

Abstract
Synapses are sites of high energy demand which are dependent on high levels of mitochondrial derived adenosine triphosphate. Mitochondria within synaptic structures are key for maintenance of functional neurotransmission and this critical biological process is modulated by energy metabolism, mitochondrial distribution, mitochondrial trafficking, and cellular synaptic calcium flux. Synapse loss is presumed to be an early yet progressive pathological event in Alzheimer disease (AD), resulting in impaired cognitive function and memory loss which is particularly prevalent at later stages of disease. Supporting evidence from AD patients and animal models suggests that pathological mitochondrial dynamics indeed occurs early and is highly associated with synaptic lesions and degeneration in AD neurons. This review comprehensively highlights recent findings that describe how synaptic mitochondria pathology involves dysfunctional trafficking of this organelle, to maladaptive epigenetic contributions affecting mitochondrial function in AD. We further discuss how these negative, dynamic alterations impact synaptic function associated with AD. Finally, this review explores how antioxidant therapeutic approaches targeting mitochondria in AD can further clinical research and basic science investigations to advance our in-depth understanding of the pathogenesis of AD.
Keywords: Mitochondria, Synaptic plasticity, Alzheimer disease, Cognitive dysfunction
INTRODUCTION
Alzheimer disease (AD) is an aging-associated neurodegenerative disorder that is clinically characterized by progressive cognitive dysfunction and eventual death. The currently established main pathological features of this devastating disease include extracellular Amyloid-beta (Aβ) plaque deposits and intracellular neurofibrillary tangles (NFTs) [1]. Although these pathological features have undergone decades of intensive basic science and clinical investigations for potential application of preclinical and clinical therapeutic strategies it remains unclear whether these cellular and molecular phenotypes drive AD pathogenesis. Recently, mitochondria have gained accepted status as a key player contributing to normal aging and AD pathogenesis [2]. Investigations of this essential organelle in AD suggest that mitochondria dysfunction and oxidative stress are early contributory events to AD progression, thus making mitochondria an appealing therapeutic target. Supporting this hypothesis, brain imaging studies have demonstrated defects in glucose metabolism in AD patients, an abnormality that may occur well before the onset of clinical symptoms [3]. Interestingly, mitochondrial abnormalities have been reported as an early and prominent feature in AD patient brains [4] and AD transgenic mouse models [5,6].
Mitochondria are double-membraned organelles responsible for large amounts of cellular energy production derived from the biochemical processes of respiration. It provides most of the cellular adenosine triphosphate (ATP) demand by oxidative phosphorylation. Unfortunately, the high demand for energy that drives ATP mediated cellular functions, also generates a significant amount of reactive oxygen species (ROS) as a byproduct of ATP biogenesis [7]. These organelles also critically regulate other essential cellular functions including fatty acid oxidation, glutamate and urea metabolism, as well as cell death via activation of cellular apoptosis [8]. Notably, several reports indicate that AD-patient derived mitochondria are differentially affected in comparison to age-matched patients without dementia [9]. These disturbances include impaired oxidative phosphorylation, energy metabolism, as well as excess generation of ROS, altered mitochondrial biogenesis and transport (Fig. 1) [10]. Not surprisingly, in areas highly associated with cognitive function, such as the hippocampus and cortex, there is evidence for high levels of impaired mitochondrial respiration, excessive ROS production, as well as aberrant disruptions in membrane potential, and cytochrome c oxidase activity. Interestingly, brain structures that are less involved in complex cognitive ability, such as the striatum or amygdala, mitochondria-regulated processes are minimally to moderately affected [11]. In this review, we will report on the association between mitochondrial dysfunction and synaptic deficits in AD, which we believe underlies critical neuropathology of this neurodegenerative disease. We will also discuss how protection of synaptic transmission by targeting mitochondrial bioenergetics processes can provide an efficacious approach toward our understanding of mitochondrial mechanisms of neurodegeneration. More importantly, this review will provide a basis for future therapeutic strategies that will advance our understanding of AD and provide novel treatment approaches for this devastating disease.
Fig. 1.
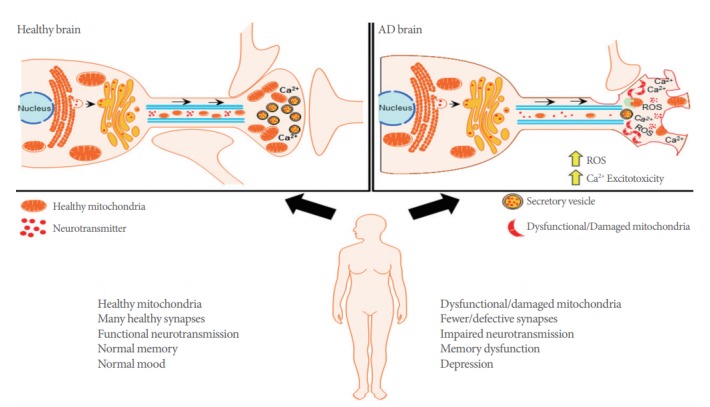
Dysfunctional mitochondria results in synaptic deficits in Alzheimer disease (AD). Dynamic mitochondrial trafficking between the neuronal soma and distal axonal synapses where Ca2+ homeostasis is critical to synaptic functionality. Healthy mitochondrial trafficking is critical to adequate mitochondrial density, control of reactive oxygen species generation (ROS), and physiological synaptic function. These critical mitochondrial functions contribute to homeostatic neurotransmission and healthy synaptic plasticity. In the AD brain, mitochondria neuropathology alters mitochondrial trafficking and density. Consequently, Ca2+ buffering is impaired, resulting in aberrant ROS generation, as well as accelerated synaptic degeneration and cognitive dysfunction.
MITOCHONDRIAL TRAFFICKING AND SYNAPTIC FUNCTION
Mitochondria derived ATP is involved in the complex regulation of synaptic plasticity and maintains the essential functions at synapses. For example, ATP generated from oxidative phosphorylation is integral for maintenance of the ion gradients required for axonal and synaptic membrane potentials [12]; mobilizing synaptic vesicles from reserve pools to release sites [13]; supporting vesicular neurotransmitter release, neurotransmitter recycling [14]; and supporting dynamic synaptic assembly and plasticity [15]. Neural presynaptic and postsynaptic terminals have high demands for ATP due to the extensive energy cost required to maintain homeostatic calcium flux during neurotransmission, and this persistent energy expenditure requires the constant presence of mitochondria [15], where they help to maintain neurotransmission by producing ATP and buffering Ca2+ at synapses [16]. Compared with nonsynaptic mitochondria, synaptic mitochondria showed a greater degree of age-dependent accumulation of Aβ, increased mitochondrial oxidative stress and impaired respiration in an AD mouse model [17]. The importance of adequate synapse function is evident in neurodegenerative disease, given that loss of synapses is one of the early pathological features of AD, and a strong correlation exists between the extent of synapse loss and the severity of dementia (Fig. 1) [18,19].
While mitochondrial biogenesis plays an essential role in homeostatic maintenance of synaptic function, efficient mitochondrial mobility is important in enabling the rapid distribution of this organelle to different areas in order to meet the high metabolic demands of neurons [20]. Motile mitochondria can become stationary but can be remobilized and redistributed in response to changes in cellular metabolic status and synaptic activity [21]. Dual-channel imaging of neuronal mitochondria in hippocampal synaptic terminals indicate several transitional motility patterns of axonal mitochondria. These include stationary mitochondria positioned outside of synapses or others that are docked at synapses. Other patterns are described as motile mitochondria that are either passing through synapses, or those that are paused at synapses for short or long time periods before re-engaging movement [22]. Mitochondrial trafficking is known to be impaired in AD and this transport defect is thought to contribute to the pathogenesis of neurodegeneration. For example, the absence of mitochondria in presynaptic terminals may reduce local ATP supplies which affect ATP-dependent vesicular neurotransmitter loading or reduce the pool of ATP available for working the myosin motors required to transport synaptic vesicles. Similarly, deficiencies in ATP can reduce the available energy to power the Na+/Ca2+ exchange process which is important for removing Ca2+ from nerve terminals, thus setting up a scenario that drives Ca2+ derived cellular excitotoxicity [23,24]. Interestingly, there are reports which suggest that Aβ and intracellular NFT are potential driving forces behind mitochondrial trafficking deficits in AD pathology. A study by Zhao et al. [25] revealed that drastic reductions in the number of axonal mitochondria was associated with a significant increase in mitochondrial size, thus suggesting that mitochondrial trafficking and mitochondrial fusion-fission are intertwined mechanisms.
Although motile mitochondria perform these essential functions, stationary docked mitochondria are also required to meet the high energy demand of axonal and synaptic functions. Docked mitochondria serve as stationary power plants for stable and continuous ATP supply necessary to maintain the activity of Na+/K+ ATPase, fast spike action potential propagation, and synaptic transmission [21]. Synaptic membrane proteins traditionally known to be involved in inhibition of SNARE vesicular neurotransmitter docking and release, have also been shown to play a role in mitochondrial anchoring. One such protein, Syntaphilin, has been identified to be the mitochondrial-anchoring protein that specifically immobilizes this organelle in axons [16]. Dysfunctional mitochondria manifest retrograde transport toward the soma, and this maladaptive behavior is typified by the selective release of syntaphilin which results in subsequent activation of PARK2/Parkin-mediated mitophagy. Notably, this syntaphilin-mediated response is robustly activated during the early disease stages of AD-related cortical neuron dysfunction [26,27].
MITOCHONDRIAL CA2+ HEMOSTASIS AND SYNAPTIC PLASTICITY
Besides supplying ATP, mitochondria also buffer Ca2+ in neuronal presynaptic terminals and dendrites [28]. The mitochondrial membrane utilizes its membrane potential to initiate and maintain Ca2+ ion flux in the mitochondrial matrix. The reversibility of this phenomenon is essential for Ca2+ storage and release in brain mitochondria [29]. Moreover, balanced Ca2+ levels in the mitochondrial matrix tightly regulate oxidative phosphorylation activity, thus adequately maintaining the rate of ATP production [30]. However, if the levels of Ca2+ accumulation overwhelm the ability for removal of Ca2+ within mitochondria, it can result in detrimental cytotoxicity. This phenomenon is often a typical byproduct of glutamate hyper-excitation during excessive excitatory neurotransmission and results in cellular excitotoxicity [29]. The ability for mitochondrial Ca2+ buffering is a critical function that must be maintained by this organelle as aberrant Ca2+ sequestration causes increased production of mitochondrial ROS. Therefore, for adequate synaptic function and plasticity, a fine balance must be achieved since ROS donors produce long-term potentiation (LTP), while conversely, ROS scavengers have been reported to block LTP [31]. Increased mitochondrial superoxide ROS can activate Ca2+-dependent protein kinases that are critical for synaptic plasticity [32]. Excessive Ca2+ uptake into mitochondria often leads to mitochondrial membrane permeabilization and induction of apoptosis. Also, Ca2+ dysregulation occurring in other cell types that support neuronal activity may contribute to degenerative neuropathology [33]. Therefore, the intracellular homeostasis of Ca2+ in neuronal mitochondria plays a significant role in energy production and maintenance of synaptic plasticity. The imbalance of Ca2+ within neuronal mitochondria is known to play an important role in the onset of AD. Modulation of mitochondrial Ca2+ is a potentially promising pharmacological therapeutic target for future AD treatment.
MITOCHONDRIA DNA METHYLATION
In humans, mitochondrial DNA (mtDNA) has approximately 37 genes, all of which are essential for normal mitochondrial function. DNA methylation is a well-known epigenetic mechanism that regulates nuclear gene transcription. Early studies showed that AD brains possessed consistent global reductions in DNA modifications of 5-methylcytosine (5mC) and 5-hydroxymethylcytosine [34]. Given the high heritability estimates for AD, there is additional supporting evidence suggest that epigenetic-mediated reductions in the expression of genes that form complex I-and-IV subunits of the electron transport chain (for example ND4), are particularly prevalent in AD brains [35,36]. Furthermore, genetic studies have identified somatic mutations in several mitochondrial encoded cytochrome-c oxidase genes, as well as in the early mitochondrial replication intermediate displacement loop (D-loop), the mutations which probably alters mitochondrial transcription, replication, and mtDNA deletions in AD brains [37,38].
In recent human studies, the mitochondrial 5mC levels were measured from the entorhinal cortex in AD patients and control brains, along with an APP/PS1 transgenic mouse model at 3, 6, and 12 months of age. The authors report the presence of mitochondrial 5mC in CpG and non-CpG sites in the hippocampal entorhinal cortex. Moreover, compared to control cases, AD patients had increased expression of 5-mC found in the Dloop region of mtDNA in the entorhinal cortex. In their APP/PS1 mouse model, they observed a dynamic pattern in the content of mitochondrial 5mC, as AD pathology progressed across 3, 6, and 12 months of age. This suggests that mtDNA epigenetic modulation in human brain is vulnerable to neurodegenerative disease states [39]. The methylation levels of the mtDNA D-loop region detected in blood DNA from 133 AD patients and 130 controls had observed a significant 25% reduction of DNA methylation in AD patients [40]. Further studies showed that when targeting, mtDNA, methylating enzymes modify cytosine in the CpG or GpC context. Interestingly, mtDNA gene expression remained unchanged upon induction of CpG methylation, whereas induction of C-methylation in the GpC context decreased mtDNA gene expression [41].
Given the important role of mitochondria in AD, exploring the impact of mtDNA methylation will add to our understanding of the role of mitochondria in AD pathogenesis. Numerous mitochondrial haplogroups and single nucleotide polymorphisms have been reported to influence risk for AD, but the majority of these studies have not been replicated, nor experimentally validated. It remains challenging to define the relationship between the mitochondrial genome and AD including small numbers of patient subjects, insufficient genetic data, and technical challenges in data analysis [42]. We suggest that with bigger sample sizes and the application of whole genome sequence data, there can be substantially beneficial advancements for the study of mitochondrial genetic variation in AD.
CONCLUSIONS
Mitochondrial dysfunction is an early pathological feature of AD development, which greatly contributes to maladaptive synaptic deficits and loss of synapses in the earlier phase of this disease. Several therapeutic studies have been conducted with promising experimental results, and these studies have identified mitochondria specific ROS scavengers, which will enable a greater concentration of antioxidants to accumulate in mitochondria and allow a more specific method for combatting mitochondrial oxidative stress. The neuroprotective role of a number of currently available agents that modulate mitochondrial Ca2+ transport pathways and buffering capacity are currently under intense investigation by the scientific community. Further, both generic and mitochondria-targeted studies should be carried out to find additional potential treatment strategies for synaptic defects and neuronal degeneration in AD.
Footnotes
Fund/Grant Support
This work was supported by NIH/NIA (R01AG058560, R01AG058560-S1), Regenerative Medicine Minnesota, Mayo Clinic Center for Regenerative Medicine, and Department of Neurologic Surgery at Mayo Clinic.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION STATEMENT
·Full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis: JT
·Study concept and design: JT
·Acquisition of data: AO
·Drafting of the manuscript: JT
·Critical revision of the manuscript for important intellectual content: AO
·Obtained funding: MHJ
·Administrative, technical, or material support: MHJ
·Study supervision: MHJ
REFERENCES
- 1.Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–8. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40:151–66. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180–95. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai Q, Tammineni P. Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J Alzheimers Dis. 2017;57:1087–103. doi: 10.3233/JAD-160726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–86. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–5. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J. 2015;29:4766–71. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 8.Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, et al. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016;1637:34–55. doi: 10.1016/j.brainres.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swerdlow RH. Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta. 2011;1812:1630–9. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hroudova J, Singh N, Fisar Z, Ghosh KK. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondrial energy metabolism. Eur J Med Chem. 2016;121:774–84. doi: 10.1016/j.ejmech.2016.03.084. [DOI] [PubMed] [Google Scholar]
- 11.Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, et al. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis. 2010;20 Suppl 2:S535–50. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- 12.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–45. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–78. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Gazit N, Vertkin I, Shapira I, Helm M, Slomowitz E, Sheiba M, et al. IGF-1 receptor differentially regulates spontaneous and evoked transmission via mitochondria at hippocampal synapses. Neuron. 2016;89:583–97. doi: 10.1016/j.neuron.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–87. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–48. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–5. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 20.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheng ZH. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–98. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013;4:413–9. doi: 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–7. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–93. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 25.Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ, et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. 2010;30:1512–22. doi: 10.1523/JNEUROSCI.3699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin MY, Cheng XT, Xie Y, Cai Q, Sheng ZH. Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy. 2017;13:1792–4. doi: 10.1080/15548627.2017.1356552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, et al. Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron. 2017;94:595–610 e6. doi: 10.1016/j.neuron.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zorzano A, Claret M. Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Front Aging Neurosci. 2015;7:101. doi: 10.3389/fnagi.2015.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–24. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeanneteau F, Arango-Lievano M. Linking mitochondria to synapses: new insights for stress-related neuropsychiatric disorders. Neural Plast. 2016;2016:3985063. doi: 10.1155/2016/3985063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee KY, Chung K, Chung JM. Involvement of reactive oxygen species in long-term potentiation in the spinal cord dorsal horn. J Neurophysiol. 2010;103:382–91. doi: 10.1152/jn.90906.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci. 2011;31:1114–27. doi: 10.1523/JNEUROSCI.5387-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brawek B, Garaschuk O. Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res. 2014;357:427–38. doi: 10.1007/s00441-014-1798-8. [DOI] [PubMed] [Google Scholar]
- 34.Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging. 2013;34:2091–9. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, et al. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol Aging. 2002;23:371–6. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 36.Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4:e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–31. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, et al. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J Alzheimers Dis. 2010;20 Suppl 2:S293–310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blanch M, Mosquera JL, Ansoleaga B, Ferrer I, Barrachina M. Altered mitochondrial DNA mthylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol. 2016;186:385–97. doi: 10.1016/j.ajpath.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Stoccoro A, Siciliano G, Migliore L, Coppede F. Decreased methylation of the mitochondrial D-loop region in late-onset Alzheimer’s disease. J Alzheimers Dis. 2017;59:559–64. doi: 10.3233/JAD-170139. [DOI] [PubMed] [Google Scholar]
- 41.van der Wijst MG, van Tilburg AY, Ruiters MH, Rots MG. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci Rep. 2017;7:177. doi: 10.1038/s41598-017-00263-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridge PG, Kauwe JSK. Mitochondria and Alzheimer’s disease: the role of mitochondrial genetic variation. Curr Genet Med Rep. 2018;6:1–10. doi: 10.1007/s40142-018-0132-2. [DOI] [PMC free article] [PubMed] [Google Scholar]