

Abstract
Purpose
This study aimed to investigate the effects of single-bout exercise on mitochondrial function, dynamics (fusion, fission), and mitophagy in cardiac and skeletal muscles.
Methods
Fischer 344 rats (4 months old) were randomly divided into the control (CON) or acute exercise (EX) group (n=10 each). The rats performed a single bout of treadmill exercise for 60 minutes. Mitochondrial function (e.g., O2 respiration, H2O2 emission, Ca2+ retention capacity), mitochondrial fusion (e.g., Mfn1, Mfn2, Opa1), mitochondrial fission (e.g., Drp1, Fis1), and mitophagy (e.g., Parkin, Pink1, LC3II, Bnip3) were measured in permeabilized cardiac (e.g., left ventricle) and skeletal (e.g., soleus, white gastrocnemius) muscles.
Results
Mitochondrial O2 respiration and Ca2+ retention capacity were significantly increased in all tissues of the EX group compared with the CON group. Mitochondrial H2O2 emissions showed tissue-specific results; the emissions showed no significant differences in the left ventricle or soleus (type I fibers) but was significantly increased in the white gastrocnemius (type II fibers) after acute exercise. Mitochondrial fusion and fission were not altered in any tissues of the EX group. Mitophagy showed tissue-specific differences: It was not changed in the left ventricle or white gastrocnemius, whereas Parkin and LC3II were significantly elevated in the soleus muscle.
Conclusions
A single bout of aerobic exercise may improve mitochondrial function (e.g., O2 respiration and Ca2+ retention capacity) in the heart and skeletal muscles without changes in mitochondrial dynamics or mitophagy.
Keywords: Acute exercise, Mitochondrial function, Dynamics, Heart, Skeletal muscle
• HIGHLIGHTS
- A single bout of aerobic exercise improved mitochondrial function (e.g., O2 respiration and Ca2+ retention capacity) in the heart and skeletal muscles.
- A single bout of aerobic exercise did not alter mitochondrial dynamics (fusion, fission) in the heart and skeletal muscles.
- A single bout of aerobic exercise did not alter mitophagy in the heart.
INTRODUCTION
Mitochondria play an important role in cellular respiration, oxidative stress, and calcium homeostasis. Previous studies suggested that mitochondrial dysfunction is associated with diverse diseases such as obesity, diabetes, and cancer, demonstrating that the underlying mechanisms are characterized by a deficiency in mitochondrial O2 respiration and a decrease in mitochondrial ATP production. Indeed, Petersen et al. [1] reported that insulin resistance results from reduced activity of oxidative phosphorylation complexes and decreased mitochondrial ATP production. Several studies reported that increased mitochondrial reactive oxygen species (ROS) are associated with the pathogenesis of the above-mentioned diseases. For example, Montaigne et al. [2] reported that elevated myocardial oxidative stress is associated with type 2 diabetes regardless of weight status. In addition, a decreased mitochondrial Ca2+ retention capacity is linked with mitochondrial permeability transition pore (mPTP) opening, which contributes to cellular apoptosis. Indeed, Anderson et al. [3] showed that atrial tissues of type 2 diabetic humans had increased sensitivity to mPTP opening resulting from Ca2+ stimulation and an abundance of active caspase 9.
Mitochondria are dynamic organelles that undergo cycles of structure and morphology, referred to as mitochondrial dynamics (fusion, fission) [4]. The dynamic interaction between mitochondrial fusion and fission controls cell survival, growth, and division as well as mitochondrial distribution during differentiation [4]. Several studies have reported the relationship between mitochondrial dynamics and insulin activity. Indeed, Weng et al. [5] showed that fusion-related molecules increased glucose uptake and insulin signaling, whereas fission-related molecules decreased glucose uptake. Mitochondria also go through a step of autophagy known as mitophagy (mitochondrial autophagy), the removal of damaged or abnormal mitochondria. Greene et al. [6] showed that mitophagy proteins (e.g., Bnip3, p62) were reduced in the skeletal muscle by obesity, suggesting that damaged or abnormal mitochondria in obesity are not well removed.
Exercise training has a protective effect on the incidence of diverse diseases, and mitochondrial function, dynamics, and mitophagy may be potential mechanisms linked to various diseases. For these reasons, previous studies established the effects of exercise training on various diseases. Indeed, many studies have investigated the changes in mitochondrial function [7], mitochondrial dynamics [8], and mitophagy [9] in skeletal muscle according to various intensities and types of exercise training. Likewise, studies have examined the changes in mitochondrial function [10] and mitochondrial dynamics [11] in the heart. However, studies on the impacts of acute exercise are scarce compared to those of chronic exercise in the cardiac and skeletal muscle. Furthermore, the results of mitochondrial oxidative stress and dynamics in skeletal muscle is contradictory among studies. Therefore, this study was performed to determine the effects of acute exercise on mitochondrial function, dynamics, and mitophagy in cardiac and skeletal muscles.
MATERIALS AND METHODS
Animals and Ethical Approval
Four-month-old Fischer 344 rats were randomly divided into the control (CON) or acute exercise (EX) group (n=10 each). We abided by the stipulation of the National Institutes of Health and the guidelines of the Korean Academy of Medical Science with respect to procedures for animal experiments. This experiment was approved by the Kyung Hee University Institutional Animal Care and Use Committee (Seoul, Korea) (KHUASP [SE]-17-089).
Exercise Design
The treadmill exercise was conducted after an adaptation period of about 10 minutes for a week. The EX group performed a single bout treadmill exercise at 20 m/min and 0% incline for 60 minutes at an intensity of approximately 60%–70% of VO2 max [12].
Mitochondrial Function
The left ventricle, soleus, and white gastrocnemius were separated in ice cold buffer X containing 7.23mM K2EGTA, 2.77mM Ca K2EGTA, 20mM imidazole, 0.5mM DTT, 20mM taurine, 5.7mM ATP, 14.3mM phosphocreatine, 6.56mM MgCl2-6H2O, and 50mM 2-(N-morpholino) ethanesulfonic acid. The separated myofibers were permeabilized using saponin. Following mitochondrial permeabilization for 30 minutes at 4°C, the myofibers were washed for at least 15 minutes before performing mitochondrial function analysis. Mitochondrial O2 consumption was measured through high-resolution respirometry (Oroboros O2K Oxygraph, Innsbruck, Austria) with an assay buffer consisting of Buffer Z with the addition of 50μM ethylene glycol tetraacetic acid (EGTA) and 20mM creatine. The mitochondrial O2 consumption protocol is as follows: 5mM glutamate+2mM malate, 4mM ADP, and 10mM succinate. Mitochondrial H2O2 emissions were measured by a Spex Fluormax 4 spectrofluorometer (HORIBA Jobin Yvon, Edison, NJ, USA) at 37°C. The experiments were conducted with buffer Z added to Amplex Red, horseradish peroxidase, and oligomycin, an inhibitor of mitochondrial ATP synthase. The mitochondrial H2O2 emission protocol is as follows: 5mM glutamate+2mM malate (complex I substrate), 10mM succinate (complex II substrate), and 10mM glycerol-3 phosphate (lipid substrate). The mitochondrial Ca2+ retention capacity was analyzed using a Spex Fluormax 4 spectrofluorometer. Overlaid traces of changes in Ca2+-induced fluorescence by Calcium Green-5N were measured continuously (ΔF/min) at 37°C during state 4 condition. After establishing background ΔF (1μM Calcium Green-5N, 80μM EGTA, 0.5M glutamate, and 0.2M malate in Buffer Z), we initiated the reaction by adding Ca2+ pulses (30μM) with excitation and emission wavelengths of 506 and 532 nm, respectively.
Western Immunoblot Analysis
Protein levels of mitochondrial fusion, fission, and mitophagy were determined. Soleus, white gastrocnemius, and left ventricle tissues were collected and immediately frozen at -80°C. Approximately 30 μg of tissues were homogenized with lysis buffer and centrifuged at 14,000 rpm for 20 minutes. Protein concentrations were quantitated by bicinchoninic assay. Protein was denatured at 95°C for 5 minutes, separated on sodium dodecyl sulfate–polyacrylamide gel with running buffer at 100 V for 2 hours and then transferred onto a nitrocellulose membrane on ice at 170 mA for 1.5 hours. After ponceau S staining, the membranes were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 for 2 hours. The membranes were incubated with primary antibodies overnight at 4°C. The membrane was then incubated at room temperature for 1 hour with secondary antibodies. The bands were detected by an enhanced chemiluminescence detection reagent kit. Protein bands were expressed using Chemidoc (Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Data are presented as mean±standard error of the mean. An independent t-test analysis of variance was performed to determine the effects of a single bout of exercise. The significance level was set at 0.05. All graphs were generated using Prism 5 (GraphPad, La Jolla, CA, USA).
RESULTS
Effects of Acute Exercise on Mitochondrial Function in Cardiac and Skeletal Muscles
Mitochondrial O2 consumption was measured with the following steps: basal, glutamate+malate (substrates of complex I, state 2), ADP (state 3), and succinate (substrate of complex II, state 3) (Fig. 1). Acute exercise significantly augmented glutamate and malate-stimulated O2 respiration added to basal respiration (GM2) by 62% in the permeabilized left ventricle (P<0.05) (Fig. 1A). In left ventricle tissues treated with acute exercise, mitochondrial O2 consumption was increased by 63% in ADP-induced O2 respiration supported by glutamate and malate (GM3, P<0.05) (Fig. 1A). Likewise, mitochondrial O2 consumption was elevated by 74% upon the addition of succinate to GM and ADP (GMS3) in the left ventricle (P<0.05) (Fig. 1A). Acute exercise significantly increased all stages including GM2, GM3, and GMS3 by 13%, 76%, and 99%, respectively, in permeabilized soleus muscle (P<0.05) (Fig. 1B). In the permeabilized white gastrocnemius muscle, acute exercise did not increase GM2 stage, but it did significantly elevate GM3, and GMS3 by 65% and 72%, respectively (P<0.05) (Fig. 1C).
Fig. 1.

The effects of acute exercise on mitochondrial O2 consumption in the left ventricle (A), soleus (B), and white gastrocnemius (C). The data are shown as mean±standard error of the mean. GM2, glutamate+malate (state 2); GM3, glutamate+malate+ADP (state 3); GMS3, glutamate+malate+succinate+ADP (state 3); CON, control group; EX, exercise group. *P<0.05 compared to the control group.
In the left ventricle and soleus fibers, a single bout of exercise did not significantly change mitochondrial H2O2 emission in GM, GMS, and GMSG3P conditions (Fig. 2A, B). However, in the white gastrocnemius muscle supported by GMS and GMS-G3P, mitochondrial H2O2 emission was significantly increased by 19% and 24% in the EX group and CON group, respectively (P<0.05) (Fig. 2C). Acute exercise significantly increased the mitochondrial Ca2+ retention capacity by 59% in the left ventricle (P<0.05) (Fig. 3A). Likewise, in the soleus and white gastrocnemius muscles, mitochondrial Ca2+ retention capacity in the EX group was higher than that in the CON group (62% and 23%, respectively) (P<0.05) (Fig. 3B, C).
Fig. 2.

The effects of acute exercise on mitochondrial H2O2 emission in the left ventricle (A), soleus (B), and white gastrocnemius (C). The data are shown as mean±standard error of the mean. GM, glutamate+malate; GM3, glutamate+malate+ADP; GMSG3P, glutam ate+malate+succinate+glycerol-3 phosphate; CON, control group; EX, exercise group. *P<0.05 compared to the control group.
Fig. 3.

The effects of acute exercise on mitochondrial Ca2+ retention capacity in the left ventricle (A), soleus (B), and white gastrocnemius (C). The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group. *P<0.05 compared to the control group.
Effects of Acute Exercise on Mitochondrial Dynamics (Fusion, Fission) and Mitophagy in Cardiac and Skeletal Muscles
In the left ventricle, none of the mitochondrial dynamics proteins differed between the groups (Fig. 4). In the soleus (Fig. 5) and white gastrocnemius (Fig. 6) muscles, no statistical differences in mitochondrial dynamics were observed in the EX compared with the CON group. In the left ventricle (Fig. 7) and white gastrocnemius muscle (Fig. 8), there were no significant intergroup differences in mitophagy-related proteins. However, in the soleus muscle, statistical increases were observed in Parkin and LC3II protein levels in the EX group compared with the CON group (Fig. 9A, C), whereas no significant intergroup differences were seen in Pink1 or Bnip3 (Fig. 9B, D).
Fig. 4.

The effects of acute exercise on mitochondrial dynamics in Mfn1 (A), Mfn2 (B), Opa1 (C), Drp1 (D), and Fis1 (E) of left ventricle. The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group.
Fig. 5.

The effects of acute exercise on mitochondrial dynamics in Mfn1 (A), Mfn2 (B), Opa1 (C), Drp1 (D), and Fis1 (E) of soleus muscle. The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group.
Fig. 6.

The effects of acute exercise on mitochondrial dynamics in Mfn1 (A), Mfn2 (B), Opa1 (C), Drp1 (D), and Fis1 (E) of white gastrocnemius muscle: The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group.
Fig. 7.

The effects of acute exercise on mitochondrial mitophagy in Parkin (A), Pink1 (B), LC3II (C), and Bnip3 (D) of left ventricle. The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group.
Fig. 8.

The effects of acute exercise on mitochondrial mitophagy in Parkin (A), Pink1 (B), LC3II (C), and Bnip3 (D) of white gastrocnemius muscle. The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group.
Fig. 9.
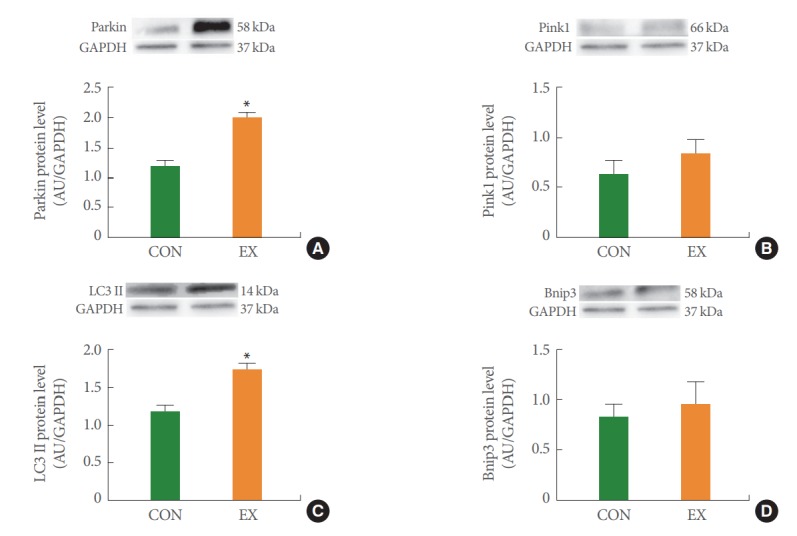
The effects of acute exercise on mitochondrial mitophagy in Parkin (A), Pink1 (B), LC3II (C), and Bnip3 (D) of soleus muscle. The data are shown as mean±standard error of the mean. CON, control group; EX, exercise group. *P<0.05 compared to the control group.
DISCUSSION
Here we observed that a single bout of exercise augmented mitochondrial O2 respiration and Ca2+ retention capacity in all tested tissues (left ventricle, soleus, white gastrocnemius). However, a single bout of exercise showed tissue-specific results in mitochondrial H2O2 emission, increasing mitochondrial H2O2 emissions in white gastrocnemius without showing statistically significant changes in the left ventricle or soleus. However, a single bout of exercise did not show any statistical differences in mitochondrial fusion and fission in any of the tissues. In particular, a single bout of exercise revealed tissue-specific mitophagy findings.
We examined whether mitochondrial function can be affected by a single bout of exercise. The current study demonstrated that mitochondrial O2 respiration was significantly increased by acute exercise in all tissues (Fig. 1). These findings of an increased mitochondrial O2 consumption following a single bout of exercise are consistent with those of previous studies and shows increased respiratory capacity of mitochondria in cardiac [13] and skeletal [14] muscles. Based on the notion that mitochondrial O2 respiration and ATP production concurrently occur, the improved mitochondrial O2 respiration may be related to activation of the mitochondrial functional capacity and ATP production.
We also analyzed mitochondrial H2O2 emission. Mitochondrial ROS is generated by products of the mitochondrial electron transfer chain during coupled respiration in complexes I and III [15]. ROS plays different biological roles depending on its concentration: (1) appropriate ROS is an essential mediator of cellular signaling and redox signals that are necessary for cellular homeostasis [16]; (2) a low level of ROS is needed for cellular differentiation, proliferation, growth, and adaptive responses [17]; (3) excessive ROS production induces both mitochondrial dysfunction and oxidative stress in proteins, nuclear DNA, mtDNA, and lipids [16]. In previous studies, regular exercise reduced ROS and elevated antioxidant enzymes in isolated heart mitochondria [10] and skeletal muscle [7]. However, several previous studies reported that acute exercise, unlike chronic exercise, augments mitochondrial oxidative stress [18]. In the current study, we observed that a single bout of exercise showed tissue-specific differences in mitochondrial H2O2 emission (Fig. 2). We conjectured that these tissue-specific differences may have been due to the number of different mitochondria depending on the tissues. Mitochondria are abundant in metabolic energy-demanding cardiac tissue, constituting 20%–40% of cellular volume [19], and are the second most plentiful in skeletal muscle because of its high metabolic need for mitochondrial ATP production in 3%–8% of skeletal muscle tissues [20]. In particular, among skeletal muscles, the number of mitochondria between red and white muscles is mainly matched by metabolic demand differences depending on exercise type [21]. Thus, we believe that mitochondrial H2O2 emissions are tissue-specific as follows: (1) mitochondrial H2O2 emission was not affected in the heart, where mitochondria are most abundant; (2) mitochondrial H2O2 emissions were not altered in the soleus, where mitochondria are mainly used during aerobic exercise; and (3) mitochondrial H2O2 emissions were significantly increased in the white gastrocnemius, which has relatively few mitochondria.
We also measured mitochondrial Ca2+ retention capacity. Mitochondrial Ca2+ plays an essential role in cellular function in terms of influencing energy production and apoptosis [22]. Mitochondria regulates energy production by activating F1F0-ATPase and the mitochondrial dehydrogenases responsible for nicotinamide adenine dinucleotide generation [23] and controlling apoptosis by regulating Ca2+ uptake [22]. According to previous studies, a damaged mitochondrial Ca2+ retention capacity is associated with programmed cell death, whereas the impacts of acute exercise on mitochondrial Ca2+ uptake have not been fully studied. In the present study, we observed that the mitochondrial Ca2+ retention capacity was significantly increased by acute exercise in all tissues (Fig. 3). We conclude that these results may be related to improved energy production and the protective role of acute exercise in mitochondria-mediated apoptosis.
Mitochondrial dynamics play an essential role in normal mitochondrial and cellular function [24]. Physical fusion between mitochondrial membranes could facilitate the exchange of mitochondrial molecules (proteins, metabolites, mtDNA), while mitochondrial fission could separate dysfunctional and unnecessary mitochondria by dividing the mitochondria into smaller components [25]. In the current study, we hypothesized that mitochondrial fusion is stimulated by energy demand due to acute exercise–induced energy deprivation. However, in this study, mitochondrial fusion proteins showed a tendency to increase without statistical significance in all tissues (Figs. 4-6). According to a previous study, the short exercise duration and absence of a recovery period may account for the insignificant difference in fusion proteins [24]. Indeed, one study in humans documented unchanged Mfn1 and Mfn2 protein levels after a single bout of exercise [26]. We conclude that energy loss due to acute exercise was insufficient to stimulate mitochondrial fusion. Meanwhile, the transcriptional regulation of mitochondrial dynamics correlated with exercise intensity [24]. Previous studies showed that exhaustive exercise-induced excessive ROS production causes mitochondrial fragmentation [27,28]. Indeed, a study in rats documented the transcriptional upregulation of Fis1 following intense exercise [29]. However, in this study, mitochondrial fission proteins tended to decrease without statistical significance in all tissues (Figs. 4-6). Thus, we conclude that mitochondrial fission is activated by high-intensity exercise; our measured fission proteins may not have been affected because of the moderate-intensity exercise protocol used in this study.
Mitochondrial autophagy (mitophagy) refers to the selective autophagic process of mitochondria. Mitophagy corresponds to mitochondrial quality control because it promotes the turnover of damaged mitochondria and inhibits the accumulation of dysfunctional mitochondria that are consequently associated with mitochondrial homeostasis [30]. A few studies reported that acute exercise activates mitophagy [31,32]. In the present study, acute exercise showed tissue-specific findings regarding mitophagy-related proteins. In the left ventricle and white gastrocnemius, mitophagy proteins tended to increase without statistical significance by acute exercise (Figs. 7, 8). In contrast, in the soleus muscle, acute exercise significantly increased Parkin and LC3II but not Pink1 and Bnip3 (Fig. 9). We speculate that these tissue-specific results may be due to the relationship between exercise type and the muscle type that is accordingly mobilized. Because we used aerobic treadmill exercise and the soleus muscle is a type 1 fiber that is mobilized by aerobic exercise, it is speculated that mitophagy increases to balance the mitochondrial biosynthesis in the soleus muscle caused by exercise. However, further studies are needed to elucidate the underlying mechanisms because our study measured mitophagy only at the protein level.
In conclusion, a single bout of aerobic exercise may improve mitochondrial function (e.g., O2 respiration and Ca2+ retention capacity) in the heart and skeletal muscle. It did not change mitochondrial dynamics (fusion and fission) or mitophagy in the heart and skeletal muscles. However, a single bout of aerobic exercise increased protein levels of Parkin and LC3II in the soleus muscle. Further studies on the impacts of acute exercise on various diseases such as obesity, diabetes, and cancer are also needed.
Footnotes
Fund/Grant Support
This work was supported by the Mid-Career Research Program through the National Research Foundation of Korea (2016R1A2B4014240, 2018R1A2A3074577).
Research Ethics
We abided by the stipulation of the National Institutes of Health and the guidelines of the Korean Academy of Medical Science with respect to procedures for animal experiments. This experiment was approved by the Kyung Hee University Institutional Animal Care and Use Committee (Seoul, Korea) (KHUASP [SE]-17-089).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: SZY
·Study concept and design: SZY, HBK
·Acquisition of data: SZY, MHN, JWH, JHK
·Analysis and interpretation of data: MHN, JWH, JHK
·Drafting of the manuscript: SZY
·Critical revision of the manuscript for important intellectual content: HBK
·Statistical analysis: SZY, MHN, JWH, JHK
·Obtained funding: HBK
·Administrative, technical, or material support: DHP, JHK, DYS, JH, SJJ
·Study supervision: HBK
REFERENCES
- 1.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–64. doi: 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- 3.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am J Physiol Heart Circ Physiol. 2011;300:H118–24. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo SZ, No MH, Heo JW, Park DH, Kang JH, Kim SH, et al. Role of exercise in age-related sarcopenia. J Exerc Rehabil. 2018;14:551–8. doi: 10.12965/jer.1836268.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weng SW, Wang PW, Lin HY, Chuang JH, Tsao CF, Lin CY, et al. The relationship between mitochondrial dynamics and insulin resistance in diabetes susceptible cybrid cells. Diabetes Res Clin Pract. 2016;120:S140. [Google Scholar]
- 6.Greene NP, Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA, et al. Mitochondrial quality control, promoted by PGC-1α, is dysregulated by Western diet-induced obesity and partially restored by moderate physical activity in mice. Physiol Rep. 2015;3(7) doi: 10.14814/phy2.12470. pii: e12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gram M, Vigelsø A, Yokota T, Helge JW, Dela F, Hey-Mogensen M. Skeletal muscle mitochondrial H2O2 emission increases with immobilization and decreases after aerobic training in young and older men. J Physiol. 2015;593:4011–27. doi: 10.1113/JP270211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konopka AR, Suer MK, Wolff CA, Harber MP. Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci. 2014;69:371–8. doi: 10.1093/gerona/glt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarpey MD, Davy KP, McMillan RP, Bowser SM, Halliday TM, Boutagy NE, et al. Skeletal muscle autophagy and mitophagy in endurance-trained runners before and after a high-fat meal. Mol Metab. 2017;6:1597–609. doi: 10.1016/j.molmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol. 2008;294:H928–35. doi: 10.1152/ajpheart.01231.2007. [DOI] [PubMed] [Google Scholar]
- 11.Zafaranieh S, Choobineh S, Soori R. The effect of 12 weeks of aerobic exercise on mitochondrial dynamics in cardiac myocytes of type 2 diabetic rats. Sport Sci Health. 2018;14:305–12. [Google Scholar]
- 12.Schefer V, Talan MI. Oxygen consumption in adult and AGED C57BL/6J mice during acute treadmill exercise of different intensity. Exp Gerontol. 1996;31:387–92. doi: 10.1016/0531-5565(95)02032-2. [DOI] [PubMed] [Google Scholar]
- 13.Ascensão A, Lumini-Oliveira J, Machado NG, Ferreira RM, Gonçalves IO, Moreira AC, et al. Acute exercise protects against calcium-induced cardiac mitochondrial permeability transition pore opening in doxorubicin-treated rats. Clin Sci. 2011;120:37–49. doi: 10.1042/CS20100254. [DOI] [PubMed] [Google Scholar]
- 14.Fernström M, Tonkonogi M, Sahlin K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J Physiol. 2004;554:755–63. doi: 10.1113/jphysiol.2003.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S, Tak E, Lee J, Rashid M, Murphy MP, Ha J, et al. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011;21:817–34. doi: 10.1038/cr.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.No MH, Heo JW, Yoo SZ, Jo HS, Park DH, Kang JH, et al. Effects of aging on mitochondrial hydrogen peroxide emission and calcium retention capacity in rat heart. J Exerc Rehabil. 2018;14:920–6. doi: 10.12965/jer.1836550.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–9. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smenes BT, Bækkerud FH, Slagsvold KH, Hassel E, Wohlwend M, Pinho M, et al. Acute exercise is not cardioprotective and may induce apoptotic signalling in heart surgery: a randomized controlled trial. Interact Cardiovasc Thorac Surg. 2018;27:95–101. doi: 10.1093/icvts/ivx439. [DOI] [PubMed] [Google Scholar]
- 19.Marín-García J, Goldenthal MJ. The mitochondrial organelle and the heart. Rev Esp Cardiol. 2002;55:1293–10. doi: 10.1016/s0300-8932(02)76802-4. [DOI] [PubMed] [Google Scholar]
- 20.Park SY, Gifford JR, Andtbacka RH, Trinity JD, Hyngstrom JR, Garten RS, et al. Cardiac, skeletal, and smooth muscle mitochondrial respiration: are all mitochondria created equal? Am J Physiol Heart Circ Physiol. 2014;307:H346–52. doi: 10.1152/ajpheart.00227.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glancy B, Balaban RS. Protein composition and function of red and white skeletal muscle mitochondria. Am J Physiol Cell Physiol. 2011;300:C1280–90. doi: 10.1152/ajpcell.00496.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams GS, Boyman L, Chikando AC, Khairallah RJ, Lederer W. Mitochondrial calcium uptake. Proc Natl Acad Sci. 2013;110:10479–86. doi: 10.1073/pnas.1300410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dirksen RT. Sarcoplasmic reticulum-mitochondrial through-space coupling in skeletal muscle. Appl Physiol Nutr Metab. 2009;34:389–95. doi: 10.1139/h09-044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picard M, Gentil BJ, McManus MJ, White K. Louis K, Gartside SE, et al. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J Appl Physiol. 2013;115:1562–71. doi: 10.1152/japplphysiol.00819.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iqbal S, Hood DA. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am J Physiol Cell Physiol. 2014;306:C1176–83. doi: 10.1152/ajpcell.00017.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol. 2010;588:4795–10. doi: 10.1113/jphysiol.2010.199448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies KJ, Quintanilha AT, Brooks GA, Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. 1982;107:1198–205. doi: 10.1016/s0006-291x(82)80124-1. [DOI] [PubMed] [Google Scholar]
- 28.Fan X, Hussien R, Brooks GA. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic Biol Med. 2010;49:1646–54. doi: 10.1016/j.freeradbiomed.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, et al. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta Gen Subj. 2010;1800:250–6. doi: 10.1016/j.bbagen.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14:1929–38. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo Verso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014;10:1883–94. doi: 10.4161/auto.32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308:C710–9. doi: 10.1152/ajpcell.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]