicates of a pfrq(c-box)-luciferase fusion control strain (verifying clock function under our experimental conditions; Figures 6B and S6G) and three transformants of the XR:LUC construct (Figures 6C and S6H). The XR:LUC fusion displayed circadian bioluminescence (Figure 6C) around an overall trend of exponential decay (Figure S6H). We confirmed by direct enzymatic assay (Yokoyama et al., 1995) that XR activity also oscillates with peak enzyme activity mirroring peak XR abundance (via luciferase tag) at around 24 hr after the transition to constant darkness (DD24 or CT13.6; compare Figures 6C and S6H to Figure 6D).
Figure 6. Circadian Post-transcriptional Regulation Impacts Xylose Metabolism.

(A)A schematic of the D-xylose to ethanol pathway, with the enzymes predicted to carry out each reaction mapped onto the pathway. Enzymes were detected at both the mRNA and protein levels. Colored boxes represent enzyme rhythmicity peak timing at the WT mRNA or protein level (p < 0.05), according to the included legend. (B and C) Luciferase traces (measured in AU) tracking (B) frq promoter activation (n = 24 wells) and (C) XYLOSE REDUCTASE protein levels (n = 36 wells), after smoothing with a moving average, rescaling, exponential detrending, and normalizing.
(D) XYLOSE REDUCTASE mean enzyme activity (SEM) from two tissue samples at each time point (two technical replicates for each). The more negative the change in absorbance at 340 nm, the higher the activity of this enzyme, which uses NADPH as a co-factor in converting D-xylose to xylitol. WT, wild-type; AU, arbitrary units; frq, Frequency; SEM, standard error of the mean.
See also Figure S6.
CSP-1 Contributes to Circadian Regulation of Metabolic Proteins
The global circadian repressor CSP-1 modulates the expression of ~800 genes whose products are predominantly involved in metabolic output (Lambreghts et al., 2009; Sancar et al., 2011) and has been implicated in the metabolic compensation mechanism that isolates the core clock from changes in glucose metabolism (Sancar et al., 2012; see, however, Olivares-Yañez et al., 2016). To examine the role of CSP-1 in regulating the metabolic proteome, we sampled fungal mats from a Δcsp-1 strain and prepared samples for TMT-MS analysis as done for the wildtype strain. 4,742 proteins were detected and imputed in all samples using LIMBR (see STAR Methods) (Crowell et al., 2018). Analysis of these data by eJTK_cycle identified 1,316 proteins (~28% of the identified Δcsp-1 proteome) as circadianly rhythmic with p < 0.05 (Hutchison et al., 2015), a proportion similar to the fraction of rhythmic wild-type proteins (1,273 or 27%). As in the WT strain, FunCat analysis of proteins cycling in the Δcsp-1 strain (Figure 7A and Table S7) showed them to be enriched (p < 0.05) in proteins with binding function, protein fate, and protein synthesis, as well as energy pathways, biogenesis, cell type differentiation, and cell fate. However, unlike the WT proteome, the overall metabolism category was not enriched in the rhythmic Δcsp-1 proteome, even when FunCat analysis was done individually on the two peak phases of rhythmic Δcsp-1 proteins (Table S8).
Figure 7. CSP-1 Contributes to Circadian Regulation of Metabolic Proteins.

(A) Breakdown of FunCat terms from rhythmic proteins detected in the Δcsp-1 strain (refer to Figure 1 for FunCat gene categories).
(B) Rhythmicity p values of all detected proteins in the Δcsp-1 strain(x axis) are plotted against p values of matching proteins in the WT strain (y axis).
(C) Heatmaps of the 1,316 rhythmic Δcsp-1 proteins showing normalized relative protein levels by circadian time of day (CT), with time points of low (blue) and high (yellow) protein levels, and proteins ordered by peak phase.
(D) A clock-style graph depicting the number of proteins that are rhythmic and peak at a given CT in the Δcsp-1 strain, with an overlay of the same information for the WT strain.
(E) Histogram depicting the number of rhythmic Δcsp-1 proteins that peak at a given CT broken down into the following sub-categories: (1) “0–2 hr phase change” relative to WT strain (blue; n = 200), (2) “>2 hr phase change” relative to WT strain (dark green; n = 302), and (3) “Only rhythmic in Δcsp-1” (light green, n = 814). CSP-1, CONIDIAL SEPARATION 1; WT, wild-type; CT, circadian time. *, p < 0.05; **, p ≤ 0.001.
See also Figure S7 and Tables S7, S8, and S9.
Comparing the rhythmicity of proteins from the WT and Δcsp-1 strains, 502 proteins were significantly rhythmic (p < 0.05) in both strains (~38% of the rhythmic proteins; area shaded by both blue and green), while more, but similar, numbers of proteins were only significantly rhythmic in the WT strain (771, area shaded only in blue) or the Δcsp-1 strain (814, area shaded only in green) (Figures 7B, S7A, and S7B). To confirm that these differences in protein rhythmicity between the two strains were not just due to the chosen p value cutoff, a total of 811 proteins had p < 0.1 in both strains (48% of all WT proteins with p < 0.1 and 44% of all Δcsp-1 proteins with p < 0.1), and a total of 1,385 proteins had p < 0.2 in both strains (55% of all WT proteins with p < 0.2 and 59% of all Δcsp-1s proteins with p < 0.2).
A heatmap (Figure 7C) shows the phase distribution of rhythmic proteins in the Δcsp-1 strain. By overlaying a densitometry “clock” graph of the number of proteins peaking at a given time of day for WT and Δcsp-1 strains, it appears that the most significant difference in the peak phases of rhythmic proteins between the WT and Δcsp-1 strains is that the peak at CT0/CT24 is larger in the Δcsp-1 strain (Figure 7D), in line with the predicted function of CSP-1 as a morning transcriptional repressor (Sancar et al., 2011). However, when we plotted a histogram of the peak phases of significantly rhythmic proteins in the Δcsp-1 strain, color coded by whether the peak protein phase was (1) maintained relative to the WT strain (“0–2 hr phase change”; n = 200), (2) changed relative to the WT strain (“>2 hr phase change”; n = 302), or (3) significantly rhythmic in the Δcsp-1 strain but not in WT (“Only rhythmic in Δcsp-1”; n = 814), it was apparent that there were overall changes in peak timing between the WT and Δcsp-1 strains (Figure 7E).
We used FunCat analysis to further investigate the differences between the WT and Δcsp-1 rhythmic proteomes (Table S9). For proteins significantly rhythmic only in the WT strain (i.e., when csp-1 is present), a high degree of FunCat enrichment was noted in metabolism (p = 0.0004), with nucleotide metabolism uniquely enriched for this category (Table S9). Energy was enriched for significantly rhythmic proteins found just in the WT strain but highly enriched for the subcategory of oxidation of fatty acids (p = 0.00054; Table S9), suggesting that CSP-1 is involved in the rhythmicity of fatty acid oxidation genes. Average modeled rhythmic Δcsp-1 protein levels were also compared to those calculated for the WT strain within each BioCyc category, using the Pathway Tools software (Figures S7C–S7F); changes were evident in peak times of pathways also identified by FunCat analysis, such as nucleotide synthesis and degradation as well as fatty acid and lipid biosynthesis and degradation. Finally, Energy pathways such as glycolysis and the pentose-phosphate pathway had overall peak phases that were shifted toward circadian morning in the Δcsp-1 strain, suggesting an increase in energy available at this time, relative to WT (Figure S7F).
DISCUSSION
Models that suggest circadian output stems from the transcriptional control of the positive arm of the clock have been challenged by evidence that post-transcriptional regulation is important in observed circadian behavior, including previously published proteomics data (e.g., Chiang et al., 2014; Kojima et al., 2011; Mauvoisin et al., 2014; Reddy et al., 2006; Robles et al., 2014; Wang et al., 2017a). A potential caveat to this conclusion has been the small numbers of rhythmic proteins identified, often less than 1% of the proteome, reflecting technical and analytical issues. Our extensive replicate time course, use of recent technological advances (TMT-MS), and improved computational techniques (Crowell et al., 2018) allowed analysis of a larger proportion of the predicted proteome in our organism (45%), from which we found 27% of detected proteins significantly cycled in abundance on a daily basis. Greater than 40% of the significantly rhythmic proteome was not associated with a corresponding significantly rhythmic transcript (Figures 2A, S4A, and S4B) and the long average phase-delay (~10-hour) between peak mRNA and protein abundance (Figure 2B) both support the importance of circadian post-transcriptional regulation in not only controlling which proteins are rhythmic but also the time of day at which these rhythmic proteins peak (Schwanhäusser et al., 2011).
The difference between the rhythmic transcriptome and proteome, more extensive than previously estimated for Neurospora (Zhou et al., 2016), prompted us to find the underlying cause of this discordance. Unlike Lück et al. (2014), we failed to detect widespread rhythmicity of E3-ubiquitin ligases (Figures 3 A and 3B) and therefore no “effective” degradation phase to explain our global proteomic timing. There is the caveat that there are additional modes of regulation on E3 ligases, such as phosphorylation (Gallagher et al., 2006); however, a lack of detailed studies on Neurospora E3 ubiquitin ligases limited our ability to include this potential factor. Rather than protein degradation, cycling protein production could give rise to protein rhythms from non-cycling transcripts. In Neurospora, peak translation elongation activity corresponds to our overall peak in protein levels and the time when the largest proportion of rhythmic mRNAs were translated without delay (Figures 1C and 3D) (Caster et al., 2016). In addition to elongation, the rhythmic abundance of ribosomal and translation proteins that we identified, such as the delta subunit of eIF-2b (NCU01468), could play a role in protein rhythms (Table S6) (Jennings and Pavitt, 2014; Wortham et al., 2014).
The large numbers of oscillating mRNAs not giving rise to significantly oscillating proteins begs the question of why an organism would regulate mRNA in a circadian manner without continuing the regulation on the protein level. Based on our work, it appears that the clock subdivides the production of mRNAs and proteins into time-specific groups that may reflect differences in optimal times of day for the processes of transcription versus translation. The expression pattern of mRNA that shows a dip in peak phase times at CT20 could relate to circadianly regulated chromatin landscapes (Aguilar-Arnal and Sassone-Corsi, 2015; Koike et al., 2012). Also, the Neurospora cell cycle is influenced by the circadian clock, and more cells undergo mitosis around CT20 when chromatin is likely less accessible (Figure 2C) (Hong et al., 2014). Given the timing of the Neurospora cell cycle, it seems plausible that rhythmic protein synthesis is timed for shortly after “dusk” as part of the preparation for cell division, with mitosis peaking later at night (Hong et al., 2014). Additionally, the circadian timing of peak protein synthesis may coincide with peak energy production in the evening due to the high energy demands of the translation process (e.g., Kafri et al., 2016).
While earlier proteomic studies in mice identified rhythmic metabolic enzymes (Chiang et al., 2014; Mauvoisin et al., 2014; Robles et al., 2014), our work demonstrates that the timing of rhythmic enzyme levels within pathways and at branch points in metabolic pathways may also be circadianly coordinated (Figure 4 and Video S1). We also found that 156 of the rhythmic metabolic proteins arose from non-cycling transcripts involved with functions such as secondary metabolism (Table S6); these would not previously have been detected as circadianly regulated. In all, the clock intensively regulates the timing of metabolic genes, presumably to ensure that the enzymes they encode are available at biologically advantageous times. An illustrative example is the post-transcriptional regulation of XR, an important enzyme for biofuel and industrial food production (Chen et al., 2010; Li et al., 2014).
Our Δcsp-1 proteomics analysis suggests that the decrease in fatty acid and lipid biosynthesis rhythms (Figure S7D) may explain previously reported changes in the lipid composition of cell membranes in a Δcsp-1 strain (Sancar et al., 2011; Sancar et al., 2012). There is also the caveat that transcriptomic changes resulting from the deletion of csp-1 are not necessarily translated to the proteomic level, suggesting a more complex role for CSP-1 in the regulation of the proteome (Figures S7B) (Sancar et al., 2015a). As the overall peak in rhythmic proteomic output occurred just after dusk in both the WT and Δcsp-1 strains (Figures 7D and 7E), energy availability and translational regulation may be more dominant than CSP-1 for metabolic regulation at the protein level.
Recent work has identified other factors impacting cross-talk between metabolic state and circadian transcription (e.g., RCO-1 and RCM-1) and metabolic genes (e.g., CRE-1) (Cupertino et al., 2015; Olivares-Yañez et al., 2016). CRE-1 (NCU08807) was significantly rhythmic at the RNA and protein (p = 0.0013) level, demonstrating a metabolic-responsive transcription factor also regulated by the clock. CPC-3 (NCU01187; functional homolog of yeast GCN2) is also significantly rhythmic at the protein level, peaking at CT16, just after the main peak in the rhythmic proteome (Figure 1C). This may provide a mechanism to repress global translation directly in response to low amino acid levels and further highlights how the clock regulates components to both anticipate and respond to its cellular environment (Harding et al., 2003; Sattlegger et al., 1998; Wek et al., 2006 ).
In summary, this deeply sampled dataset confirms the importance of the clock in regulating the metabolic output of an organism (Hurley et al., 2014; Sancar et al., 2015b), including post-transcriptional circadian regulation. Moreover, this work shows the importance of studying the output of the clock beyond the level of the transcriptome, as a significant amount of proteins appear to be rhythmic at the protein level only, meaning that their clock regulation would be missed in a standard transcriptomic analysis. As this work supports the importance of circadian regulation of metabolism and the potential importance of translation in this process, it highlights the need for further research into circadian control over translation and the inter-relationships between the clock and metabolism.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Jennifer M. Hurley (hurlej2@rpi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
The Neurospora strains FGSC2489 (WT) and FGSC11348 (Δcsp-1) were used for all proteomic analyses and transformations were performed as previously described using Neurospora strains FGSC9718 and FGSC2489 (Bardiya and Shiu, 2007; Colot et al., 2006) and screened using a MicroBeta TriLux liquid scintillation and luminescence counter (Perkin Elmer). For Neurospora proteomics time-course culture, conidia were inoculated into Bird medium (Metzenberg, 2004) containing 1.8% glucose. A separate time-course using Neurospora strain 536–1 (frhV5H6::bar+; ras-1bd), grown in Liquid Culture Medium (LCM) containing 1×Vogel’s, 0.5% arginine, and 50 ng/mL biotin with 2% glucose and 0.001M QA (pH 5.75) was completed with sampling every 4 hrs. over 24 hrs, yielding tissue for the xylose reductase enzyme activity assay. Tissue was prepared for circadian time series analysis as previously described (Hurley et al., 2014; Loros et al., 1989).
METHOD DETAILS
Cellular Homogenization of Fungal Mats
For each fungal mat, 100 mg pressed, wet weight tissue ground in liquid nitrogen was transferred to an Eppendorf Safe-Lock tube and homogenized using a Qiagen TissueLyser II with a 2×24 adapter (chilled to −20°C) following the vendor suggested protocol for tissue samples, with modifications. Briefly, a 3-mm stainless steel bead was added to each tube prior to processing twice in TissueLyser II for 5 min at 30 Hz. After confirming homogenization, the samples were again chilled to −20°C prior to transferring to a chilled Sorenson MμlTI™ SafeSeal™ microcentrifuge tube.
Chloroform-Methanol Extraction of Fungal Mats
To isolate the aqueous proteins, each homogenate was first mixed with a 1.2 mL of chilled 2:1 chloroform/methanol and then mixed with 0.24 mL chilled nanopure water (Deatherage Kaiser et al., 2013). The mixture was then incubated at −20°C for 20 min before separating the layers via centrifugation (12k × g, 15 min, 4°C). The protein interlayer was dried completely using a vacuum concentrator.
Tryptic Digestion
The protein interlayer was tryptically digested (Callister et al., 2006) after reconstituting into an 8M urea solution (in 50mM NH4HCO3, pH 8.0) and estimating the protein concentration using a BCA protein assay. Prior to digestion, the proteins were reduced in a thermomixer with 5mM dithiothreitol (60°C, 30 min), alkylated with 40mM iodoacetamide (37°C, 60 min), and diluted ten-fold with 50mM NH4HCO3, pH 8.0 before adding a final concentration of 1 mM CaCl2. For the digestion, enough trypsin (1 mg/mL in 5mM acetic acid, UBX) was added to result in an enzyme-to-protein ratio of 1:50. To quench the digestion, trifluoroacetic acid was added at a concentration of 0.1%. The peptides were subsequently desalted using Supelco DSC-18 solid phase extraction columns. A final concentration was measured using a BCA protein assay prior to normalizing the concentrations and isobaric labeling.
Isobaric Labeling of Peptides
For each peptide sample, 25μg was isobarically labeled using the TMT10plex Isobaric Mass Tag Labeling Reagents Sets (ThermoFisher) following the vendor protocol (Dayon et al., 2008; Thompson et al., 2003). Due to the quantity and complexity of samples for this experiment, two universal pooled samples were created to be used across each of the nineteen TMT10plex sets used. One pool was generated from 25μg from each of the wild type samples and the other from 25μg of the Δcsp-1 mutant. The individual time points for each cell type were then randomly distributed between the nineteen TMT10plex sets, such that for each time point both cell types were within the same set. After labeling and pooling individual labels into a single sample for each set, they were desalted as before using C-18 SPE, and then fractionated on an HPLC using high pH reverse phase chromatography (Wang et al., 2011). The resulting fraction sets were each pooled (using concatenation) into 12 total fractions per set.
LC-MS Processing of Proteins
These samples were analyzed by reverse phase LC-MS/MS using a Waters NanoEquity UPLC system (Millford, MA) coupled with a QExactive-Plus (batch 4064) or an LTQ Velos Pro Orbitrap mass spectrometer (batch 4144), both from Thermo Fisher Scientific (San Jose, CA). Sample was loaded on a solid phase extraction (SPE) column followed by separation on a C18 analytical column. Analytical column was packed in-house by slurry packing 3-μm Jupiter C18 stationary phase (Phenomenex, Torrence, CA) into a 70-cm long, 360 μm OD × 75 μm ID fused silica capillary tubing (Polymicro Technologies, Phoenix, AZ). The SPE column (360 μm OD × 150 μm ID) of 5cm length was similarly made with 3.6-μm Aries C18 particles. Mobile phases consisted of 0.1% formic acid in water (MP- A) and 0.1% formic acid in acetonitrile (MP- B). Samples were made at a concentration of ~0.1 μg/μL and 6 μL sample volume was injected on the SPE column via a 5μL sample loop for 30 min at a flow rate of 3μL per minute and then separated by the analytical column using a 110-minute gradient from 99% A to 5% A at a flow rate of 0.3μL per minute. Mass spectrometry analysis was started 15 min after the sample was moved to the analytical column and mass spectra were recorded for 100 min. After the gradient was completed, column was washed with 100% MP- B first and then reconditioned with 99% MP- A for 30 min.
The effluents from the LC column were ionized by electrospray ionization and mass analyzed with a QExactive-Plus or an LTQ Velos Pro Orbitrap hybrid mass spectrometer operated in the data-dependent analysis mode. A voltage of 2.2 kV was used for electrospray ionization and inlet capillary to the mass spectrometer was maintained at a temperature of 350°C for ion de-solvation. A primary survey scan was performed in the mass range of 400 to 2000 Daltons at a resolution of 70,000 (QExactive Plus, defined at m/z 200) or 120,000 resolution (Velos Pro) and automatic gain control (AGC) setting of 1e6 ions for both instruments. Top 10 highest intensity ions from the survey scan were selected for MS/MS analysis. On QExactive-Plus, a quadrupole mass filter was used for the isolation of ions for high energy collision dissociation (HCD) in an octopole collision cell with nitrogen and mass analyzed by the Orbitrap at a resolution of 17,500. An isolation window of 2 Daltons was used for the isolation of ions for HCD at a normalized collision energy of 28% (a Thermo nomenclature for ion energy for dissociating ions by collision) was used for HCD with AGC setting of 1e5 ions. Mass spectra were recorded for 100 min by repeating this process with a dynamic exclusion of previously selected ions for 30 seconds. On the Velos Pro Orbitrap mass spectrometer, top 10 ions were isolated (isolation width of 2 m/z) by the linear ion trap in the higher vacuum region and transferred to the linear ion trap in the lower vacuum region for collision-induced dissociation by multiple collisions with helium gas present in the trap at a normalized collision energy of 35% and AGC setting at 1E4 ions. The resulting fragment ions and remaining parent ions were transferred back to the higher vacuum linear ion trap for their mass analysis and detection by a pair of conversion dynode/electron multiplier placed on both sides of the linear trap. Mass spectra were recorded for 100 min by repeating the above process with a dynamic exclusion of 30 seconds.
Western Blots
Protein lysates were prepared on a small scale with a protease inhibitor mixture (P9599; Sigma). For western blot analysis, 10 μg of total protein was loaded per lane. Anti-FRQ antibody was diluted 1:250 (Garceau et al., 1997) and the SuperSignal West Femto ECL (Pierce) was used for signal development.
Real-Time CCD Recording
An electronically cooled camera from Princeton Instruments (PIXIS 1024) was used to follow luminescence using the Lightfield program. Camera runs were completed at 25°C, with cultures grown in black, flat-bottomed 96-well plates from Eppendorf. Liquid suspensions of conidia (O.D. of 0.5 at 600nm) were plated onto QA medium (0.03% glucose, 0.05% arginine, 0.001 M QA, pH 5.75) containing 25mM luciferin and covered with a Breathe-Easy strip (USAScientific). Inoculated plates were then subjected to 48 h of 12:12 dark:light cycle conditions at 25°C:28°C before starting the camera trial in constant-dark and 25°C conditions. Signals were accumulated for 15 min every hour. A custom ImageJ Macro called “Toolset Image Analysis Larrondo’s Lab v. 1.0” was used to process the images (Larrondo et al., 2012) within FIJI v.2.0.0 (Schindelin et al., 2012). Raw data arising from each time series was smoothed using a 3-point moving average, and then processed using custom-written software to detrend (by removing the exponential decay signal), rescale, and normalize amplitudes.
Xylose Reductase Enzyme Activity Assay
Ground tissue from each time point was lysed with 250 mM potassium phosphate buffer (pH 6.8) and a protease/phosphatase inhibitor (1861282; Thermo Scientific), vortexed and centrifuged at 14,000 rpm for 10 min. at 4°C. The total soluble protein fraction for each time point sample was measured using a Bradford Assay with BSA standards and extracted protein for each set of time course samples was standardized to 5 mg/ml total protein. Similar to Yokoyama et al. (Yokoyama et al., 1995), a paired kinetic time scan at 340nm was used to measure xylose reductase activity. A decrease in absorbance at 340 nm represents a decrease in NADPH levels, which is a co-factor used by XR in converting D-xylose to xylitol. Specifically, for each time point, a reference cuvette containing 600 μL of 250 mM potassium phosphate buffer (pH 6.8), 100 μL of 100 μM b-mercaptoethanol, 150 μL of distilled/deionized H2O, 50 μL of 3.4 mM NADPH (NADPH-RO; Sigma) and 100 μL of the standardized protein lysate from a given time point was mixed and kept on ice. Each matching experimental time point cuvette contained the same reagents as above, except the amount of distilled/deionized H2O was decreased to 50 μL so that a final volume of 1 mL was maintained in the final assay. Enzyme activity assays used a Hitachi U-2910 Spectrophotometer with the program UV Solutions 2.2. Reference and experimental cuvettes were brought to room temperature for 10 min, before auto-zeroing and starting the kinetic time scan at 340 nm. After 5 sec., 100 μL of the substrate, 0.5 M D-xylose (Sigma #X1500–500G), was added to the experimental cuvette and the time scan completed. The slope of the initial velocity of enzyme activity (during the linear phase) was measured immediately after mixing (Dataset S3). Protein was extracted from two sets of time course samples, and a technical replicate of the enzyme assay completed on a different day, after storing the extracted protein samples at −20°C (except for one of the last time point samples, where only one round of the enzyme assay could be completed due to limited sample size).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Processing, eJTK and FunCat Analyses
Peptides for which fewer than 30% of observations were missing had those missing values imputed (inferred from observations of other non-missing peptides) by the K nearest neighbors method with K=10, within LIMBR ver. “pre-alpha” as of Oct. 3 2016 (Crowell et al., 2018). In this approach, a missing value of a peptide in a given sample is replaced by the average value in that sample of the K (10) other peptides which had the most similar expression across other samples for which the peptide with the missing value was observed; i.e. taking advantage of the fact that there were three replicates for each point. This method of imputation is well established and has been employed successfully across multiple bioinformatics data types recently showing top tier performance in the analysis of differential expression in MS data (Bastista and Monard, 2003; Schmitt et al., 2015; Wang et al., 2017b). The imputed data for each sample is then divided by the corresponding pooled control to account for variability induced by the individual MS runs. Prior to learning batch effects, each sample is quantile normalized and then the abundances for each peptide are scaled to zero mean and unit variance. Batch effects were then removed with a time series specific algorithm within LIMBR (Crowell et al., 2018), based on Surrogate Variable Analysis (SVA). In brief, this method applies to the data a non-parametric LOWESS (Locally Weighted Scatterplot Smoothing) model, which captures the expected agreement of replicates, adjacent time points and time points with similar CTs. A singular value decomposition is applied to the residuals of this fit and the significance of the resulting components is determined by a permutation analysis (10,000 permutations used). Components deemed significant are then removed from the data. Python code for LIMBR is available for download at https://github.com/aleccrowell/LIMBR and the package can be downloaded and installed with dependencies using ‘pip install limbr’.
Circadian rhythms were assessed by eJTK_cycle analysis (Hutchison et al., 2015), with updates as of Aug 3, 2016 (Hutchison et al., 2018; Hutchison et al., 2015). In this updated version of eJTK, instead of calculating empirical p values, computational time has been reduced by using a Gamma distribution to yield corrected p values for a given gene, termed GammaP in the output and referred to in this paper as p values. While Benjamini-Hochberg adjustments are available in this version of eJTK, they are more conservative than true FDRs (Hutchison et al., 2015), therefore any protein with a (Gamma) p value <0.05 was considered a rhythmic protein in this study. For reference, in our datasets a p value of 0.05 roughly corresponded to the overly conservative Benjamini-Hochberg-adjusted p value of 0.19. Functional annotation of the differently regulated proteins was performed according to Functional Categories (FunCat) (Ashburner et al., 2000) and N. crassa Genome Database (Galagan et al., 2003) within the FungiFun Ver. 0.5 interface (Priebe et al., 2011) and a value of p<0.05 was used as a cut-off for significantly enriched terms, while p<0.001 was considered highly enriched.
Phase Set Enrichment Analysis (PSEA)
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways for Neurospora genes were downloaded from FunCat ver. 0.5 (KEGG, ver. Apr 26, 2011) and converted to gene matrix transpose (GMT) file format. Using the PSEA enrichment Java package (ver. 1.1) (Zhang et al., 2016), we input the converted Neurospora GMT file and a list of rhythmic WT proteins (n=1273) with their eJTK-identified peak phases in CT. We required a minimum of 10 genes per gene set (n=89 gene sets met the criteria), utilized 10,000 simulations in the PSEA Kuiper test, and defined phase enrichment as q<0.05 (Benjamini-Hochberg adjusted p value). The Kuiper test is a non-parametric aggregate score method that can be used on cyclic data (Kuiper, 1960). As implemented in PSEA, the Kuiper test evaluates how closely a cumulative probability distribution for the sample matches a given background distribution (Zhang et al., 2016). In this case, the phase enrichment was completed relative to a uniform background distribution, to summarize any overall synchronization of peak phases within gene sets.
Categorizing Noise Using Residual Variance
We normalized the proteomics data, in order for both the transcriptomics and proteomics data to be on the same scale. Proteomics expressions were normalized in the following manner:
where zi is the resulting normalized expression for time point i = 1,.,24 and gene j = 1,.,4747, xijis the original proteome expression, μj is the mean expression, and σj is the standard deviation of expression for gene j.
We found the residuals, or the difference between the fitted and actual expression, for only significantly rhythmic (p<0.05) proteins or transcripts:
where rij is the residual, is the fitted expression from eJTK, and yij is the actual expression.
We then calculated the variance for each expression’s residuals and used a two-sided F-test to determine whether the distributions of residual variances for the rhythmic transcriptome and the rhythmic proteome were significantly different, using a 0.05 cutoff.
Clock-Controlled Expression Simulation Based on Peak Phase for Pathway Mapping
Using the peak phase data, we simulated expression of each clock-controlled mRNA and protein over circadian time, starting at CT12. Expression was simulated using arbitrary amplitude units that vary between 0 and 2, with 2 being the “peak phase” (θ) according to the following function:
where the period T=24 circadian hours, the amplitude A and the offset D both equal 1. Time t is simulated every 2.13 circadian hours from CT12 to CT11.5.
Metabolic Overview Poster and Omics Dashboard Figures
The cellular overview poster of metabolic pathways was generated using the BioCyc program (ver. 21.5), as in Paley and Karp (2006) and omics dash board figures as in Paley et al., 2017.
DATA AND SOFTWARE AVAILABILITY
The accession number for the mass spectrometry proteomics data reported in this paper is ProteomeXchange Consortium: PXD009682, 10.6019/PXD009682 (Vizcaino et al., 2016). RNA-seq data from Hurley et al. (2014), was previously deposited in the National Center for Biotechnology Information Short Read Archive (SRA): SRP045821, SRP046458 (Hurley et al., 2014). Additionally, raw and processed TMT-MS and RNA-seq data used in this paper are available at Mendeley: [https://doi.org/10.17632/8mzwdr9sxc.1]. Genes sets used for FunCat analyses in this paper are accessible at Mendeley: [https://doi.org/10.17632/r68j3rnxhw.1]. Finally, raw XR enzyme activity assay data reported in this paper are at Mendeley: [https://doi.org/10.17632/x3c66jk53m.1]. For software availability, please refer to the STAR Methods Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FRQ (rabbit) | Jay Dunlap (Garceau et al., 1997) | N/A |
| Bacterial and Virus Strains | ||
| E.coli, DH5alpha, Electro-competent | New England Bio | C2989K |
| Chemicals, Peptides, and Recombinant Proteins | ||
| USB TPCK treated Trypsin | Affymetrix | 22725 250 MG |
| Protease Inhibitor Cocktail | Sigma | P9599 |
| Protease Inhibitor Cocktail | ThermoScientific | 1861282 |
| SuperSignal West Femto ECL (Pierce) | ThermoFisher | 34094 |
| NADPH Tetrasodium salt (Roche) | Sigma | NADPH-RO |
| D-(+)-Xylose, ≥99% | Sigma | X1500–500G |
| Critical Commercial Assays | ||
| BCA protein quantification kit (Pierce) | ThermoFisher | 23225 |
| TMT10plex Isobaric Mass Tag Labeling Reagents Sets | ThermoFisher | 90406 |
| Deposited Data | ||
| Mass spectrometry proteomics data: ProteomeXchange Consortium via the PRIDE partner repository | This paper | PXD009682 and 10.6019/PXD009682 |
| RNA-seq data: National Center for Biotechnology Information Short Read Archive (SRA) | Hurley et al. (2014) | SRP045821 and SRP046458 |
| Mendeley dataset: Raw and processed TMT-MS and RNA-seq data. | This paper | [https://doi.org/10.17632/8mzwdr9sxc.1] |
| Mendeley dataset: Genes sets used for FunCat analyses. | This paper | [https://doi.org/10.17632/r68j3rnxhw.1] |
| Mendeley dataset: Raw XR enzyme activity assay data. | This paper | [https://doi.org/10.17632/x3c66jk53m.1] |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: FY834 | Fungal Genetics Stock Left (Colot et al., 2006) | FGSC #9721 |
| N. crassa: WT: 74-OR23–1VA | Fungal Genetics Stock Left | FGSC #2489 |
| N. crassa: Δcsp-1: delta csp-1::hph+ | Fungal Genetics Stock Left | FGSC #11348 |
| N. crassa: FGSC9718: delta mus-51::bar+ | Fungal Genetics Stock Left | FGSC #9718 |
| N. crassa: 536–1: frhV5H6::bar+;ras-1bd | Jen Hurley (Hurley et al., 2013) | 536 |
| N. crassa: Pfrq:luc: Pfrq(c-box)::luc::hph+ | Jay Dunlap | WT-1 |
| N. crassa: XR:LUC: XR::luc::hph+ | This paper | Clones #4, 5, 10 |
| Oligonucleotides | ||
| Primer: pRS426 to NCU08384, -1000 bp from stop codon, forward: GTAACGCCAGGGTTTTCCCAGTCACGAC/ ATTTTAAGGACACGAGGAGCAG | This paper | SFQ7 |
| Primer: NCU08384 (end of ORF minus stop codon) to luciferase/hph,forward: GAGAACCTCTGGATTTTCGGT/ GGCGGAGGCGGCGGAGGCGG | This paper | SFQ8 |
| Primer: NCU08384 (end of ORF minus stop codon) to luciferase/hph,reverse: CCGCCTCCGCCGCCTCCGCC/ ACCGAAAATCCAGAGGTTCTC | This paper | SFQ9 |
| Primer: Luciferase/hph to NCU08384, just after stop codon, forward: GAAGTTATGGATCCGAGCTCG/ GTCAACCTTTTTCCGCAGCTTAG | This paper | SFQ10 |
| Primer: Luciferase/hph to NCU08384, just after stop codon, reverse: CTAAGCTGCGGAAAAAGGTTGAC/ CGAGCTCGGATCCATAACTTC | This paper | SFQ11 |
| Primer: NCU08384, +1000 bp from stop codon to pRS426, reverse: GCGGATAACAATTTCACACAGGAAACAGC/ GCCGGCATTGTATAGGAACAAG | This paper | SFQ12 |
| Recombinant DNA | ||
| Plasmid: pRS426: Yeast episomal vector with URA3 marker, and Ampicillin resistance | Fungal Genetics Stock Left (Colot et al., 2006) | pRS426 |
| Software and Algorithms | ||
| LIMBR v. “pre-alpha”, Oct 3, 2016 | Crowell et al. (2018) | https://github.com/aleccrowell/LIMBR |
| eJTK_cycle, v. Aug 3, 2016 | Hutchison et al. (2015); Hutchison et al. (2018) | https://github.com/alanlhutchison/empirical-JTK_CYCLE-with-asymmetry |
| FunCat v. 8/12/2010, within FungiFun v. 0.5 | Priebe et al. (2011) | https://elbe.hki-jena.de/fungifun/ |
| PSEA, v. 1.1 | Zhang et al. (2016) | https://github.com/ranafi/PSEA |
| Pathway Tools software within the BioCyc Pathway/ Genome Database (PGDB), v. 21.5 | Caspi et al. (2016) | https://biocyc.org/ |
| Toolset Image Analysis Larrondo’s Lab v. 1.0, within FIJI (ImageJ) v. 2.0.0 | Jen Hurley (Larrondo et al., 2012; Schindelin et al., 2012) | https://fiji.sc/ |
Supplementary Material
Highlights.
Circadian output is dominated by post-transcriptional regulation
Circadian post-transcriptional regulation is broadly imparted by translational control
Metabolic processes are highly impacted by circadian post-transcriptional regulation
CSP-1 plays a complex role in the post-transcriptional regulation of circadian output
ACKNOWLEDGMENTS
The proteomic data was generated at the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. DOE OBER and located at PNNL. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under contract DE-AC05–76RLO 1830. We thank the Fungal Genetics Stock Center at the University of Missouri for Neurospora strains. This work is supported by NIH-National Institute of Biomedical Imaging and Bioengineering grant EB022546 (J.M.H., J.C.D., and W.R.C.); DOE BER project 69513 (W.R.C.); NIH-National Institute of General Medical Sciences grants GM118021 (J.C.D.), GM128586 (J.M.H.) and GM118022 (J.J.L.); DOE-EMSL grant 47818 (J.M.H., J.J.L., and J.C.D.), and Rensselaer Polytechnic Startup funds (J.M.H.). We thank Emily Collins, Jackie Pelham, Joel Morgan, Calvin Chen, Jason Hicken, Rose Perry, Alan Hutchison, and Ron Anafi for experimental advice and assistance.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, nine tables, and one video and can be found with this article online at https://doi.org/10.1016/j.cels.2018.10.014.
REFERENCES
- Aguilar-Arnal L, and Sassone-Corsi P (2015). Chromatin landscape and circadian dynamics: spatial and temporal organization of clock transcription. Proc. Natl. Acad. Sci. USA 112, 6863–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardiya N, and Shiu PK (2007). Cyclosporin A-resistance based gene placement system for Neurospora crassa. Fungal Genet. Biol 44, 307–314. [DOI] [PubMed] [Google Scholar]
- Bastista G, and Monard M (2003). A Study of K-Nearest Neighbour as an Imputation Method (Healthcare Infection Society).
- Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, and Zoran MJ (2005). Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet 6, 544–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callister SJ, Dominguez MA, Nicora CD, Zeng XH, Tavano CL, Kaplan S, Donohue TJ, Smith RD, and Lipton MS (2006). Application of the accurate mass and time tag approach to the proteome analysis of sub-cellular fractions obtained from Rhodobacter sphaeroides 2.4.1. Aerobic and photosynthetic cell cultures. J. Proteome Res 5, 1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 44, D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caster SZ, Castillo K, Sachs MS, and Bell-Pedersen D (2016). Circadian clock regulation of mRNA translation through eukaryotic elongation factor eEF-2. Proc. Natl. Acad. Sci. USA 113, 9605–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Jiang ZH, Chen S, and Qin W (2010). Microbial and bioconversion production of D-xylitol and its detection and application. Int. J. Biol. Sci 6, 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CK, Mehta N, Patel A, Zhang P, Ning Z, Mayne J, Sun WY, Cheng HY, and Figeys D (2014). The proteomic landscape of the suprachiasmatic nucleus clock reveals large-scale coordination of key biological processes. PLoS Genet. 10, e1004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, and Dunlap JC (2006). A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 103, 10352–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell AM, Greene CS, Loros JJ, and Dunlap JC (2018). Learning and Imputation for Mass-spec Bias Reduction (LIMBR). Bioinformatics 29, 2804–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupertino FB, Virgilio S, Freitas FZ, Candido, Tde S., and Bertolini, M.C. (2015). Regulation of glycogen metabolism by the CRE-1, RCO-1 and RCM-1 proteins in Neurospora crassa. The role of CRE-1 as the central transcriptional regulator. Fungal Genet. Biol 77, 82–94. [DOI] [PubMed] [Google Scholar]
- Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, and Sanchez JC (2008). Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- Dayon L, and Sanchez JC (2012). Relative protein quantification by MS/MS using the tandem mass tag technology. Methods Mol. Biol 893, 115–127. [DOI] [PubMed] [Google Scholar]
- Deatherage Kaiser BL, Li J, Sanford JA, Kim YM, Kronewitter SR, Jones MB, Peterson CT, Peterson SN, Frank BC, Purvine SO, et al. (2013). A multi-omic view of host-pathogen-commensal interplay in Salmonella-mediated intestinal infection. PLoS One 8, e67155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap JC, and Loros JJ (2018). Just-so stories and origin myths: phosphorylation and structural disorder in circadian clock proteins. Mol. Cell 69, 165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel-Mahan K, and Sassone-Corsi P (2013). Metabolism and the circadian clock converge. Physiol. Rev 93, 107–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller KK, Hurley JM, Loros J, and Dunlap J (2014). Photobiology and circadian clocks in Neurospora. In The Mycota, Nowrousian M, ed. (Springer; ), pp. 121–148. [Google Scholar]
- Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, et al. (2003). The genome sequence of the filamentous fungus Neurospora crassa. Nature 422, 859–868. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, and Karin M (2006). Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc. Natl. Acad. Sci. USA 103, 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garceau NY, Liu Y, Loros JJ, and Dunlap JC (1997). Alternative initiation of translation and time-specific phosphorylation yield multiple forms of the essential clock protein FREQUENCY. Cell 89, 469–476. [DOI] [PubMed] [Google Scholar]
- Gooch VD, Mehra A, Larrondo LF, Fox J, Touroutoutoudis M, Loros JJ, and Dunlap JC (2008). Fully codon-optimized luciferase uncovers novel temperature characteristics of the Neurospora clock. Eukaryot. Cell 7, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hong CI, Zámborszky J, Baek M, Labiscsak L, Ju K, Lee H, Larrondo LF, Goity A, Chong HS, Belden WJ, et al. (2014). Circadian rhythms synchronize mitosis in Neurospora crassa. Proc. Natl. Acad. Sci. USA 111, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, and Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JM, Dasgupta A, Emerson JM, Zhou X, Ringelberg CS, Knabe N, Lipzen AM, Lindquist EA, Daum CG, Barry KW, et al. (2014). Analysis of clock-regulated genes in Neurospora reveals widespread posttranscriptional control of metabolic potential. Proc. Natl. Acad. Sci. USA 111, 16995–17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JM, Larrondo LF, Loros JJ, and Dunlap JC (2013). Conserved RNA helicase FRH acts nonenzymatically to support the intrinsically disordered neurospora clock protein FRQ. Mol. Cell 52, 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JM, Loros JJ, and Dunlap JC (2016). Circadian oscillators: Around the transcription-translation feedback loop and on to output. Trends Biochem. Sci 41, 834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison AL, Allada R, and Dinner AR (2018). Bootstrapping and empirical Bayes methods improve rhythm detection in sparsely sampled data. J. Biol. Rhythms 33, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison AL, Maienschein-Cline M, Chiang AH, Tabei SM, Gudjonson H, Bahroos N, Allada R, and Dinner AR (2015). Improved statistical methods enable greater sensitivity in rhythm detection for genome-wide data. PLoS Comput. Biol 11, e1004094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings MD, and Pavitt GD (2014). A new function and complexity for protein translation initiation factor eIF2B. Cell Cycle 13, 2660–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri M, Metzl-Raz E, Jona G, and Barkai N (2016). The cost of protein production. Cell Rep. 14, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Shingle DL, and Green CB (2011). Post-transcriptional control of circadian rhythms. J. Cell Sci 124, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper NH (1960). Tests concerning random points on a circle. Indagationes Mathematicae (Proceedings) 63, 38–47. [Google Scholar]
- Lambreghts R, Shi M, Belden WJ, Decaprio D, Park D, Henn MR, Galagan JE, Bastürkmen M, Birren BW, Sachs MS, et al. (2009). A high-density single nucleotide polymorphism map for Neurospora crassa. Genetics 181, 767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrondo LF, Loros JJ, and Dunlap JC (2012). High-resolution spatiotemporal analysis of gene expression in real time: in vivo analysis of circadian rhythms in Neurospora crassa using a FREQUENCY-luciferase translational reporter. Fungal Genet. Biol 49, 681–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lin L, Li H, Tian C, and Ma Y (2014). Transcriptional comparison of the filamentous fungus Neurospora crassa growing on three major monosaccharides D-glucose, D-xylose and L-arabinose. Biotechnol. Biofuels 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loros JJ, Denome SA, and Dunlap JC (1989). Molecular cloning of genes under control of the circadian clock in Neurospora. Science 243, 385–388. [DOI] [PubMed] [Google Scholar]
- Lück S, Thurley K, Thaben PF, and Westermark PO (2014). Rhythmic degradation explains and unifies circadian transcriptome and proteome data. Cell Rep. 9, 741–751. [DOI] [PubMed] [Google Scholar]
- Mauvoisin D, Wang J, Jouffe C, Martin E, Atger F, Waridel P, Quadroni M, Gachon F, and Naef F (2014). Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc. Natl. Acad. Sci. USA 111, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzenberg RL (2004). Bird Medium: an alternative to Vogel Medium. Fungal Genetics Reports 51, 19–20. [Google Scholar]
- Olivares-Yañez C, Emerson J, Kettenbach A, Loros JJ, Dunlap JC, and Larrondo LF (2016). Modulation of circadian gene expression and metabolic compensation by the RCO-1 corepressor of Neurospora crassa. Genetics 204, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley S, Parker K, Spaulding A, Tomb JF, O’Maille P, and Karp PD (2017). The Omics Dashboard for interactive exploration of gene-expression data. Nucleic Acids Res. 45, 12113–12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley SM, and Karp PD (2006). The Pathway Tools cellular overview diagram and Omics Viewer. Nucleic Acids Res. 34, 3771–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Green CB, and Takahashi JS (2014). Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priebe S, Linde J.r., Albrecht D, Guthke R, and Brakhage AA (2011). FungiFun: A web-based application for functional categorization of fungal genes and proteins. Fungal Genet. Biol 48, 353–358. [DOI] [PubMed] [Google Scholar]
- Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O’Neill JS, Wong GK, Chesham J, Odell M, Lilley KS, et al. (2006). Circadian orchestration of the hepatic proteome. Curr. Biol 16, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Robles MS, Cox J, and Mann M (2014). In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet. 10, e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar C, Ha N, Yilmaz R, Tesorero R, Fisher T, Brunner M, and Sancar G (2015a). Combinatorial control of light induced chromatin remodeling and gene activation in Neurospora. PLoS Genet. 11, e1005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar C, Sancar G, Ha N, Cesbron F, and Brunner M (2015b). Dawnand dusk-phased circadian transcription rhythms coordinate anabolic and catabolic functions in Neurospora. BMC Biol. 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar G, Sancar C, Brügger B, Ha N, Sachsenheimer T, Gin E, Wdowik S, Lohmann I, Wieland F, Höfer T, et al. (2011). A global circadian repressor controls antiphasic expression of metabolic genes in Neurospora. Mol. Cell 44, 687–697. [DOI] [PubMed] [Google Scholar]
- Sancar G, Sancar C, and Brunner M (2012). Metabolic compensation of the Neurospora clock by a glucose-dependent feedback of the circadian repressor CSP1 on the core oscillator. Genes Dev. 26, 2435–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattlegger E, Hinnebusch AG, and Barthelmess IB (1998). cpc-3, the Neurospora crassa homologue of yeast GCN2, encodes a polypeptide with juxtaposed eIF2 alpha kinase and histidyl-tRNA synthetase-related domains required for general amino acid control. J. Biol. Chem 273, 20404–20416. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt P, Mandel J, and Guedj M (2015). A comparison of six methods for missing data imputation. J. Biometr. Biostat 6, 10.4172/2155-6180.1000224. [DOI] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, and Hamon C (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Tseng SSL, Weaver PL, Liu Y, Hitomi M, Tartakoff AM, and Chang TH (1998). Dbp5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export. EMBO J. 17, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet D, Voet J, and Pratt C (2013). Fundamentals of Biochemistry: Life at the Molecular Level, Fourth Edition (John Wiley and Sons, Inc; ). [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mauvoisin D, Martin E, Atger F, Galindo AN, Dayon L, Sizzano F, Palini A, Kussmann M, Waridel P, et al. (2017a). Nuclear proteomics uncovers diurnal regulatory landscapes in mouse liver. Cell Metab. 25, 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Li LW, Chen T, Ma J, Zhu YP, Zhuang JJ, and Chang C (2017b). In-depth method assessments of differentially expressed protein detection for shotgun proteomics data with missing values. Sci Rep. 7, 3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Yang F, Gritsenko MA, Wang YC, Clauss T, Liu T, Shen YF, Monroe ME, Lopez-Ferrer D, Reno T, et al. (2011). Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, and Anthony TG (2006). Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans 34, 7–11. [DOI] [PubMed] [Google Scholar]
- Wortham NC, Martinez M, Gordiyenko Y, Robinson CV, and Proud CG (2014). Analysis of the subunit organization of the eIF2B complex reveals new insights into its structure and regulation. FASEB J. 28, 2225–2237. [DOI] [PubMed] [Google Scholar]
- Yokoyama SI, Suzuki T, Kawai K, Horitsu H, and Takamizawa K (1995). Purification, characterization and structure analysis of Nadph-dependent D-xylose reductases from Candida tropicalis. J. Ferment. Bioeng 79, 217–223. [Google Scholar]
- Zhang R, Podtelezhnikov AA, Hogenesch JB, and Anafi RC (2016). Discovering biology in periodic data through phase set enrichment analysis (PSEA). J. Biol. Rhythms 31, 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Dang Y, Zhou M, Li L, Yu CH, Fu J, Chen S, and Liu Y (2016). Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 113, E6117–E6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the mass spectrometry proteomics data reported in this paper is ProteomeXchange Consortium: PXD009682, 10.6019/PXD009682 (Vizcaino et al., 2016). RNA-seq data from Hurley et al. (2014), was previously deposited in the National Center for Biotechnology Information Short Read Archive (SRA): SRP045821, SRP046458 (Hurley et al., 2014). Additionally, raw and processed TMT-MS and RNA-seq data used in this paper are available at Mendeley: [https://doi.org/10.17632/8mzwdr9sxc.1]. Genes sets used for FunCat analyses in this paper are accessible at Mendeley: [https://doi.org/10.17632/r68j3rnxhw.1]. Finally, raw XR enzyme activity assay data reported in this paper are at Mendeley: [https://doi.org/10.17632/x3c66jk53m.1]. For software availability, please refer to the STAR Methods Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FRQ (rabbit) | Jay Dunlap (Garceau et al., 1997) | N/A |
| Bacterial and Virus Strains | ||
| E.coli, DH5alpha, Electro-competent | New England Bio | C2989K |
| Chemicals, Peptides, and Recombinant Proteins | ||
| USB TPCK treated Trypsin | Affymetrix | 22725 250 MG |
| Protease Inhibitor Cocktail | Sigma | P9599 |
| Protease Inhibitor Cocktail | ThermoScientific | 1861282 |
| SuperSignal West Femto ECL (Pierce) | ThermoFisher | 34094 |
| NADPH Tetrasodium salt (Roche) | Sigma | NADPH-RO |
| D-(+)-Xylose, ≥99% | Sigma | X1500–500G |
| Critical Commercial Assays | ||
| BCA protein quantification kit (Pierce) | ThermoFisher | 23225 |
| TMT10plex Isobaric Mass Tag Labeling Reagents Sets | ThermoFisher | 90406 |
| Deposited Data | ||
| Mass spectrometry proteomics data: ProteomeXchange Consortium via the PRIDE partner repository | This paper | PXD009682 and 10.6019/PXD009682 |
| RNA-seq data: National Center for Biotechnology Information Short Read Archive (SRA) | Hurley et al. (2014) | SRP045821 and SRP046458 |
| Mendeley dataset: Raw and processed TMT-MS and RNA-seq data. | This paper | [https://doi.org/10.17632/8mzwdr9sxc.1] |
| Mendeley dataset: Genes sets used for FunCat analyses. | This paper | [https://doi.org/10.17632/r68j3rnxhw.1] |
| Mendeley dataset: Raw XR enzyme activity assay data. | This paper | [https://doi.org/10.17632/x3c66jk53m.1] |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: FY834 | Fungal Genetics Stock Left (Colot et al., 2006) | FGSC #9721 |
| N. crassa: WT: 74-OR23–1VA | Fungal Genetics Stock Left | FGSC #2489 |
| N. crassa: Δcsp-1: delta csp-1::hph+ | Fungal Genetics Stock Left | FGSC #11348 |
| N. crassa: FGSC9718: delta mus-51::bar+ | Fungal Genetics Stock Left | FGSC #9718 |
| N. crassa: 536–1: frhV5H6::bar+;ras-1bd | Jen Hurley (Hurley et al., 2013) | 536 |
| N. crassa: Pfrq:luc: Pfrq(c-box)::luc::hph+ | Jay Dunlap | WT-1 |
| N. crassa: XR:LUC: XR::luc::hph+ | This paper | Clones #4, 5, 10 |
| Oligonucleotides | ||
| Primer: pRS426 to NCU08384, -1000 bp from stop codon, forward: GTAACGCCAGGGTTTTCCCAGTCACGAC/ ATTTTAAGGACACGAGGAGCAG | This paper | SFQ7 |
| Primer: NCU08384 (end of ORF minus stop codon) to luciferase/hph,forward: GAGAACCTCTGGATTTTCGGT/ GGCGGAGGCGGCGGAGGCGG | This paper | SFQ8 |
| Primer: NCU08384 (end of ORF minus stop codon) to luciferase/hph,reverse: CCGCCTCCGCCGCCTCCGCC/ ACCGAAAATCCAGAGGTTCTC | This paper | SFQ9 |
| Primer: Luciferase/hph to NCU08384, just after stop codon, forward: GAAGTTATGGATCCGAGCTCG/ GTCAACCTTTTTCCGCAGCTTAG | This paper | SFQ10 |
| Primer: Luciferase/hph to NCU08384, just after stop codon, reverse: CTAAGCTGCGGAAAAAGGTTGAC/ CGAGCTCGGATCCATAACTTC | This paper | SFQ11 |
| Primer: NCU08384, +1000 bp from stop codon to pRS426, reverse: GCGGATAACAATTTCACACAGGAAACAGC/ GCCGGCATTGTATAGGAACAAG | This paper | SFQ12 |
| Recombinant DNA | ||
| Plasmid: pRS426: Yeast episomal vector with URA3 marker, and Ampicillin resistance | Fungal Genetics Stock Left (Colot et al., 2006) | pRS426 |
| Software and Algorithms | ||
| LIMBR v. “pre-alpha”, Oct 3, 2016 | Crowell et al. (2018) | https://github.com/aleccrowell/LIMBR |
| eJTK_cycle, v. Aug 3, 2016 | Hutchison et al. (2015); Hutchison et al. (2018) | https://github.com/alanlhutchison/empirical-JTK_CYCLE-with-asymmetry |
| FunCat v. 8/12/2010, within FungiFun v. 0.5 | Priebe et al. (2011) | https://elbe.hki-jena.de/fungifun/ |
| PSEA, v. 1.1 | Zhang et al. (2016) | https://github.com/ranafi/PSEA |
| Pathway Tools software within the BioCyc Pathway/ Genome Database (PGDB), v. 21.5 | Caspi et al. (2016) | https://biocyc.org/ |
| Toolset Image Analysis Larrondo’s Lab v. 1.0, within FIJI (ImageJ) v. 2.0.0 | Jen Hurley (Larrondo et al., 2012; Schindelin et al., 2012) | https://fiji.sc/ |