

Abstract
Purpose
Transient global ischemia arising in human due to cardiac arrest causes selective, delayed neuronal death in hippocampal CA1 and cognitive impairment. Growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) is a wellknown molecule in both DNA damage-related pathogenesis and therapies. Emerging evidence suggests that Gadd45b is an anti-apoptotic factor in nonneuronal cells and is an intrinsic neuroprotective molecule in neurons. However, the mechanism of Gadd45b pathway is not fully examined in neurodegeneration associated with global ischemia.
Methods
Rats were subjected to transient global ischemia by the 4-vessel occlusion or sham operation. The animals were sacrificed at 24 hours, 48 hours, and 7 days after ischemia. The hippocampal CA1 was microdissected and processed to examine mRNA and protein level. To assess neuronal death, tissue sections were cut and processed for Fluoro-Jade and Nissl staining.
Results
Here we show that ischemic insults increase abundance of Gadd45b and brain-derived neurotrophic factor, a known target of Gadd45 mediated demethylation, in selectively-vulnerable hippocampal CA1 neurons. We further show that knockdown of Gadd45b increases abundance of a pro-apoptotic Bcl-2 family member Bax while decreasing the antiapoptotic protein Bcl-2, which together promote neuronal death.
Conclusions
These findings document a protective role of Gadd45b against neuronal insults associated with global ischemia and identify Gadd45b as a potential therapeutic target for the amelioration of hippocampal neurodegeneration.
Keywords: Global ischemia, Gadd45b, Mitochondrial dysfunction, Brain-derived neurotrophic factor, Neuroprotection
• HIGHLIGHTS
- Global ischemia induces DNA damage and apoptotic neuronal death in hippocampal CA1.
- Gadd45b plays a role in neuroprotection by regulating Bcl-2, Bax, and mitochondrial function.
- Global ischemia increases abundance of BDNF, a target of Gadd45b-mediated DNA demethylation in hippocampal CA1 neurons.
INTRODUCTION
Ischemic stroke is the leading cause of death worldwide and a major cause of severe disability among adults. Surviving patients demonstrate a remarkable decrease in brain plasticity and restoration of normal cognitive function [1]. Despite several therapeutics that have been undertaken, most Phase III clinical trials conducted to date have failed [2]. It is thus critical to investigate molecular mechanisms to identify potential therapeutic targets and to bring them into the translational arena with the goal of developing novel treatment strategies for neuroprotection. Global ischemia which is a severe form of stroke arises as a consequence of cardiac arrest, cardiac surgery, profuse bleeding, near-drowning and carbon monoxide poisoning [3-6]. Global ischemia in humans or induced experimentally in animals causes a selective, delayed death of hippocampal CA1 pyramidal neurons and severe cognitive deficits [4-7]. Early events include mitochondrial dysfunction, cytochrome c release, alteration of Bcl-2 family proteins and caspase activation [8-11].
Four-vessel occlusion (4-VO) is a well-established animal model for studying of global ischemia. Although the 4-VO model induced neuronal death in the hippocampal CA1 at 7 days after occlusion, the onset of histologically-detectable cell death does not manifest until 48 hours [4-6,12]. The substantial delay between neuronal insult and cell death is consistent with a role for alterations in gene expression. This affords a defined window of time to examine molecular and cellular alterations which would allow us to understand mechanisms underlying the pathophysiology of global ischemic stroke.
Growth arrest and DNA-damage inducible protein 45 is a highly homologous family of proteins consisting of growth arrest and DNA-damage-inducible protein 45 alpha (Gadd45a), beta (Gadd45b), and gamma (Gadd45g) Gadd45 plays an essential role in various cellular functions including DNA repair, cell survival, apoptosis and DNA methylation [13-15]. In this study, we investigated the role of Gadd45b in global ischemiainduced neuronal death. Brain-derived neurotrophic factor (BDNF) has been shown to be a target of Gadd45b-induced DNA demethylation in neural activity-induced proliferation of neural progenitors and dendritic growth of newborn neurons in the adult hippocampus [16]. This study showed that Gadd45b binds the bdnf exon IX promoter and increases DNA demethylation which in turn, activates BDNF gene expression in response to electroconvulsive treatment in wild type but not in Gadd45b knockout mice. Subsequent studies have shown that Gadd45b regulates BDNF expression in neurodegeneration induced by focal ischemia [13,17].
The present study was undertaken to decipher the molecular pathways that mediate the neuroprotective functions of Gadd45b against ischemic insults. Here we show that Gadd45b is increased in hippocampal CA1 neurons transiently at 24 and 48 hours after global ischemia. The increase in Gadd45b coincides with an increase in its target BDNF, indicating induction of an intrinsic neuroprotective factor in neurons destine to die at early time points after ischemia, prior to the onset of histologically detected neuronal death. We further show that knockdown of Gadd45b activates a pro-apoptotic protein Bax while decreasing the antiapoptotic protein Bcl-2, which together accelerate neuronal death. These findings demonstrate a causal relation between increase in Gadd45b and neuroprotection in neuronal death associated with global ischemia. Taken together, this study provides new insights into the molecular mechanism of Gadd45b as a protective effector underlying the pathophysiology of global ischemia.
MATERIALS AND METHODS
Animal Model
Rats were subjected to transient global ischemia by means of the 4-VO method as previous described [12,18]. For sham surgery, animals were subjected to the same anesthesia and surgical conditions, except that the carotid arteries were not occluded, and the animals were killed at 24 hours, 48 hours, and 7 days. Body temperature was maintained at 37.5°C±0.5°C by external warming until recovery from anesthesia. Animals that failed to show complete loss of the righting reflex and dilation of the pupils 2 minutes after occlusion were initiated and the rare animals that exhibited behavioral manifestations such as abnormal vocalization when handled, generalized convulsions, loss of >20% body weight by 3–7 days, or hypoactivity were excluded from the study. All rats were housed in standard Plexiglas cages under a 12 hours light/dark cycle. Food and water were provided ad libitum. Animal care and handling procedures were approved by the Albert Einstein Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines.
Tissue Preparation, Immunofluorescence, Confocal Imaging
All rats (sham, 24 hours, 48 hours, and 7 days after 4-VO, n=4 for each) were killed, and brains were collected for immunofluorescence. Samples were prepared as 18-μm-thick sections. Sections were submerged in 4% para-formaldehyde for 30 minutes, and antigen retrieval procedure was performed as previously described [19]. Brain sections were stained with Gadd45b, phospho H2A.X, cleaved caspase-3 specific antibodies (Table 1). Images were acquired on a Zeiss LSM 880 confocal system using a z-track configuration.
Table 1.
Antibodies information
| Name | Dilution | Company | Catalog number |
|---|---|---|---|
| Gadd45b | 1:500 | Invitrogen | PA5-43160 |
| Phospho-H2A.X | 1:500 | Millipore | 05-636-I |
| Cleaved Caspase-3 | 1:250 | Cell Signaling | 9664 |
| Cdk1 | 1:5,000 | Abcam | ab18 |
| BDNF | 1:1,000 | Abcam | ab88901 |
| Cytochrome c | 1:1,000 | BD bioscience | 556433 |
| Bcl-2 | 1:2,000 | BD bioscience | 610538 |
| Bax | 1:2,000 | Santacruz | sc-7480 |
| Cox IV | 1:2,000 | Abcam | ab16056 |
| β-actin | 1:10,000 | Sigma | A5316 |
Histology and Fluoro-Jade staining
Neuronal cell death was assessed by histological examination from animals killed at 7 days after sham or 4-VO surgery. Animals were perfused using 0.9% saline with heparin followed by ice-cold 4% paraformaldehyde in 0.1M phosphate buffered saline (pH, 7.4). Coronal sections (20 μm) were cut at the level of the dorsal hippocampus with a cryostat and processed for staining with cresyl violet or Fluoro-Jade C. All analysis was conducted with blind test.
Primary Neuron Culture
Primary cortical cultures were prepared from E18 rat embryo tissue. Cortices were dissected and dissociated with trypsin. Cells were plated at a density 8,000 cells/cm2 on poly-L lysine coated plates and maintained in Neurobasal medium with B-27 supplement, and GlutaMAX.
Immunoblotting
Sham or 4-VO treated hippocampi were separated from the brains and CA1 subfield was rapidly microdissected. Each specimen was homogenized in lysis buffer as described in [18]. Equal concentrations of protein (30–50 μg) were separated on 4%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, transferred to nitrocellulose membranes and incubated with GADD45b, cleaved caspase-3, cycline dependent kinase 1 (CDK1), phospho-H2A.X, Bcl-2, Bax, cytochrome c, CoxIV, BDNF, and β-actin specific antibodies followed by chemiluminescent detection (Table 1). Band densities were normalized to β-actin or tubulin. Mean band densities for samples from experimental animals were normalized to the corresponding samples from control neurons or sham animals.
Gadd45b-shRNA and Neuronal Transfection
Lentiviral vectors encoding Gadd45b RNAi were generated and evaluated for specificity and efficacy of Gadd45b knockdown. In this study, we used 2 RNAi antisense sequences targeted to Gadd45b: shRNA-Gadd45b #1 (TCAGGGTATCAGGCATGCAAAGGCTGGCG), and shRNA-Gadd45b #2 (CCTGGTCACGAACTGTCATACAGATTCCT). A nontargeting (NT) control shRNA (GGGTGAACTCACGTCAGAA) was used for negative control. All viral vectors were purchased from Applied Biological Materials, Richmond, BC, Canada. These lentivectors are based on the same plasmid backbone (piLenti-siRNAGFP). HEK-293T cells were transfected with lentiviral transfer construct piLenti-siRNA-GFP and packaging constructs pMDLg-pRRE and pRSV-REV and pMD2.G envelope protein construct by means of calcium phosphate method.
LDH Cytotoxicity Measurement
Cell death was quantified by measuring lactate dehydrogenase (LDH) activity released into the culture medium from damaged cells. LDH was measured using a LDH-Cytotoxicity Assay Kit II (Abcam, Cambridge, MA, USA) according to the manufacturer’s instructions. LDH concentrations were normalized to the mean concentration of cells exposed to 400μM H2O2 (100%, full kill) after subtracting the mean value of sham-washed control cells (0%) [20].
Quantitative RT-PCR
RNA was extracted using PureLink microto-midi total RNA purification system (Invitrogen, Carlsbad, CA, USA) from hippocampal CA1 region. RNA was reverse-transcribed to cDNA with SuperScript III Reverse Transcriptase (Invitrogen). Quantitative real time-polymerase chain reaction (qRT-PCR) for the detection of Gadd45b, Wnt1, Wnt3a, Wnt4, Wnt5a, Wnt7a, Wnt7b, Wnt9a, Bdnf, and Gapdh as a housekeeping gene of mRNA was performed using the power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) (Table 2).
Table 2.
Primer sequence list for real time quantitative polymerase chain reaction
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| Gadd45b | GTTCTGCTGCGACAATGACA | TTGGCTTTTCCAGGAATCTG |
| Wnt1 | TTCTGCTACGTTGCTACTGGCACT | CATTTGCACTCTTGGCGCATCTCA |
| Wnt3a | GGTGGTAGAGAAACACCGAGA | CAGCTGACATAACAGCACCAG |
| Wnt4 | GAACCGGCGCTGGAACT | GGTCCCTTGTGTCACCACCTT |
| Wnt5a | GCGTGGCTATGACCAGTTTAAGA | TTGACATAGCAGCACCAGTGAAA |
| Wnt7a | GCCCACCTTTCTGAAGATCAAG | TGGGTCCTCTTCACAGTAATTGG |
| Wnt7b | CAAGGAGAAGTACAACGCAGC | CACTTGACGAAGCAACACCAG |
| Wnt9a | GCTTTCCTCTACGCCATCTCT | CACGAGGTTGTTGTGGAAGTC |
| Bdnf | GCCTTTGGAGCCTCCTCT | CTGTCACACACGCTCAGCTC |
Statistical Analysis
Data are expressed as mean±standard error of the mean. We conducted the data analysis using Student t-test (2-tails) for comparative assessment. Values were considered significant if P≤0.05.
RESULTS
Neuronal Death Is Focused on Pyramidal Neurons in Hippocampal CA1 in Global Ischemia
We subjected adult rats to global ischemia or sham operation (Fig. 1A) and validated the pattern of neuronal death. Global ischemia induced a loss of neurons and a significant number of damaged cells with pyknotic nuclei specifically in the hippocampal CA1, but not CA3 or dentate gyrus as assessed by Nissl staining (Fig. 1B, C). We next performed Fluoro-Jade (FJ) staining, an indicator of compromised neurons, to assess neuronal death. Visualization of FJ positive neurons were observed only in the hippocampal CA1 in ischemia, not in sham animals (Fig. 1B, C). Thus, these results reveal that the 4-VO model in rat induces selective cell death that is largely restricted to the pyramidal neurons of the hippocampal CA1.
Fig. 1.
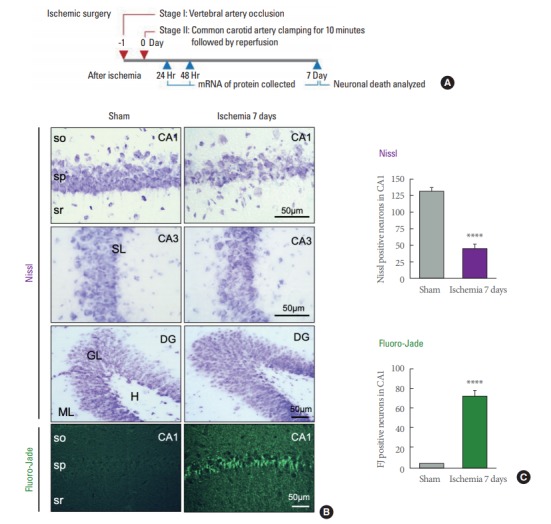
Histological evidence showing 4-vessel occlusion (4-VO) model induces neuronal death in the hippocampal CA1 by 7 days. (A) Time line of ischemic surgery and molecular assay. (B) Representative images of brain sections stained with Nissl and Fluoro-Jade from animals subjected to global ischemia or sham operation 7 days after surgery. (C) Summary data showing the number of living CA1 pyramidal neurons as assessed by Nissl staining (upper), and injured CA1 pyramidal neurons by Fluoro-Jade staining (lower) (n=7 for sham, 4 for ischemic animals). so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; ML, molecular layer; GL, granular layer; H, hilus; SL, stratum lacunosum-moleculare. ****P<0.0001. Scale bar is 50 μm.
Apoptotic Neuronal Cell Death and DNA Damage Arises After Ischemia
To identify molecular mechanisms underlying delayed, selective neuronal cell death after global ischemia, we examined the protein abundance of the pro-apoptotic marker cleaved-caspase 3 and phospho-H2A.X, a maker for a DNA damage. Immunoblot analysis showed that cleaved-caspase 3 was increased in hippocampal CA1 at 24 and 48 hours after ischemia which are the time points we assessed (Fig. 2A left). This finding is in corroboration of the previous studies [8,21]. In addition, phospho-H2A.X expression was increased at 24 and 48 hours after ischemia (Fig. 2A right). Immunofluorescence labeling revealed that cleaved-caspase3 and phospho-H2A.X are increased at 24 and 48 hours after ischemia in hippocampal CA1 (Fig. 2B). Consistent with this, a major cell cycle G2/M regulator, the CDK1 was time dependently decreased (Data is not shown). Taken together, these data indicate that global ischemia triggers DNA-damage, which induces apoptosis in hippocampal CA1 neurons.
Fig. 2.

Apoptosis and DNA damage induced by ischemic insults. (A) Representative immunoblot images of apoptotic maker, cleavedcaspase-3 and DNA damage marker, phospho-H2A.X. Quantifications were normalized by Tubulin expression (n=7 for sham, 4 for ischemia 24- and 48-hour animals). Values represent mean±standard error of the mean (*P<0.05. **P<0.01). (B) Representative confocal imaging showing, upper, cleaved-caspase-3 (∆Cas3, red) and lower, phospho-H2A.X (red) and counterstained with 4’-6-diamidino-2-phenylindole (DAPI) (blue). Scale bar is 20 µm.
Activation of Bax Leads to Mitochondrial Dysfunction and Cytochrome c Release in Ischemia
Insufficient oxygen triggers the mitochondrial dysfunction in global ischemia [8-11]. For more detailed analysis, we isolated mitochondrial and cytosolic fractions of hippocampal CA1 from both sham and ischemia. We found cytochrome c is reduced in the mitochondrial fraction and increased in cytosolic fraction at 24 hours after ischemia (Fig. 3A). These findings suggest that compromise of the integrity of the outer mitochondrial membrane promotes cytochrome c release from the mitochondria to cytosol in response to ischemic insults, an initial step in the apoptotic pathway. Next, we examined whether apoptosis related factors such as Bcl-2 and Bax are involved. Anti-apoptotic factor Bcl-2 was reduced, and pro-apoptotic factor Bax was increased at 24 hours after ischemia (Fig. 3B, C). These data strongly suggest that global ischemia induced oxidative stress triggers a reduction in Bcl-2 and increase in Bax abundance, indicative of the progression of apoptotic cell death.
Fig. 3.

Mitochondrial and cytosolic cytochorome c, and anti- or pro-apoptotic protein in ischemia. (A) Cytochrome c abundance in mitochondrial (left) and cytosolic fraction (right) from hippocampal CA1 at 24 hours after ischemia. (B) Antiapoptotic protein, Bcl2. (C) Pro-apoptotic protein, Bax expression levels in whole cell lysate form hippocampal CA1 at 24 hours after ischemia. Quantifications were normalized by Cox IV for mitochondrial fraction and β-actin for cytosolic fraction and whole cell lysate expression (n=4 for each group). *P<0.05. ***P<0.001.
Wnt Family and Gadd45b are Upregulated in Ischemia-Induced Neuronal Death
The Wnt signaling pathway is involved in growth of neural progenitors during neuronal repair [22]. Gadd45b has been known to be involved in the Wnt signaling cascade by regulating Dickkopf-1, a negative modulator of Wnt signaling [23]. Moreover, Dickkopf-1 is increased in hippocampal CA1 neurons and is required for the development of ischemic neuronal damage [24]. In this study, we examined the gene expression of several Wnt family members during different time periods after ischemia using qRT-PCR. Wnt1, 5a and 7b are all significantly increased at 24 hours after ischemia (Fig. 4A). We hypothesized that an increase in Wnt signaling would result in a concomitant upregulation of Gadd45b. Interestingly, gene expression of Gadd45b was increased at 24 hours after ischemia (Fig. 4B), moreover, Gadd45b protein expression was increased both at 24 and 48 hours after ischemia (Fig. 4C). The images of Gadd45b immunostaining in hippocampal CA1 supported the protein level pattern at 24 and 48 hours after ischemia (Fig. 4D). These data show that a set of Wnt family genes and Gadd45b expression are increased in response to ischemic insults possibly to act as a neuroprotective factor against ischemic insults.
Fig. 4.

Changes of neuroprotective molecules after global ischemia. (A) Wnt family mRNA expression showing time-dependent alterations in ischemia hippocampal CA1 regions as determined by quantitative real time-polymerase chain reaction (qRT-PCR). (B) Growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) mRNA expression quantified by qRT-PCR. (C) Representative immunoblot images of Gadd45b in hippocampal CA1 with complementary quantifications normalized by tubulin (n=7 for sham, 4 for ischemia 24- and 48-hour animals). Values represent mean ±standard error of the mean (*P <0.05. **P <0.01. ***P <0.001. ****P<0.0001). (D) Confocal imaging depicting Gadd45b (red), counterstained with 4’-6-diamidino-2-phenylindole (DAPI) (blue). Scale bar is 20 µm.
Knockdown of Gadd45b Accelerates Neuronal Death
Gadd45b is thought to play an important role in apoptosis signaling. We designed 2 different shRNA constructs directed against Gadd45b (see MATERIALS AND METHODS Section). First, we validated knockdown efficiency of Gadd45b-shRNAs by immunoblot analysis which showed Gadd45b abundance was decreased about 40% by Gadd45b-shRNA #1 and 55% by shRNA #2 (Fig. 5A). Using these 2 Gadd45b shRNAs, we subjected cortical neurons to H2O2 (50μM) or vehicle and assessed cell death as a function of LDH activity in the medium. LDH assay is a well-established method to monitor cell damages [20]. Interestingly, LDH release was markedly increased in rats treated with sh-Gadd45b compared to that of rats treated with NT shRNA in a H2O2-induced model of neuronal death (Fig. 5B) indicating that knockdown of Gadd45b accelerated neuronal death. Moreover, protein levels of apoptotic factors involved in mitochondrial dysfunction were changed in sh-Gadd45b-treated group. The anti-apoptotic factor Bcl-2 was further decreased, and the pro-apoptotic factor Bax was further increased in the sh-Gadd45b-treated group compare to sh-NT treated group in H2O2-induced model of neuronal death. These results indicate that knockdown of Gadd45b is critical to the neuronal death associated with mitochondrial dysfunction. Collectively these findings strongly suggest that Gadd45b acts as a protective effector in neurons against neural insults.
Fig. 5.

Knockdown of growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) accelerates neuronal cell death and mitochondrial dysfunction. (A) Validation of knockdown efficiency for Gadd45b shRNA in cortical neurons by immunoblotting. (B) Summary data of lactate dehydrogenase (LDH) activity assay for cortical neurons treated with 50µM H2O2 (2 hours) in the presence of nontargeting (NT) or Gadd45b shRNA. LDH activity values are normalized to the value of cells exposed to 400μM H2O2 (100%, FK; full kill) after subtracting the value of control cells (0%). (C) Representative immunoblot images showing Bcl-2. (D) Bax expression level in cortical neurons treated with 50µM H2O2 (2 hours) in the presence of NT or Gadd45b shRNA. (E) Quantitative real timepolymerase chain reaction data showing brain-derived neurotrophic factor (BDNF) mRNA expression at indicated time periods after ischemia. (F) Representative immunoblot images Gadd45b in hippocampal CA1 and summary data. (n=7 for sham, 4 for ischemia 24- and 48-hour animals). Values represent mean±standard error of themean. *P<0.05. **P<0.01. ***P<0.001.
Global Ischemia Upregulates Gadd45b Downstream Target BDNF
The findings thus far have indicated that Gadd45b is upregulated in insulted hippocampal neurons and that the increase in Gadd45b plays an important role in for neuroprotection, but do not address whether Gadd45b downstream mechanisms critical to neuroprotection can be activated. To address this issue, we examined the abundance of BDNF, a known target of Gadd45b-mediated DNA demethylation and an anti-apoptotic protein. Both mRNA and protein levels of BDNF are significantly increased at 24 and 48 hours after ischemia (Fig. 5E, F) while BDNF mRNA level is decreased at day 7 after ischemia (Fig. 5E). These results demonstrate that upregulation of BDNF and are consistent with the concept that Gadd45b affords a neuroprotective role at the times of after ischemic insults.
DISCUSSION
Global cerebral ischemia is characterized by the critical reduction of cerebral blood flow to the brain. Global ischemia can be modeled in rodents by several different approaches. In this study, we used the 4-VO method, leading to selective neuronal cell death in hippocampal CA1 and cognitive dysfunction [4-7]. In this study, we examined gene expression and protein abundance over time of neuroprotective regulators after ischemic insults. Our comparative analyses of the time-course expression profiles suggest a defined window of opportunity to not only study molecular changes resulting from ischemia, but also potential therapeutic targets prior to irreversible neuronal cell death.
Cell death facilitated by DNA-damaged plays a critical role in ischemia. Numerous studies have shown that DNA damage in neurons is a prominent event during ischemia [25]. After ischemia followed by reperfusion, we show that DNA damage occurred at 24 and 48 hours, and correlated with the onset of apoptosis in hippocampal CA1 neurons; A majority of dying neurons leads to increase expression of cleaved-caspase-3 and phospho-H2A.X after brain injury (Fig. 2). In addition, neuronal injury of ischemia leads to impairment of mitochondrial function which plays an essential role in normal statement of neurons through the calcium homeostasis [8-11]. Mitochondrial dysfunction results in loss of cytochrome c from mitochondria and this process impairs cellular oxygen level and activates caspase cascade [8-10]. In our study, we showed cytochrome c release and abnormal changes of apoptotic proteins involved in mitochondrial function after global ischemia (Fig. 3). This is significant in that our findings suggest a novel target not only for global ischemia, but also for other neurological disorders and diseases in which mitochondrial function is impaired.
Gadd45 family members are involved in regulation of stress related pathways such as p53, TGFb, MAPKs/NF-kB, and Wnt signaling [26-28]. It also has been reported that the Wnt/β-catenin signaling pathway is involved in neural survival and function in neurodegeneration associated with ischemic stroke [29-31]. Our results in global ischemia showed that expression of Wnt1, 5a, 7b genes are significantly increased at 24 and 48 hours after neural insults, indicating that regulation of Wnt signaling is probably involved in the pathogenesis of neuronal cell death after global ischemia. While we only showed gene expression patterns of a subset of Wnt family members, further studies are required to elucidate the interaction with Wnt signaling in ischemia-induced neuronal death and examine how Gadd45b is involved in or regulate the Wnt signaling pathway.
The most vital mechanisms that decide between neuroprotective and apoptosis remain a point of controversy. In this study, we hypothesized that neuroprotective molecules could promote the rate of neural survival after ischemic damage. The Gadd45 family had been previously identified as stress-response genes in physiological condition [27,28]. Among this family, Gadd45b had been implicated as an anti-apoptotic factor [32-34]. BDNF is well known neuroprotective factor. BDNF inhibits caspase-3 cleavage and activation following hypoxic-ischemic injury [35]. Here we showed that ischemia significantly, but transiently increased Gadd45b mRNA at 24 hours and protein at 24 and 48 hours which are early time points of neural insults, and declined by 7 days after ischemia in hippocampal CA1 neurons. We further showed that knockdown of Gadd45b decreases anti-apoptotic factor Bcl-2 and increases pro-apoptotic factor Bax abundance which together accelerate H2O2-induced neuronal death in primary cultured neurons suggesting a causal relation between Gadd45b and neuroprotection. Our findings suggest Gadd45b as a neuroprotective effector against mitochondrial dysfunction by regulating Bcl-2 and Bax. Consistent with increase in Gadd45b, neuroprotective effector BDNF gene and protein levels are increased at 24 and 48 hours after ischemia (Fig. 5). This is significant in that BDNF is implicated as a DNA demethylation target of Gadd45b. Gadd45b removes methylation in the promoter of bdnf exon IX which promotes BDNF gene expression [16]. Consistent with the neuroprotective role of BDNF, Gadd45b thus attenuates apoptosis by increasing expression of BDNF in the cortex in a model of focal ischemia [13]. Moreover, Gadd45b shRNAs downregulated BDNF expression after focal ischemia [13]. Thus, the two models of ischemic stroke, global and focal ischemia, exert similar effect on Gadd45b and BDNF expression, despite the fact that they induce cell death via different mechanisms and target different populations of neurons.
We have begun to screen for promising targets in understanding the neuroprotective effects in our ischemic model. There is an abundance of studies describing the molecular mechanisms of antiapoptotic processes after brain injury. Notably, this research has elucidated the profiling of gene expression and protein level in ischemia model. We demonstrate that Gadd45b and BDNF have a key neuroprotective effect after brain injury. To better understanding the protective effect against ischemic insults, overexpression of Gadd45b studies will be required to investigate the mechanism of the protective function.
In summary, we suggest the time-window and key contributors, Gadd45b and BDNF, for neural protection in global ischemia by regulating mitochondrial function (Fig. 6). These findings represent a previously unappreciated role for Gadd45b in neuronal death associated pathways with global ischemia and identify a novel therapeutic target for amelioration of the neurodegeneration.
Fig. 6.

Model depicting a hypothetical mechanism by which growth arrest and DNA-damage-inducible protein 45 beta (Gadd45b) protects neurons against ischemic insults and activates its DNA methylation target brain-derived neurotrophic factor (BDNF). To compensate neuronal damages, global ischemic insults promote activation of Gadd45b and its binding to the promoter of BDNF exon IX. This, in turn, removes DNA methylation of cytosine residues at its promoter, allowing for activation of BDNF expression. Gadd45b can promote neuroprotective mechanisms in which indirectly inhibits Bax activation while increases Bcl-2 abundance. In consequence, the mitochondrial function is restored and it allows to block caspase activation.
Footnotes
Fund/Grant Support
This work was supported by NIH NS100047, AHA Scientist Development Grant 16SDG31500001, NARSAD Young Investigator Grant 25369 and LB692 Nebraska Tobacco Settlement Biomedical Research Development Funds to JYH; NIH NS046742, HD083828, NS100047 and the generous grant from the F.M. Kirby Foundation to RSZ. RSZ is the F.M. Kirby Chair in Neural Repair and Protection.
Research Ethics
Animal care and handling procedures were approved by the Albert Einstein Institutional Animal Care and Use Committee (IACUC) in accordance with National Institutes of Health guidelines.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: CHC, HRB, RSZ, JYH
·Study concept and design: CHC, JYH
·Acquisition of data: CHC, HRB, TJ
·Drafting of the manuscript: CHC, HRB, JYH
·Critical revision of the manuscript for important intellectual content: CJ, ARC, TJ, RSZ, JY
·Obtained funding: RSZ, JYH
·Administrative, technical, or material support: RSZ, JYH
·Study supervision: JYH
REFERENCES
- 1.Di Filippo M, Tozzi A, Costa C, Belcastro V, Michela T, Picconi B, et al. Plasticity and repair in the post-ischemic brain. Neuropharmacology. 2008;55:353–62. doi: 10.1016/j.neuropharm.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Xu SY, Pan SY. The failure of animal models of neuroprotection in acute ischemic stroke to translate to clinical efficacy. Med Sci Monit Basic Res. 2013;19:37–45. doi: 10.12659/MSMBR.883750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann JT, Cohan CH, Dave KR, Wright CB, Perez-Pinzon MA. Global cerebral ischemia: synaptic and cognitive dysfunction. Curr Drug Targets. 2013;14:20–35. doi: 10.2174/138945013804806514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103–42. doi: 10.1016/s0301-0082(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 5.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–98. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Ofengein D, Shi Y, Zhang F, Hwang JY, Chen J, et al. Molecular and cellular mechanisms of ischemia-induced neuronal death. In: Grotta JC, Albers GW, Broderik JP, Kasner SE, Lo EH, Mendelow AD, et al., editors. Stroke: pathophysiology, diagnosis and management. 6th ed. Philadelphia (PA): Churchill Livingstone Elsevier; 2016. pp. 60–79. e5. [Google Scholar]
- 7.Bacigaluppi M, Comi G, Hermann DM. Animal models of ischemic stroke. Part two: modeling cerebral ischemia. Open Neurol J. 2010;4:34–8. doi: 10.2174/1874205X01004020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–9. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:4892–7. doi: 10.1073/pnas.0800628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, et al. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci. 2012;15:574–80. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H, et al. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci. 2006;26:6851–62. doi: 10.1523/JNEUROSCI.5444-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang JY, Kaneko N, Noh KM, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol. 2014;426:3454–66. doi: 10.1016/j.jmb.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B, Zhang YH, Jiang Y, Li LL, Chen Q, He GQ, et al. Gadd45b is a Novel mediator of neuronal apoptosis in ischemic stroke. Int J Biol Sci. 2015;11:353–60. doi: 10.7150/ijbs.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, DiStefano PS, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 15.Engelmann A, Speidel D, Bornkamm GW, Deppert W, Stocking C. Gadd45 beta is a pro-survival factor associated with stress-resistant tumors. Oncogene. 2008;27:1429–38. doi: 10.1038/sj.onc.1210772. [DOI] [PubMed] [Google Scholar]
- 16.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Li LL, Tan XD, Zhang YH, Jiang Y, He GQ, et al. Gadd45b mediates axonal plasticity and subsequent functional recovery after experimental stroke in rats. Mol Neurobiol. 2015;52:1245–56. doi: 10.1007/s12035-014-8909-0. [DOI] [PubMed] [Google Scholar]
- 18.Hwang JY, Gertner M, Pontarelli F, Court-Vazquez B, Bennett MV, Ofengeim D, et al. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017;24:317–29. doi: 10.1038/cdd.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussaini SM, Jun H, Cho CH, Kim HJ, Kim WR, Jang MH. Heatinduced antigen retrieval: An effective method to detect and identify progenitor cell types during adult hippocampal neurogenesis. J Vis Exp. 2013;78:e50769. doi: 10.3791/50769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 21.Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, et al. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–24. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bond AM, Ming GL, Song H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell. 2015;17:385–95. doi: 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, et al. DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene. 2004;23:8520–6. doi: 10.1038/sj.onc.1207892. [DOI] [PubMed] [Google Scholar]
- 24.Cappuccio I, Calderone A, Busceti CL, Biagioni F, Pontarelli F, Bruno V, et al. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J Neurosci. 2005;25:2647–57. doi: 10.1523/JNEUROSCI.5230-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu W, et al. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology. 2018;134(Pt B):208–17. doi: 10.1016/j.neuropharm.2017.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moskalev AA, Smit-McBride Z, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, et al. Gadd45 proteins: relevance to aging, longevity and age-related pathologies. Ageing Res Rev. 2012;11:51–66. doi: 10.1016/j.arr.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvador JM, Brown-Clay JD, Fornace AJ., Jr Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol. 2013;793:1–19. doi: 10.1007/978-1-4614-8289-5_1. [DOI] [PubMed] [Google Scholar]
- 28.Liebermann DA, Hoffman B. Gadd45 in stress signaling. J Mol Signal. 2008;3:15. doi: 10.1186/1750-2187-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libro R, Bramanti P, Mazzon E. The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sci. 2016;158:78–88. doi: 10.1016/j.lfs.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Caraci F, Busceti C, Biagioni F, Aronica E, Mastroiacovo F, Cappuccio I, et al. The Wnt antagonist, Dickkopf-1, as a target for the treatment of neurodegenerative disorders. Neurochem Res. 2008;33:2401–6. doi: 10.1007/s11064-008-9710-0. [DOI] [PubMed] [Google Scholar]
- 31.Sultan FA, Sweatt JD. The role of the Gadd45 family in the nervous system: a focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv Exp Med Biol. 2013;793:81–119. doi: 10.1007/978-1-4614-8289-5_6. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Suyeoka G, Papa S, Franzoso G, Neufeld AH. Growth arrest and DNA damage protein 45b (Gadd45b) protects retinal ganglion cells from injuries. Neurobiol Dis. 2009;33:104–10. doi: 10.1016/j.nbd.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta M, Gupta SK, Hoffman B, Liebermann DA. Gadd45a and Gadd45b protect hematopoietic cells from UV-induced apoptosis via distinct signaling pathways, including p38 activation and JNK inhibition. J Biol Chem. 2006;281:17552–8. doi: 10.1074/jbc.M600950200. [DOI] [PubMed] [Google Scholar]
- 34.Gupta SK, Gupta M, Hoffman B, Liebermann DA. Hematopoietic cells from gadd45a-deficient and gadd45b-deficient mice exhibit impaired stress responses to acute stimulation with cytokines, myeloablation and inflammation. Oncogene. 2006;25:5537–46. doi: 10.1038/sj.onc.1209555. [DOI] [PubMed] [Google Scholar]
- 35.Chen A, Xiong LJ, Tong Y, Mao M. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed Rep. 2013;1:167–76. doi: 10.3892/br.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]