

CONSPECTUS
Natural products are significant therapeutic agents and valuable drug leads. This is likely owing to their three-dimensional structural complexity, which enables them to form complex interactions with biological targets. Enzymes from natural product biosynthetic pathways show great potential to generate natural product-like compounds and libraries. Many challenges still remain in biosynthesis, such as how to rationally synthesize small molecules with novel structures, and how to generate maximum chemical diversity.
In this account, we describe recent advances from our laboratory in the synthesis of natural product-like libraries using natural biosynthetic machinery. Our work has focused on the pat and tru biosynthetic pathways to patellamides, trunkamide, and related compounds from cyanobacterial symbionts in marine tunicates. These belong to the cyanobactin class of natural products, which are part of the larger group of Ribosomally synthesized and Posttranslationally modified Peptides (RiPPs).
These results have enabled the synthesis of rationally designed small molecules and libraries covering more than 1 million estimated derivatives. Because the RiPPs are translated on the ribosome and then enzymatically modified, they are highly compatible with recombinant technologies. This is important because it means that the resulting natural products, their derivatives, and wholly new compounds can be synthesized using the tools of genetic engineering. The RiPPs also represent possibly the most widespread group of bioactive natural products, although this is in part because of the broad definition of what constitutes a RiPP. In addition, the underlying ideas may form the basis for broad-substrate biosynthetic pathways beyond the RiPPs. For example, some of the ideas about kinetic ordering of broad-substrate pathways may apply to polyketide or nonribosomal peptide biosynthesis as well.
While making these products, we have sought to understand what makes biosynthetic pathways plastic, and whether or not there are any rules that might generally apply to plastic biosynthetic pathways. We present three principles of diversity-generating biosynthesis: 1) Substrate evolution, in which the substrates change while enzymes remain constant; 2) Pairing of recognition sequences on substrates with biosynthetic enzymes; 3) An inverse metabolic flux in comparison to canonical pathways.
If these principles are general, they may enable the design of unimagined derivatives using biosynthetic engineering. For example, it is possible to discover substrate evolution directly by examining sequencing data. By shuffling appropriate recognition sequences and biosynthetic enzymes, it has already been possible to make new hybrid products of multiple pathways. While cases so far have been limited, if this is more general designed synthesis will become routine. Finally, biosynthesis of natural products is regulated in elaborate ways that are just beginning to be understood. If the inverse metabolic flux model is widespread, it potentially informs on what the timing and relative production level of each enzyme in a designer pathway should be in order to optimize the synthesis of new compounds in vivo.
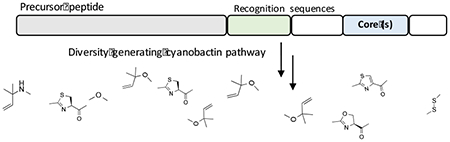
Introduction
Natural products and their derivatives comprise the majority of approved drugs.1 They possess diverse chemical structures that might interact with a variety of targets. Synthetic chemists have long sought to imitate this structural complexity in the design of synthetic libraries with target-oriented synthesis (TOS) or diversity-oriented synthesis (DOS) methods.2 TOS focuses on the key structural elements of the final products that might probe a known target. DOS, however, first focuses on expanding structural diversity and complexity and then finding new targets. Instead of chemical synthesis, a widespread goal is to engineer natural biosynthetic pathways to create desired compounds, such as “unnatural” natural products. Thus, in principle target-oriented or diversity-oriented compounds might be synthesized using synthetic biology. One advantage of biosynthesis compared to chemical synthetic methods is that it is integrated with biology, enabling selection methods to discover and improve compounds. Biosynthetic methods can also be used in living cells.
Secondary metabolites (natural products) show more chemical diversity compared to primary metabolites.3–5 In the canonical view of primary metabolism, each step requires the product from the previous step. The process is often efficient and focused on one or just a few compounds. By contrast, in secondary metabolism the enzymes are often relatively relaxed in specificity and can accept the incomplete products from previous steps or other analogs. As a result, the enzymes in some secondary metabolic pathways can generate a series of congeners in a metabolic grid fashion. It has not been fully understood why specialized metabolism more easily generates diversity, but it is hypothesized that this catalytic promiscuity helps evolve and select more potent compounds for different targets. A similar system is the immune system in humans, where the variable regions of antibodies help target different antigens.6 It should be emphasized that there is a range of plasticity in specialized metabolism, from pathways that are relatively selective for a single product to very relaxed specificity pathways.
There are different ways to generate chemical diversity biosynthetically using metabolic engineering, mainly focused on two aspects: how individual biosynthetic enzymes function and how modules of multiple enzymes can be rearranged.7 Enzyme specificity is usually tested with substrate analogs. Individual enzymes can be further engineered to increase promiscuity by directed evolution and site mutagenesis. Besides focusing on enzymes, biosynthetic assembly lines can also be manipulated to generate new molecules by genetic engineering, precursor-directed synthesis or other methods.8 While many pathways have enzymes or modules that accept different substrates, the property of diversity generating biosynthesis and its application to rational engineering are still poorly defined. Here, we present our application of cyanobactin biosynthesis to understanding this problem. Using this model system, we have generated libraries to produce millions of compounds.
Cyanobactin biosynthetic pathways as a model system
The program started out in our lab with the observation that many macrocyclic peptides with similar structures were isolated from marine tunicates. These included peptides with different sequences, different macrocycle sizes, and different chemical modifications to the standard 20 proteinogenic amino acids. These compounds were all found in animals that are superficially at least quite similar. Because of this close relationship between organisms, we hypothesized that a naturally plastic biosynthetic pathway was involved. If so, such a pathway would be very useful in learning how to rationally produce compounds and libraries using biosynthetic machinery.
At the time, most progress in this field was obtained by comparing bacterial biosynthetic pathways, which are usually quite different from each other in ways that confound generating a simple model. In previous work with bacterial symbionts from sponges, we noticed that there is sometimes a very straightforward phylogenetic relationship between bacterial strains and chemistry that might provide a more obvious, simple model of how chemical diversity is generated.9 By contrast, at the time the relationships between pathways in cultivated bacteria were usually more complex. We thus collected tunicate samples from around the tropical Pacific Ocean, with the goal of understanding the biosynthesis and the generation of chemical diversity.
In 2005, in collaboration with Jacques Ravel and others we used the first metagenome sequencing approach to natural products to obtain the patellamides (pat) pathway from uncultivated symbionts of tunicates. We then expressed this pathway in Escherichia coli.10 Subsequently, we discovered the related pathway tru (after the natural product trunkamide) also from tunicates.11 These compounds are made not by the animals but by symbiotic cyanobacteria, Prochloron spp. In addition, there are many related pathways more recently discovered in free-living cyanobacteria.12 In 2008, we named the products of these pathways “cyanobactins” because of their common features.11 Much work has been done to identify and functionally characterize cyanobactin pathways and enzymes by our lab and by groups including those of Dittmann, Jaspars, Nair, Naismith, Sivonen, Suga, and others, which is beyond the scope of this manuscript.
The pat and tru products are made on the ribosome via a RiPP pathway.11,13–14 In RiPPs, the mature natural product is encoded on a core peptide within a precursor peptide. Associated enzymes in the same pathway act on the core peptide to introduce posttranslational modifications. Subsequently, the precursor peptide is proteolytically processed to convert the core peptide into a mature, bioactive natural product (Figure 1).
Figure 1.
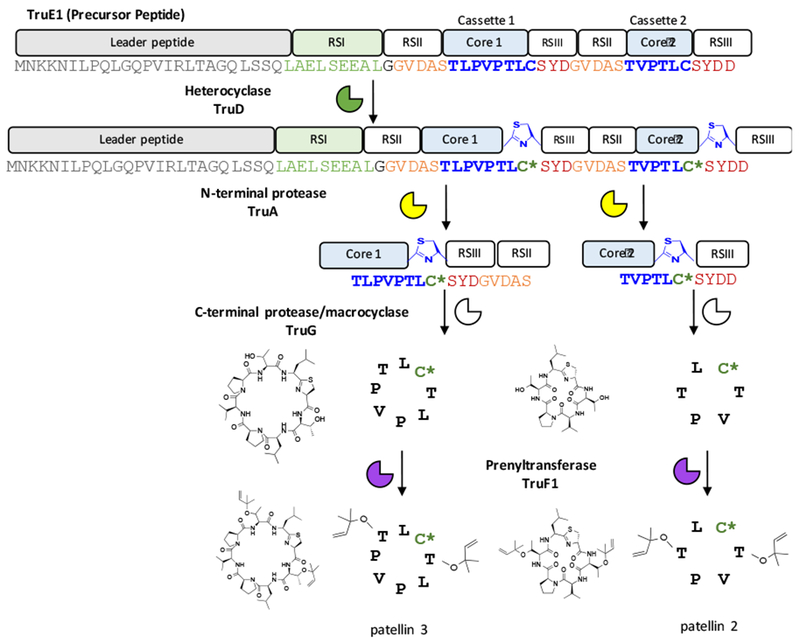
Biosynthesis of cyanobactins. In this example from the tru pathway, patellins are encoded in core sequences within the precursor peptide TruE (blue). Heterocyclase TruD (green) is paired with RSI (green) and heterocyclizes the cysteine residue. N-terminal protease TruA (yellow) recognizes RSII (yellow/orange) and removes the N-terminus of the core. TruG (pink) paired with RSIII (pink/red) then proteolyzes the C-terminus of the core and macrocyclizes the substrate in a head-to-tail manner. Finally, prenyltransferase TruF1 (purple) prenylates the threonine residues.
What was striking about the discovered pat pathway was that the generation of chemical diversity was immediately explained. Instead of encoding one core peptide and one product, the pathway encoded two highly different products on a single precursor. Multiple precursor peptides encoding different cores were also discovered, predicted to generate at least seven patellamide-like products. Each core was flanked by highly conserved elements that we named “recognition sequences” because they were most likely markers for enzyme recognition to catalyze specific biochemical transformations.14 This led to over a decade of research in our lab in which we sought to understand the nature and scope of broad substrate tolerance in whole biosynthetic pathways.
This is the story of what we have learned about what makes biosynthesis plastic, and how we have applied the resulting discoveries to the (bio)synthesis of large libraries and designed compounds, in which we learned three principles. These principles apply specifically to pathways from symbiotic Prochloron, since we have much less experimental data about diversity generation in other cyanobactin pathways from free-living cyanobacteria. For example, within specific symbiotic pathways such as pat, the modifying enzymes are nearly 100% sequence identical in animals obtained throughout the tropical Pacific, even if vastly different products are synthesized.15 By contrast, when comparing free-living cyanobacteria there are often many mutations in the enzymes as well as products.
Principle 1: Substrate evolution
In cyanobactin pathways, the precursor peptide consists of the core peptide, encoding the mature natural product, recognition sequences before and after the core peptide enabling enzyme recognition of the precursor peptide, and an N-terminal leader sequence that appears to be required in vivo but not in vitro. Within the symbiotic pathways, such as pat and tru, the precursor peptide is nearly 100% conserved, except that the core peptides are hypervariable.11,14 Natural circular peptides from 6-8 amino acids can be produced, and every position of the macrocycle has been modified in natural analogs with the exception of a required C-terminal cysteine that is modified to thiazoline/thiazole. While the core peptides are hypervariable, all of the enzymes in each pathway are also nearly 100% identical to their homologues (Figure 2). Thus, these natural pathways demonstrate a situation in which the substrates, but not the enzymes, are evolving.
Figure 2.

Substrate evolution and recognition sequence- (RS)-enzyme pairing in cyanobactin pathways. The precursor peptides from the same pathway are often 100% identical, except for the core peptides (blue), which are hypervariable. RS elements in the precursor pathway pair with their cognate modifying enzymes, as indicated by paired colors. Enzymes are also commonly 100% identical within pathways, even though the core peptides are hypervariable. By pairing with conserved recognition sequence, enzymes are able to modify hypervariable cores.
This extreme substrate flexibility confers each cyanobactin pathway the potential to generate millions of peptide products, a feature which we have experimentally verified.16,17 Diverse precursor peptides can be easily generated by mutating the precise position of the pathway encoding the core peptide. A compound library of the tru pathway was biosynthesized with size the of 204 by random quadruple mutation of the precursor gene sequence in Escherichia coli.16 Rationally designed compounds were also generated in vivo using the tru pathway, showing that the pathway can accept even nonproteinogenic amino acids in living cells.17 In vitro, both single step and multi-step enzyme reactions confirmed that substrate evolution is highly permissive in the cyanobactin pathways, generating new compounds and libraries.18–20 Nonribosomal peptide- and polyketide-like products were also achieved using the macrocyclase / circulase enzyme, PatG.21 Thus, each enzyme is highly permissive, and notably the pathway as a whole constituting multiple different enzymatic steps is capable of generating millions of analogs.
These data represented the first experimental evidence of a multi-step natural diversity-generating pathway. There are many pathways from which a few analogs had been developed, and there exist exceptionally broad-substrate single enzymes. A good example of the latter is the discovery of the RiPP pathway to prochlorosins, consisting of the precursor ProcA and the extremely flexible lanthionine synthetase ProcM.22–26 Interestingly, these also originate in cyanobacteria.
We considered that this combination of multiple, extremely-relaxed substrate steps was highly promising for engineering. Some (but not all, or perhaps not even most) RiPP pathways also likely undergo similar processes, while others may be relatively more fixed and directed at synthesizing single products. For example, in other RiPPs mutations have been made to generate artificial libraries, including lasso peptide microcin J25, 27,28 lanthipeptides nukacin ISK-1,29 mersacidin,30 and nisin.31 Similarly, duramycin,32,33 cinnamycin,33 and bottromycin34 derivatives have been synthesized by mutagenesis. This field is growing rapidly, and we have listed a limited subset of representative examples. Whether these representative examples comprise true diversity-generating pathways awaits further experimentation.
One feature that is found in broad-substrate pathways, and that may be inherent to diversity generation, is the presence of multiple copies of the core peptide within a single precursor peptide, or the presence of multiple similar precursor peptides in the same genome. This might be due to module exchanges between different cyanobactin-producing strains.35 Multiple copies of similar precursor peptides have been found in the case of both cyanobactins and prochlorosins, for example. Sometimes, multiple core sequences are found within a single precursor peptide. When this happens, they are usually flanked by enzyme recognition sequences that are proteolytically removed prior to the synthesis of the mature natural product. We named these core-recognition sequence combinations “cassettes”, since they are features that can be duplicated from one to up to 16 times in the most extreme case known. Multi-cassette pathways include those to pheganomycin,36 cyclotides37 and orbitides,38 microviridin,39,40 phomopsin,41 and ustiloxin.42 Existing data indicate that many of these pathways are likely to be diversity generating in the same sense as cyanobactins, although experimental evidence is needed. The functional reason underlying the presence of multiple cassettes (evolutionary, biochemical, yield increasing, etc.) is still not known.
The presence of substrate evolution can be directly identified by sequencing multiple (meta)genomes and directly observing that the core peptide sequence is variable, while other elements are conserved.43,44 In this situation of substrate evolution, there is a constraint that makes the enzymes maintain their broad substrate selectivity and high plasticity. This stands in contrast to cases in which the core peptide is more conserved than the enzymes, in which case the pathway may be under selection to maintain a single product, and the enzymes may become more selective.
Principle 2: Pairing of recognition sequences with cognate enzymes
Given that enzymes in the pat and tru pathways are so relaxed in specificity, we asked the question of how the enzymes maintain this plasticity. One of the earliest clues was found in the cassettes of cyanobactins. Repeating sequence elements flanking each core peptide were clearly visible, which we proposed were recognition sequences for proteolytic enzymes for an N-terminal protease and a macrocyclizing protease.10 These two elements, later renamed recognition sequences II and III, were borne out in biochemical experiments demonstrating the necessity and sufficiency of short recognition sequence elements. In turn, these elements are proteolytically cleaved and not found in the final products.21,45 Using this simple mechanism, enzymes can recognize numerous substrates without sacrificing hypervariability in the core (Figure 2).
Subsequently, we identified a short sequence in the leader peptide, now called recognition sequence I, that is essential for recognition by the first biosynthetic enzyme in the pathway, heterocyclase.12 This element was later demonstrated in our lab using biochemical evidence and by Naismith and Jaspars using crystallographic, biochemical, and bioengineering methodologies.20,43, 46–49 The only pat or tru enzyme that so far has no obvious recognition sequence is the prenyltransferase, TruF1.17,50 This enzyme acts after the peptide has been macrocyclized and all recognition sequences have been excised. Earlier, a homologous prenyltransferase LynF was also verified biochemically with the same characteristics.51 Recently, we reported a crystal structure of another homologous enzyme PagF that explains how the prenyltransferase can exhibit relatively relaxed selectivity in the absence of a recognition sequence.52
The portability of recognition sequence elements, when paired with enzymes, is highly useful to biotechnology. For example, we have shown multiple examples in which enzymes from very different cyanobactin biosynthetic pathways, can be paired, as long as the right recognition sequence elements are in place (summarized in reference 18). We have used this in the synthesis of designer compounds and in the total (bio)synthesis of natural products.18
The recognition sequence concept has since been broadly validated across a wide swath of RiPPs. Although much work has been done in this area, perhaps the best demonstration is the finding of a RiPP precursor peptide recognition element (RRE) within many RiPP enzymes that binds to conserved elements in the precursor peptides.53 Examples of potential enzyme-recognition pairs include the conserved FXLD motif in nisin biosynthesis, which has been proved to pair with lantibiotic dehydratase NisB and cyclase NisC.54,55 In microviridin biosynthetic pathway, conserved PFFARFL motif is paired with two ATP grasp type ligases.56,57 One application has been to fuse recognition sequences with the cognate enzymes, and then feed core peptides in trans. Examples include the LctM from lacticin 481 biosynthesis,58 LynD from a cyanobactin pathway,48 and TruD from the tru pathway.18 Two ATP-grasp ligases fused with microviridin leader peptide were also shown to catalyze the reaction with high yield.40,59 These engineered broad substrate enzymes have important applications in in vitro biosynthesis tool kits. Very recently, Mitchell and coworkers have used RiPP precursor peptide recognition elements to pair RiPP enzymes from broadly divergent biosynthetic pathways, showing that the scope of this methodology may be truly broad.60 There is usually a sequence preference within the core peptide as well, for example in the requirement of a heterocycle (proline or azoline) at the P1 residue in PatG/TruG substrates.
Principle 3: Metabolic flux is inverted from expectation
Clearly, substrate evolution and recognition sequence-enzymes pairs are crucial to broad-substrate RiPP pathways. We have long sought to understand what it is about multiple enzymes in a pathway that allows them to exhibit extreme plasticity. A model for enzyme promiscuity has been proposed by Arnold and co-workers in which there is a sweet spot where enzymes are relatively plastic; in this event they often exhibit decreased catalytic rates.61 This finding, rigorously examined using artificially produced plastic enzymes, might also be applicable to plastic natural product pathways. Indeed, in studying ProcM, the van der Donk group showed that the first ring is built most quickly, while later steps catalyzed by ProcM are increasingly slow.22 The starting materials are linear peptides that are fairly similar to each other, while as different rings are built the structures are increasingly different from each other. Thibodeaux et al. called this a “kinetic price” for broad substrate selectivity.22
While much has been done with single-enzyme promiscuity, we have long wondered how promiscuity can be maintained over multiple steps in a biosynthetic pathway. We recently made some serendipitous inroads into this area.50 In producing trunkamide derivatives using the tru pathway, we accidentally discovered that the addition of cysteine and mevalonate (co-expressed with mevalonate pathway) to cultures increases the yield of product by about 3,000-fold in E. coli culture. In trying to understand the mechanism causing this increase, we directly measured the amounts of tru pathway intermediates in E. coli. We found that the first biochemical step is the fastest, and the further steps are increasingly slow until the last prenylation by TruF1. While the first step is complete in E. coli within a few hours, the last prenylation event is still measurably increasing for up to >5 days, even though transcription and translation of enzymes is complete within about one day. Using this and evidence from biochemical experiments, we proposed that the tru pathway undergoes an inverted kinetic order from what is expected in primary metabolism: instead of the rate limiting step being first or early, it is late or last (Figure 3).
Figure 3.

Metabolic model for diversity generating pathways. As shown with the example of the tru pathway,50 earlier steps are faster than late steps.
Interestingly, tru substrates are increasingly different as the pathway proceeds. While the substrates for the first enzymatic event are simple linear peptides that are highly conserved except for the cores, as the pathway proceeds they are increasingly differentiated as recognition sequences are proteolyzed. This structural differentiation parallels the decreasing speed of late steps in the pathway. Thus, it appears that cyanobactin pathways also may pay the “kinetic price” for broad-substrate tolerance.
We hypothesize that in diversity-generating biosynthesis even beyond the RiPP family, an inverted kinetic order is common. For example, many tailoring enzyme modifications which are relatively broad substrate are rate-limiting steps. In penicillin biosynthesis the tailoring isopenicillin N-acyltransferase catalyzes the rate-limiting step and incorporates either a phenyl- or phenoxyacetyl group to yield penicillin G or V.62 In the RiPP polytheonamide (poy) biosynthetic pathway, the N-methyltransferase PoyE inserts eight methyl groups to the Asn at different positions. It takes at least three days to obtain eight methylated products after co-expressing precursor peptide with PoyE.63 Compared to the early stage epimerization reaction in poy, it takes much longer time to obtain the late stage fully methylated compound. There are many other examples where late tailoring is relatively slow, and many derivatives with different tailoring modifications can be isolated from microbes.
Application to synthesis and synthetic biology
In this account, we have introduced three general principles for diversity-generating pathways: substrate evolution, enzyme-recognition element pairing and inverse kinetic order. We hypothesize that other diversity-generating pathways share similar principles. Such pathways may readily be identified by comparing genomics and chemical products, as described for cyanobactin and prochlorosin pathways.
Crucially, diversity-generating biosynthesis can be applied to create rationally engineered compounds and large libraries. The three rules we define above will be very useful in the design of such pathways (Figure 4). For example, pairing recognition elements with cognate enzymes enables production of designer derivatives that are unconstrained by natural evolutionary space. We also predict that design of pathways will need to keep account of metabolic flux, where instead of optimizing efficiency of each step it will be more appropriate to pair increasingly slow enzymes with increasingly late steps in the pathway. Since the rate-limiting step may be late in the pathways, it may also be appropriate to increase the concentration of enzymes or precursors for slower, late-stage enzymes. We are currently in the process of testing the broad application of these rules to engineering production of diverse compounds in vivo.
Figure 4.

Applying the principles of diversity-generating biosynthesis to make novel compounds. A) In the precursor peptide, different recognition sequences are inserted before or after a designed core sequences, based upon the principle of substrate evolution. These recognition sequences are paired with cognate enzymes, following the principle of enzyme-RS pairs. Expression of the enzymes should follow the rule of inverted kinetic order. B) Example of manipulating RS elements in the total biosynthesis of the natural product, aeruginosamide B. RSI is fused to the N-terminus of TruD and heterocyclizes the cysteine residue of a short precursor peptide to thiazoline (green). AgeG recognizes and removes RSIII (red) from the tru pathway. AgeMTPT recognizes the N-terminus and C-terminus of the substrate and adds a prenyl and methyl group, respectively (purple).
Finally, it is interesting that nature has developed strategies not only to efficiently produce single compounds as found in primary metabolism, but also to produce diversity. It will be of great interest to understand the underlying evolutionary mechanisms that preserve broad-substrate tolerance within large populations of bacteria.
Acknowledgments
Funding
NIH GM102602, GM122521.
Biographies
Biographical Information
Wenjia Gu is a graduate student in the Department of Medicinal Chemistry at the University of Utah. She received her B.S. in Chemistry at Zhejiang University in 2014 and joined Prof. Eric W. Schmidt’s lab in 2015. Her current research focuses on the diversity-generating biosynthesis of natural products.
Eric W. Schmidt is Professor of Medicinal Chemistry and Adjunct Professor of Biology at the University of Utah. He obtained his Ph.D. at the University of California, San Diego in 1999 under the supervision of Prof. D. John Faulkner and was a postdoctoral fellow with Prof. Craig A. Townsend. His research focuses on the biosynthesis and chemistry of natural products, particularly in animal-bacteria symbioses.
Footnotes
Conflict of Interest
EWS co-founded Synthetic Biodesign using this technology.
REFERENCES
- (1).Newman DJ; Cragg GM Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- (2).Schreiber SL Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [DOI] [PubMed] [Google Scholar]
- (3).Weng J -K.; Noel, J. P. The Remarkable Pliability and Promiscuity of Specialized Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 309–320. [DOI] [PubMed] [Google Scholar]
- (4).Weng J-K; Philippe RN; Noel JP The Rise of Chemodiversity in Plants. Science 2012, 336, 1667–1670. [DOI] [PubMed] [Google Scholar]
- (5).Firn RD; Jones CG Natural Products - a Simple Model to Explain Chemical Diversity. Nat. Prod. Rep. 2003, 20, 382. [DOI] [PubMed] [Google Scholar]
- (6).Honjo T; Habu S Origin of Immune Diversity: Genetic Variation and Selection. Annu. Rev. Biochem. 1985, 54, 803–830. [DOI] [PubMed] [Google Scholar]
- (7).Kim E; Moore BS; Joon Yoon Y Reinvigorating Natural Product Combinatorial Biosynthesis with Synthetic Biology. Nat. Chem. Biol. 2015, 11, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cane DE; Kudo F; Kinoshita K; Khosla C Precursor-Directed Biosynthesis: Biochemical Basis of the Remarkable Selectivity of the Erythromycin Polyketide Synthase toward Unsaturated Triketides. Chem. Biol. 2002, 9, 131–142. [DOI] [PubMed] [Google Scholar]
- (9).Haygood MG; Schmidt EW; Davidson SK; Faulkner DJ Microbial Symbionts of Marine Invertebrates: Opportunities for Microbial Biotechnology. J. Mol. Microbiol. Biotechnol. 1999, 1, 33–43. [PubMed] [Google Scholar]
- (10).Schmidt EW; Nelson JT; Rasko DA; Sudek S; Eisen JA; Haygood MG; Ravel J Patellamide A and C Biosynthesis by a Microcin-like Pathway in Prochloron Didemni, the Cyanobacterial Symbiont of Lissoclinum Patella. Proc. Natl. Acad. Sci. U S A 2005, 102, 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Donia MS; Ravel J; Schmidt EW A Global Assembly Line for Cyanobactins. Nat. Chem. Biol. 2008, 4, 341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Donia MS; Schmidt EW Linking Chemistry and Genetics in the Growing Cyanobactin Natural Products Family. Chem. Biol. 2011, 18, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian K-D; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Donia MS; Hathaway BJ; Sudek S; Haygood MG; Rosovitz MJ; Ravel J; Schmidt EW Natural Combinatorial Peptide Libraries in Cyanobacterial Symbionts of Marine Ascidians. Nat. Chem. Biol. 2006, 2, 729–735. [DOI] [PubMed] [Google Scholar]
- (15).Donia MS; Fricke WF; Ravel J; Schmidt EW Variation in Tropical Reef Symbiont Metagenomes Defined by Secondary Metabolism. PLoS One 2011, 6, e17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ruffner DE; Schmidt EW; Heemstra JR Assessing the Combinatorial Potential of the RiPP Cyanobactin tru Pathway. ACS Synth. Biol. 2015, 4, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tianero MDB; Donia MS; Young TS; Schultz PG; Schmidt EW Ribosomal Route to Small-Molecule Diversity. J. Am. Chem. Soc. 2012, 134, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).S Sardar D; Hao Y; Lin Z; Morita M; Nair SK; Schmidt EW Enzymatic N- and C-Protection in Cyanobactin RiPP Natural Products. J. Am. Chem. Soc. 2017, 139, 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Goto Y; Ito Y; Kato Y; Tsunoda S; Suga H One-Pot Synthesis of Azoline-Containing Peptides in a Cell-Free Translation System Integrated with a Posttranslational Cyclodehydratase. Chem. Biol. 2014, 21, 766–774. [DOI] [PubMed] [Google Scholar]
- (20).Houssen WE; Bent AF; McEwan AR; Pieiller N; Tabudravu J; Koehnke J; Mann G; Adaba RI; Thomas L; Hawas UW; Liu H; Schwarz-Linek U; Smith MCM; Naismith JH; Jaspars M An Efficient Method for the in Vitro Production of Azol(in)e-Based Cyclic Peptides. Angew. Chem. Int. Ed. 2014, 53, 14171–14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McIntosh JA; Robertson CR; Agarwal V; Nair SK; Bulaj GW; Schmidt EW Circular Logic: Nonribosomal Peptide-like Macrocyclization with a Ribosomal Peptide Catalyst. J. Am. Chem. Soc. 2010, 132, 15499–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Thibodeaux CJ; Ha T; Van Der Donk WA A Price to Pay for Relaxed Substrate Specificity: A Comparative Kinetic Analysis of the Class II Lanthipeptide Synthetases ProcM and HalM2. J. Am. Chem. Soc. 2014, 136, 17513–17529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mukherjee S; van der Donk WA Mechanistic Studies on the Substrate Tolerant Lanthipeptide Synthetase ProcM. J. Am. Chem. Soc. 2014, 136, 10450–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tang W; Van Der Donk WA Structural Characterization of Four Prochlorosins: A Novel Class of Lantipeptides Produced by Planktonic Marine Cyanobacteria. Biochemistry 2012, 51, 4271–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li B; Sher D; Kelly L; Shi Y; Huang K; Knerr PJ; Joewono I; Rusch D; Chisholm SW; van der Donk WA Catalytic Promiscuity in the Biosynthesis of Cyclic Peptide Secondary Metabolites in Planktonic Marine Cyanobacteria. Proc. Natl. Acad. Sci. U S A 2010, 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Q; Yang X; Wang H; Van Der Donk WA High Divergence of the Precursor Peptides in Combinatorial Lanthipeptide Biosynthesis. ACS Chem. Biol. 2014, 9, 2686–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pan SJ; Link AJ Sequence Diversity in the Lasso Peptide Framework: Discovery of Functional Microcin J25 Variants with Multiple Amino Acid Substitutions. J. Am. Chem. Soc. 2011, 133, 5016–5023. [DOI] [PubMed] [Google Scholar]
- (28).Pavlova O; Mukhopadhyay J; Sineva E; Ebright RH; Severinov K Systematic Structure-Activity Analysis of Microcin J25. J. Biol. Chem. 2008, 283, 25589–25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Islam MR; Shioya K; Nagao J; Nishie M; Jikuya H; Zendo T; Nakayama J; Sonomoto K Evaluation of Essential and Variable Residues of Nukacin ISK-1 by NNK Scanning. Mol. Microbiol. 2009, 72, 1438–1447. [DOI] [PubMed] [Google Scholar]
- (30).Appleyard AN; Choi S; Read DM; Lightfoot A; Boakes S; Hoffmann A; Chopra I; Bierbaum G; Rudd BAM; Dawson MJ; Cortes J Dissecting Structural and Functional Diversity of the Lantibiotic Mersacidin. Chem. Biol. 2009, 16, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rink R; Wierenga J; Kuipers A; Kluskens LD; Driessen AJM; Kuipers OP; Moll GN Production of Dehydroamino Acid-Containing Peptides by Lactococcus lactis. Appl. Environ. Microbiol. 2007, 73, 1792–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huo L; Okesli A; Zhao M; van der Donk WA Insights into the Biosynthesis of Duramycin. Appl. Environ. Microbiol. 2017, 83, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Widdick DA; Dodd HM; Barraille P; White J; Stein TH; Chater KF; Gasson MJ; Bibb MJ Cloning and Engineering of the Cinnamycin Biosynthetic Gene Cluster from Streptomyces cinnamoneus cinnamoneus DSM 40005. Proc. Natl. Acad. Sci. U S A 2003, 100, 4316–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hou Y; Tianero MDB; Kwan JC; Wyche TP; Michel CR; Ellis GA; Vazquez-Rivera E; Braun DR; Rose WE; Schmidt EW; Bugni TS Structure and Biosynthesis of the Antibiotic Bottromycin D. Org. Lett. 2012, 14, 5050–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lin Z; Torres JP; Tianero MD; Kwan JC; Schmidt EW Origin of Chemical Diversity in Prochloron-Tunicate Symbiosis. Appl. Environ. Microbiol. 2016, 82, 3450–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Noike M; Matsui T; Ooya K; Sasaki I; Ohtaki S; Hamano Y; Maruyama C; Ishikawa J; Satoh Y; Ito H; Morita H; Dairi T A Peptide Ligase and the Ribosome Cooperate to Synthesize the Peptide Pheganomycin. Nat. Chem. Biol. 2015, 11, 71–76. [DOI] [PubMed] [Google Scholar]
- (37).Craik DJ; Malik U Cyclotide Biosynthesis. Curr. Opin. Chem. Biol. 2013, 17, 546–554. [DOI] [PubMed] [Google Scholar]
- (38).Shim YY; Young LW; Arnison PG; Gilding E; Reaney MJT Proposed Systematic Nomenclature for Orbitides. J. Nat. Prod. 2015, 78, 645–652. [DOI] [PubMed] [Google Scholar]
- (39).Ziemert N; Ishida K; Liaimer A; Hertweck C; Dittmann E Ribosomal Synthesis of Tricyclic Depsipeptides in Bloom-Forming Cyanobacteria. Angew. Chem. Int. Ed. 2008, 47, 7756–7759. [DOI] [PubMed] [Google Scholar]
- (40).Ahmed MN; Reyna-González E; Schmid B; Wiebach V; Süssmuth RD; Dittmann E; Fewer DP Phylogenomic Analysis of the Microviridin Biosynthetic Pathway Coupled with Targeted Chemo-Enzymatic Synthesis Yields Potent Protease Inhibitors. ACS Chem. Biol. 2017, 12, 1538–1546. [DOI] [PubMed] [Google Scholar]
- (41).Ding W; Liu W; Jia Y; Li Y; van der Donk WA; Zhang Q Biosynthetic Investigation of Phomopsins Reveals a Widespread Pathway for Ribosomal Natural Products in Ascomycetes. Proc. Natl. Acad. Sci. U S A 2016, 113, 3521–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ye Y; Minami A; Igarashi Y; Izumikawa M; Umemura M; Nagano N; Machida M; Kawahara T; Shin-ya K; Gomi K; Oikawa H Unveiling the Biosynthetic Pathway of the Ribosomally Synthesized and Post-Translationally Modified Peptide Ustiloxin B in Filamentous Fungi. Angew. Chem. Int. Ed. 2016, 55, 8072–8075. [DOI] [PubMed] [Google Scholar]
- (43).Sardar D; Pierce E; McIntosh JA; Schmidt EW Recognition Sequences and Substrate Evolution in Cyanobactin Biosynthesis. ACS Synth. Biol. 2015, 4, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cubillos-Ruiz A; Berta-Thompson JW; Becker JW; van der Donk WA; Chisholm SW Evolutionary Radiation of Lanthipeptides in Marine Cyanobacteria. Proc. Natl. Acad. Sci. U S A 2017, 114, E5424–E5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lee J; Mcintosh J; Hathaway BJ; Schmidt EW Using Marine Natural Products to Discover a Protease That Catalyzes Peptide Macrocyclization of Diverse Substrates. J. Am. Chem. Soc. 2009, 131, 2122–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McIntosh JA; Schmidt EW Marine Molecular Machines: Heterocyclization in Cyanobactin Biosynthesis. ChemBioChem 2010, 11, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mcintosh JA; Donia MS; Schmidt EW Insights into Heterocyclization from Two Highly Similar Enzymes. J. Am. Chem. Soc. 2010, 132, 4089–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Koehnke J; Mann G; Bent AF; Ludewig H; Shirran S; Botting C; Lebl T; Houssen WE; Jaspars M; Naismith JH Structural Analysis of Leader Peptide Binding Enables Leader-Free Cyanobactin Processing. Nat. Chem. Biol. 2015, 11, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Koehnke J; Bent AF; Zollman D; Smith K; Houssen WE; Zhu X; Mann G; Lebl T; Scharff R; Shirran S; Botting CH; Jaspars M; Schwarz-Linek U; Naismith JH The Cyanobactin Heterocyclase Enzyme: A Processive Adenylase That Operates with a Defined Order of Reaction. Angew. Chem., Int. Ed. 2013, 52, 13991–13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Tianero MD; Pierce E; Raghuraman S; Sardar D; McIntosh JA; Heemstra JR; Schonrock Z; Covington BC; Maschek JA; Cox JE; Bachmann BO; Olivera BM; Ruffner DE; Schmidt EW Metabolic Model for Diversity-Generating Biosynthesis. Proc. Natl. Acad. Sci. U S A 2016, 113, 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).McIntosh JA; Donia MS; Nair SK; Schmidt EW Enzymatic Basis of Ribosomal Peptide Prenylation in Cyanobacteria. J. Am. Chem. Soc. 2011, 133, 13698–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hao Y; Pierce E; Roe D; Morita M; McIntosh JA; Agarwal V; Cheatham TE; Schmidt EW; Nair SK Molecular Basis for the Broad Substrate Selectivity of a Peptide Prenyltransferase. Proc. Natl. Acad. Sci. U S A 2016, 113, 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Burkhart BJ; Hudson GA; Dunbar KL; Mitchell DA A Prevalent Peptide-Binding Domain Guides Ribosomal Natural Product Biosynthesis. Nat. Chem. Biol. 2015, 11, 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Abts A; Montalban-Lopez M; Kuipers OP; Smits SH; Schmitt L NisC Binds the FxLx Motif of the Nisin Leader Peptide. Biochemistry 2013, 52, 5387–5395. [DOI] [PubMed] [Google Scholar]
- (55).Ortega MA; Hao Y; Zhang Q; Walker MC; van der Donk WA; Nair SK Structure and Mechanism of the tRNA-Dependent Lantibiotic Dehydratase NisB. Nature 2014, 517, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Li K; Condurso HL; Li G; Ding Y; Bruner SD Structural Basis for Precursor Protein–directed Ribosomal Peptide Macrocyclization. Nat. Chem. Biol. 2016, 12, 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Weiz AR; Ishida K; Makower K; Ziemert N; Hertweck C; Dittmann E Leader Peptide and a Membrane Protein Scaffold Guide the Biosynthesis of the Tricyclic Peptide Microviridin. Chem. Biol. 2011, 18, 1413–1421. [DOI] [PubMed] [Google Scholar]
- (58).Oman TJ; Knerr PJ; Bindman NA; Velásquez JE; Van Der Donk WA An Engineered Lantibiotic Synthetase That Does Not Require a Leader Peptide on Its Substrate. J. Am. Chem. Soc. 2012, 134, 6952–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Reyna-González E; Schmid B; Petras D; Süssmuth RD; Dittmann E Leader Peptide-Free In Vitro Reconstitution of Microviridin Biosynthesis Enables Design of Synthetic Protease-Targeted Libraries. Angew. Chem., Int. Ed. 2016, 55, 9398–9401. [DOI] [PubMed] [Google Scholar]
- (60).Burkhart BJ; Kakkar N; Hudson GA; van der Donk WA; Mitchell DA Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent. Sci. 2017, 3, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Renata H; Wang ZJ; Arnold FH Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chemie Int. Ed. 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Weber SS; Polli F; Boer R; Bovenberg RAL; Driessen AJM Increased Penicillin Production in Penicillium Chrysogenum Production Strains via Balanced Overexpression of Isopenicillin N Acyltransferase. Appl. Environ. Microbiol. 2012, 78, 7107–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Freeman MF; Helf MJ; Bhushan A; Morinaka BI; Piel J Seven Enzymes Create Extraordinary Molecular Complexity in an Uncultivated Bacterium. Nat. Chem. 2016, 9 (4), 387–395. [DOI] [PubMed] [Google Scholar]