

Abstract
At present there are no drugs for the treatment of chronic liver fibrosis that have been approved by the Food and Drug administration of the United States. Telmisartan, a small-molecule antihypertensive drug, displays antifibrotic activity, but its clinical use is limited because it causes systemic hypotension. Here, we report the scalable and convergent synthesis of macromolecular telmisartan prodrugs optimized for preferential release in diseased liver tissue. We optimized the release of active telmisartan in fibrotic liver to be depot-like (that is, a constant therapeutic concentration) through the molecular design of telmisartan brush-arm star polymers, and show that these lead to improved efficacy and to the avoidance of dose-limiting hypotension in both metabolically and chemically induced mouse models of hepatic fibrosis, as determined by histopathology, enzyme levels in the liver, intact-tissue protein markers, hepatocyte necrosis protection, and gene-expression analyses. In rats and dogs, the prodrugs are retained long-term in liver tissue and have a well-tolerated safety profile. Our findings support the further development of telmisartan prodrugs that enable infrequent dosing in the treatment of liver fibrosis.
Liver cirrhosis, the final stage of hepatic fibrotic disease, is a rising global health issue claiming over 1 million lives (2% of global deaths) in 2010;1,2 within 5 years of being diagnosed with decompensated cirrhosis, the overall patient survival rate is 45%.3 This disease, for which there are currently no FDA-approved therapies, can result from several insults, including non-alcoholic steatohepatitis (NASH), alcoholic liver damage, viral hepatitis forms, and toxic damage.4 The pathogenesis involves deregulated extracellular matrix production including activated hepatic stellate cells (HSCs) and stromal fibroblasts. In preclinical studies, inhibition of components of the renin-angiotensin system (RAS) with small molecule angiotensin II antagonists has been shown to limit stromal cell activation and proliferation, inflammation, and oxidative stress associated with liver fibrosis.5–7 For example, telmisartan (TEL), a known angiotensin II type 1 receptor blocker (ARB) and a widely used antihypertensive drug, has been reported to ameliorate hepatic fibrosis.8 Despite this potential application, the clinical translation of ARBs, including TEL, for non-cardiovascular applications has been hindered by dose-limiting hypotension.9,10 Such limits are even more problematic in advanced liver fibrosis where systemic hypotension often develops and is further accentuated by systemic drug accumulation due to decreased liver metabolism.11
In order to tackle these challenges, we set out to design a macromolecular TEL prodrug with optimized biodistribution (BD) to fibrotic tissue and tunable pharmacokinetic (PK) profiles to ensure preferential release of active TEL in the diseased microenvironment. We envisioned that a macromolecular-conjugate-mediated ARB delivery strategy may overcome the poor therapeutic index often associated with therapies for chronic fibrotic disorders, and provide a clinically translatable therapeutic technology for liver fibrosis.12 Macromolecular drug carriers can alter the biodistribution of small-molecule drugs, with the liver being a major reservoir and clearance organ for neutrally charged ~10-50 nm objects.13,14 Moreover, macromolecules in this size range are able to pass the leaky vasculature associated with chronic inflammatory disease and accumulate in the fibrotic microenvironment.15–17 Despite these advantages, there is often an inverse relationship between scalability and the potential for modularity and complexity of macromolecular prodrugs that limits the rational and modular optimization of material properties in vivo and ultimately hinders their clinical translation (Fig. 1a). Materials that physically encapsulate their therapeutic payload have achieved clinical success in specific disease contexts; however, their formulation depends on the chemical properties of the drug, which limits the ability to vary properties such as drug identity, hydrodynamic diameter (Dh), and drug release kinetics (Fig. 1a, top left).18 In contrast, the reproducible scaling of advanced material architectures that rely on linear multi-step synthetic routes is difficult and may preclude rapid property optimization (Fig. 1a, bottom right).17 Several recent discussions have highlighted the fact that most nanomedicines reported in the literature do not translate to clinical applications.19,20 Though such a phenomenon may be expected for experimental drugs, in our view, this dearth of examples is in part due to a lack of modular synthesis strategies for macromolecular prodrugs that are both versatile and scalable. Here, we present such a strategy for the rational design and optimization of a TEL-based convergent prodrug that reverses liver fibrosis in vivo. Our synthesis is demonstrated to be highly reproducible up to the 100 g scale, which establishes a benchmark for the scalable synthesis of complex macromolecular prodrugs required for drug development and in vivo safety pharmacology studies.
Figure 1. Rational design of TEL-x-MM and semi-batch synthesis of TEL-x-BASP.
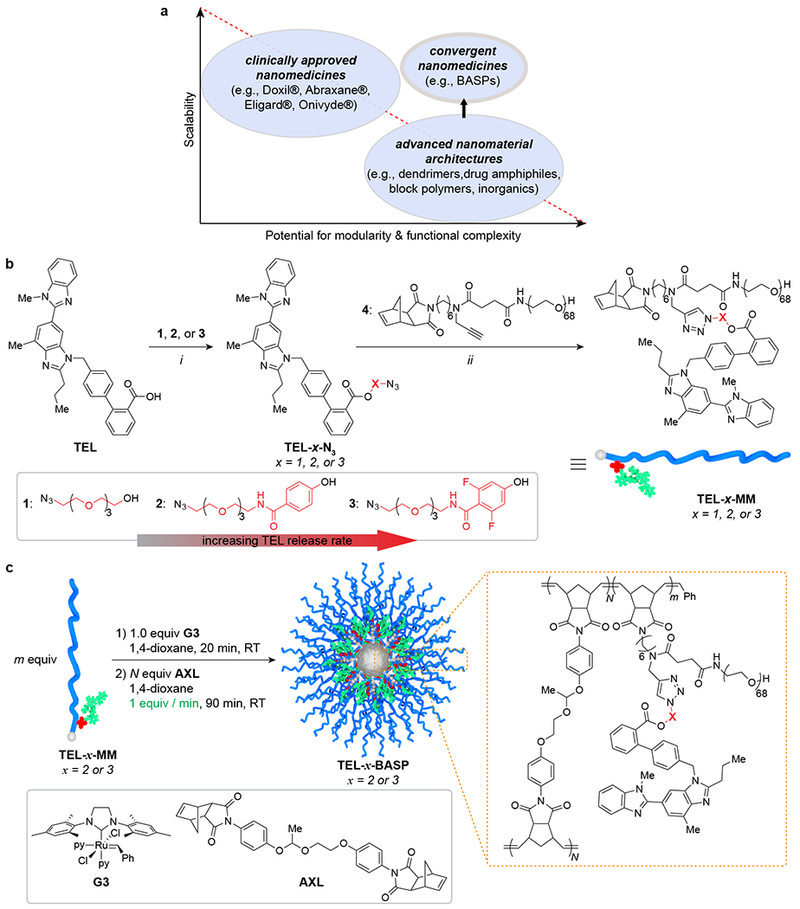
a. Diagram depicting the inverse relationship between scalability and potential for modularity and functional complexity in carriers for drug delivery. Convergent macromolecular prodrugs can potentially satisfy both demands. b. Synthesis of telmisartan macromonomers (TEL-MM) via esterification and CuAAC: i: EDC·HCl, DMAP, CH2Cl2, 16 h, 73% - 93%; ii: CuOAc, CH2Cl2, 1 h, 43% - 62%. Ester linkages between TEL and polymer increase in lability due to stronger electron-withdrawing groups (left to right). c. BASP synthesis via polymerization of TEL-x-MM (1) followed by semi-batch addition of AXL to induce crosslinking (2).
BASPs have Chemically Tunable Release Kinetics in vitro.
Our approach involves a unique macromolecular architecture – the Brush-Arm Star Polymer (BASP) – the synthesis of which was intended to be modular and scalable.21–26 BASPs are prepared in a convergent ring-opening metathesis polymerization (ROMP) process whereby norbornene-based drug-conjugated macromonomers (MMs) are polymerized to generate living bottlebrush polymer “arms” that are subsequently cross-linked.23 The composition and size of BASPs depends on the macromonomer and cross-linker structures and the ratio of polymerization initiator to macromonomer and cross-linker, respectively, all of which can be optimized in parallel for specific applications. Moreover, this approach enables rational control of drug release kinetics through linker design. For example, TEL contains a carboxylic acid functional group that we reasoned could serve as a linker site for conjugation via esterification (Fig. 1b). To identify the ester structure with optimal TEL release kinetics and therapeutic index in vivo, we began our study with the synthesis of three azide-functionalized (N3) TEL-esters – TEL-1-N3, TEL-2-N3, and TEL-3-N3 – from alcohols 1, 2, and 3, respectively (Fig. 1b). These compounds were devised to provide increasing rates of esterase-mediated hydrolysis (i.e., TEL release) via successive stabilization of the alkoxide hydrolysis products through both resonance and inductive effects. To test the validity of this design, TEL-1-N3, TEL-2-N3, and TEL-3-N3 were conjugated to alkyne-containing norbornene-polyethylene glycol (PEG) macromonomer 4 via copper-catalyzed azide alkyne cycloaddition to yield water-soluble macromonomers TEL-1-MM, TEL-2-MM, and TEL-3-MM (Fig. 1b). The pseudo-first order rate constants (± SD) for release of TEL from these macromonomers in the presence of porcine liver esterase (100 units/mL) at 37 °C in phosphate-buffered saline (PBS) solution at pH = 7.6 were 2.9 ± 0.1 × 10−3 min−1 (TEL-1-MM), 4.6 ± 0.4 × 10−3 min−1 (TEL-2-MM), and 8.8 ± 0.7 × 10−3 min−1 (TEL-3-MM) (Fig. S1). Thus, the electron-deficient difluorophenyl-based linker (TEL-3-MM) provides a 3-fold greater release rate than the tetraethylene glycol linker (TEL-1-MM) under these conditions. Moreover, while all of these MMs showed nearly complete TEL release after ~1 day in the presence of esterase in vitro, they showed minimal release (< 15%) after nearly a week in PBS buffer at 37 °C in the absence of esterase. Given that esterases are typically present at lower levels in plasma and at higher concentrations in intracellular lysosomal compartments of inflamed tissue,27 our strategy could favorably preclude the dose-limiting blood-pressure lowering systemic effects of TEL by a preferential esterase-mediated release of TEL in the fibrotic liver microenvironment.
Next, we turned our attention to the synthesis of TEL-x-BASPs (where x corresponds to linker 1, 2, or 3) via ROMP of these TEL-ester macromonomers followed by cross-linking with a degradable acetal-based crosslinker (AXL) (Fig. 1c). With the ultimate goal of clinical translation of these materials, we developed an operationally simple and scalable semi-batch BASP synthesis protocol. In a typical reaction, Grubbs 3rd-generation catalyst (G3, Fig. 1c) was added to a 0.05 M – 0.1 M solution of TEL-x-MM (20 mg – 2 g) in 1,4-dioxane (G3:TEL-x-MM = 1:N) to generate a living TEL-bottlebrush polymer with an average degree of polymerization of N. Next, a 0.1 M solution of AXL (m equiv) in 1,4-dioxane was added by syringe pump at a rate of 1 equiv/min (Fig. 1b). After completion of the cross-linking process as determined by gel permeation chromatography (GPC) (Fig. S2), ethyl vinyl ether (EVE, ca. 100 equivalents) was added to quench the reaction. We screened m and N values to identify optimal ratios for the synthesis of TEL-BASPs with narrow size distributions and hydrodynamic diameters (Dh) of ~25-30 nm, as this size range is considered optimal for passage through the leaky vasculature of fibrotic diseased tissues.16,28,29 We were particularly interested in understanding the impact of m and N on BASP homogeneity, both in terms of size and size distribution, as well as bottlebrush-to-BASP percent conversion (p). Using TEL-2-MM as a test macromonomer, TEL-2-BASPs were prepared using m values of 7, 10, and 13; for each m value a variety of N values were evaluated (Table 1). GPC was used to assess p (Fig. S3), while dynamic light scattering (DLS) was used to measure Dh as functions of m and N. For the range of m and N studied, p varied from 82% to 97% (Table 1). Larger p values were obtained with higher N and lower m values. Dh was fairly consistent ranging from ~20-30 nm; however, for a given m value Dh increased with N (Fig. S8 – S16). Though it is possible to achieve p values as high as 97% in this system, this increased conversion comes at the expense of the TEL mass fraction, which generally decreases with increased N (Table 1). We conducted control experiments to confirm that the semi-batch protocol described herein is superior (as assessed by p value and dispersity) to our previous methods for BASP synthesis (Fig. S4). Based on these results, we proceeded with the m = 10, N = 9 stoichiometry, as it provided an optimal combination of p (84% w/w) and TEL loading (11% w/w theoretical; 10.4 ± 1.1% w/w experimental, see SI section H for details).
Table 1.
Effect of precursor stoichiometry on BASP properties.
| m / TEL-2-MM : G3 | N / AXL : G3 | p / % by weighta | Dh / nmb | theoretical TEL % by weight |
|---|---|---|---|---|
| 7 | 7 | 82 | 22 ± 3 | 11 |
| 7 | 14 | 90 | 24 ± 7 | 9.4 |
| 7 | 21 | 95 | 27 ± 10 | 8.4 |
| 7 | 30 | 97 | 28 ± 7 | 7.6 |
| 10 | 9 | 84 | 24 ± 5 | 11 |
| 10 | 20 | 88 | 28 ± 11 | 9.5 |
| 10 | 30 | 91 | 28 ± 11 | 8.5 |
| 13 | 13 | 82 | 24 ± 7 | 11 |
| 13 | 24 | 87 | 27 ± 8 | 9.6 |
| 13 | 30 | 87 | 36 ± 16 | 9.2 |
| 13 | 40 | 90 | 36 ± 18 | 8.5 |
Conversion from brush to BASP determined by gel permeation chromatography (DMF)
Hydrodynamic diameter determined by dynamic light scattering in H2O; error is reported as polydispersity (PD) from a regularization fitting method.
TEL-2-BASP Displays Optimal Liver BD and PK.
To select the optimal TEL linker for scale-up, the BD and PK profiles of TEL-2-BASP and TEL-3-BASP were investigated; TEL-1-BASP was not pursued further based on the sluggish TEL release kinetics observed for TEL-1-MM. Fluorophore-conjugated TEL-2-BASP and TEL-3-BASP were prepared from TEL-2-MM and TEL-3-MM, respectively (m = 10), a Cyanine7.5-fluorophore-labeled (Cy7.5) macromonomer (0.01 equiv, see SI section B for synthetic details), and AXL (N = 9 equiv) following our optimized semi-batch protocol described above. After dialysis, sterile filtration, and lyophilization, TEL-2-BASP and TEL-3-BASP were examined in vivo. The lyophilized BASP product was reconstituted in PBS (40 mg/ml), sterile filtered, and administered intravenously (i.v., 200 mg/kg) to BALB/c mice (n = 15) with blood and liver tissue samples collected for longitudinal LC-MS/MS measurements of both released and polymer-conjugated TEL (see SI section I.2 for additional details). As shown in Fig. 2a, over the first 10 days following administration, TEL-3-BASP released significantly more TEL in blood compared to TEL-2-BASP. Given our goal of reducing the concentration of free TEL in blood, we selected TEL-2-BASP for analysis of liver PK (Fig. 2b). The results from this study consistently showed ~100-fold greater concentration of released TEL in the liver compared to blood samples. In addition, the concentration of polymer-conjugated TEL was >100-fold greater than released TEL in the liver even 10 days post i.v. administration. The concentration of released TEL reached a steady state over this time period. These data suggest that TEL-2-BASP acts as TEL-releasing depot in the liver, which we hypothesized would enable infrequent dosing compared to generic TEL. Lastly, ex vivo, near-infrared fluorescence images (Cy7.5 fluorophore: λex = 700 nm, λem = 800 nm) of organs harvested from a representative animal sacrificed 7 days post TEL-2-BASP administration revealed preferential accumulation of fluorescence in the liver and the spleen (Fig. 2c).
Figure 2. Pharmacokinetic (PK) and biodistribution (BD) data for TEL-x-BASPs in healthy mice.
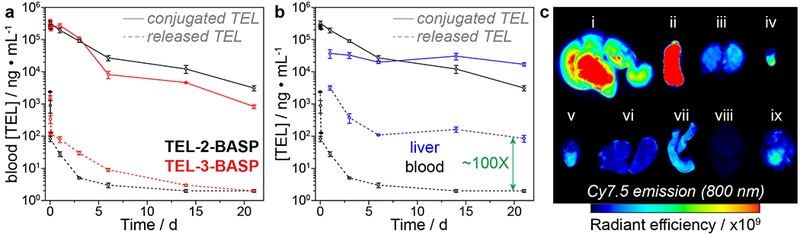
a. Longitudinal whole blood PK measurements of released and conjugated TEL following a single i.v. dose (200 mg/kg BASP) of TEL-2-BASP and TEL-3-BASP (n = 3 animals per time point). b. Comparison of blood and liver tissue PK of released and conjugated TEL following TEL-2-BASP administration (200 mg/kg i.v.). c. Ex vivo fluorescence imaging (800 nm) of mouse organs collected 7 days post administration (300 mg/kg i.v.) of Cy7.5-conjugated TEL-2-BASP; i: liver, ii: spleen, iii: kidney, iv: bladder, v: heart, vi: lung, vii: gut, viii: brain, ix: skeletal muscle. Units of radiant efficiency: (p/sec/cm2/sr)/(μW/cm2). All data reported as mean ± SEM.
TEL-2-BASP Synthesis is Scalable.
Having identified TEL-2-BASP as our optimal candidate for further translation, we turned our attention to scaling-up the synthesis of this macromolecular prodrug. We first investigated the performance of our optimized protocol in the context of a 2.0 g scale TEL-2-BASP synthesis. DLS and transmission electron microscopy (TEM) analysis of TEL-2-BASP prepared on the 0.02 g scale (Fig. 3a) versus the 2 g scale (Fig. 3b) revealed very similar particle sizes and size distributions; p was also similar for these batches. Next, the same procedure was repeated on a 100 g scale (a 5000-fold increase over the 0.02 g scale reaction). Here again, the TEL-2-BASP produced from this batch had the same size and size distribution as the smaller-scale batches (Fig. 3c); however, in this reaction p was improved to 93%. We believe that the improved conversion in the larger scale process is due to superior mixing achieved by the use of a shaft-driven mechanical stirrer rather than a magnetic stir bar. Altogether, these results clearly demonstrate the scalability of our brush-first ROMP TEL-2-BASP synthesis protocol.
Figure. 3. Scalable synthesis of TEL-2-BASP.
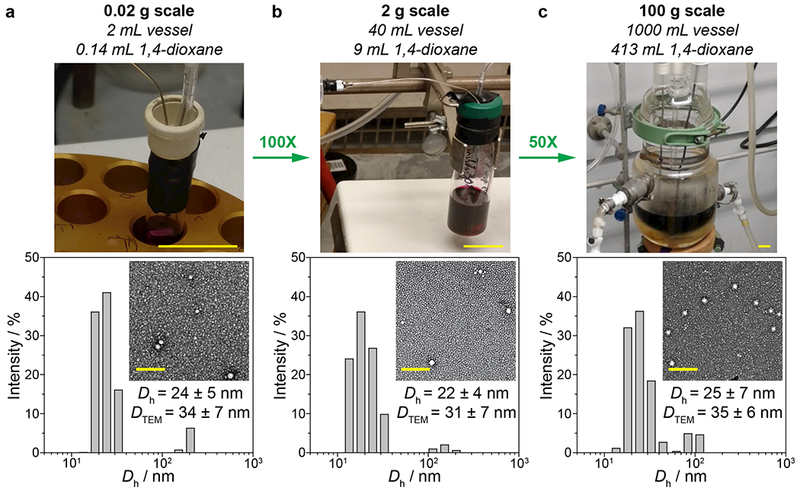
Photographs of reactions, dynamic light scattering histograms, and transmission electron micrographs (uranyl acetate) of 20 mg (a), 2 g (b), and 100 g (c) batches. Scale bar for reaction photographs: 2 cm. Scale bar for micrographs: 200 nm. See Fig. S5 – S7 for GPC traces of TEL-2-BASP.
TEL-2-BASP is Well Tolerated in Mice.
To determine safety and efficacy of TEL-2-BASP, we conducted single and repeat dose safety and toxicology studies in mice. First, to benchmark the potential blood pressure-lowering effects of TEL-2-BASP versus generic TEL (administered at the equivalent dose to the human therapeutic blood pressure-controlling dosage of 10 mg/kg perorally), three groups of mice (n = 8 per group) were dosed with either vehicle control, generic TEL, or TEL-2-BASP at three times the human therapeutic dosage equivalent to generic TEL (300 mg/kg BASP based on 10% TEL loading). As expected, administration of generic TEL led to a statistically significant, transient drop in both systolic (Fig. 4a) and diastolic (Fig. 4b) blood pressures (two-tailed Student’s t test, P < 0.0001) 2 h after administration and returned to normal physiological levels within 24 h, consistent with a reported t1/2 of 6 h and once daily dosing. In normotensive patients, such systemic blood pressure lowering limits the more widespread usage of generic ARBs to treat fibrotic disorders. In addition, blood pressure lowering is prohibitive for usage in patients with advanced liver fibrosis/cirrhosis that have developed systemic hypotension. In contrast to generic TEL, even at 3× the concentration of generic TEL, TEL-2-BASP did not produce a drop in systolic or diastolic blood pressures after 2 h (1st dose) or following a repeat dosing (2nd dose) given to the same animals 7 days after the first dose. These results agree with the blood PK data for TEL-2-BASP (Fig. 2), and support our hypothesis that a macromolecular TEL prodrug with appropriate linker structure can prevent the undesired blood pressure-lowering effects of generic TEL concomitant with achieving significant TEL exposure in fibrotic liver tissue.
Figure 4. Safety and pharmacokinetic analysis of TEL-2-BASP in rodents.
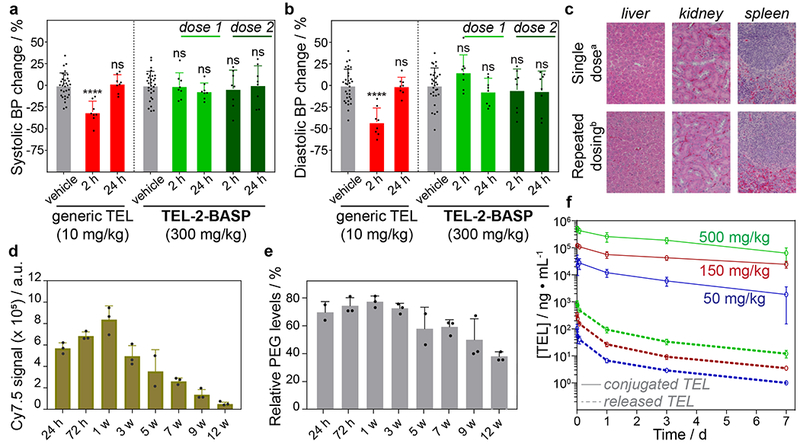
a. Systolic blood pressure measurements in mice following administration of generic TEL (red: single peroral dose, 1X, 10 mg/kg) or TEL-2-BASP (green: repeat i.v. dosing, 3X =300 mg/kg BASP equivalent to 30 mg/kg of telmisartan i.v.) highlighting the blood pressure-sparing TEL-2-BASP formulation. b. Diastolic blood pressure measurements in mice following administration with generic telmisartan (red: single dose, 1X, 10 mg/kg) or TEL-2-BASP (green: repeat dosing, 3X =300 mg/kg BASP equivalent to 30 mg/kg TEL) highlighting the blood pressure-sparing TEL-2-BASP formulation. c. Representative H&E stained photomicrographs (from 1 out of 3 total animals per group) of mouse liver, kidney, and spleen 24 hours after single dose (a300 mg/kg BASP = 30 mg/kg telmisartan) and seven weeks after repeat dosing (b300 mg/kg BASP once a week for 3 weeks) showing no acute or chronic microscopic lesions or PEG-induced vacuolation. d. Quantification of Cy7.5 emission in SDS-PAGE resolved liver tissue homogenates at different times points (24 h – 12 w) following a single dose of TEL-2-BASP (300 mg/kg BASP, n = 3 animals per time point). e. Anti-PEG ELISA assay of liver tissue homogenate at different time points (24 h – 12 w) following a single dose of TEL-2-BASP (300 mg/kg BASP, n = 3 animals per time point) to evaluate PEG clearance. f. Dose-range finding study in rats showing TEL-2-BASP dose proportionality between 50 mg/kg BASP and 500 mg/kg BASP (Maximum Feasible Dosage; MFD) with dose proportional pharmacokinetics. All data reported as mean ± SD; statistical analyses performed with two-tailed Student’s t test (****: P < 0.0001)
To further assess the safety of TEL-2-BASP, mice were given one or multiple doses of the Cy7.5-fluorescently labeled compound. After a single dose, 25 different tissues were harvested from each mouse at day 1, day 3, or week 1, 3, 5, 7, 9, or 12 post dosing. Mice receiving 3 repeat doses weekly had their tissues harvested at week 3, 7, or 12 post dosing. The emission of the Cy7.5 label in the various tissues was assessed by whole organ fluorescence; liver, spleen, and kidney were the tissues with the highest levels of fluorescence. From the mice receiving a single dose of TEL-2-BASP, liver tissues were homogenized (n = 3 animals per time point), resolved on an SDS-PAGE gel, and imaged (Fig. S17). For each time point, the relative Cy7.5 signal was quantified; the results revealed a 7 to 8-fold decrease in overall Cy7.5 signal over a 12 week period (Fig. 4d). Anti-PEG enzyme-linked immunosorbent assay (ELISA) on the same liver homogenate samples also showed a trend of PEG clearance during the course of the study (Fig. 4e and Fig. S18). Combined, these results provide support for eventual clearance of TEL-2-BASP (and smaller polymeric degradation products derived from TEL-2-BASP) despite the seemingly long t1/2. Moreover, hematoxylin and eosin (H&E)-stained sections of the formalin-fixed tissues were submitted for blinded analysis by a Board-Certified Veterinary Pathologist, which included assessment of the presence or absence of cellular cytoplasmic vacuoles that have been previously associated with PEG-ylated test articles in other settings. No PEG-induced vacuoles were observed in any tissues examined (Fig. 4c).30,31
TEL-2-BASP is Well Tolerated in Higher Species.
Having established that TEL-2-BASP is well-tolerated in mice, we next conducted dose-escalation and safety toxicology studies in Sprague Dawley rats, a species commonly used for drug safety/toxicity assessment as part of IND-enabling studies. Following the M3(R2) regulatory guidance,32 rats were first dosed up to the maximum feasible dose (MFD), which is 5 mL/kg for a slow-push bolus i.v. injection. This dose corresponds to 500 mg/kg of TEL-2-BASP, which based on solubility and viscosity considerations is prepared at 100 mg/mL. Importantly, no test article-related clinical observations or effects on body weight, food consumption, hematology, or organ weights in any of the tested dose groups (50, 150, and 500 mg/kg i.v.) were found after a single dose. At necropsy (day 7 post dosing), there were no test article-related macroscopic or microscopic findings in the tissues examined (liver, kidney, spleen, brain, choroid plexus, lung, and heart), and there were no statistically significant differences in serum chemistry parameters between the control group and the test article-treated groups (Tables S1 – S2). Likewise, the differential blood cell counts were normal, showing no evidence of immune activation or blood coagulation as has been reported for some PEG-based materials. Importantly, blood PK analysis over the course of 7 days showed dose-proportionality for both the conjugated and released drug (n = 12; 6 male and 6 female) and revealed no sex differences in the drug release profiles (Fig. 4f). Finally, the toxicity and tolerability of TEL-2-BASP was studied in Beagle dogs, a species widely recognized to be appropriate for repeat-dose toxicity studies. Briefly, TEL-2-BASP was first administered to a single Beagle (slow-push bolus i.v. injection, 5 mL/kg) of each sex (n = 1 animal per sex) at escalating doses of 36 mg/kg, 100 mg/kg, and 500 mg/kg. Injections were spaced approximately 1 week apart. In addition, repeat injections were made at the MFD (500 mg/kg) such that a group of male dogs and a group of female dogs (n = 2 animals per sex) received two 500 mg/kg injections of TEL-2-BASP one week apart. All dogs were telemetrized to enable monitoring of EKG, including blood pressure, pulse pressure, and pulse. The clinical observations and post-necropsy investigations were similar to those determined for rats, confirming the safety of TEL-2-BASP in higher species. Remarkably, there were neither significant differences in serum chemistry values (Tables S3 – S4) nor observed blood pressure changes even at the repeat MFDs; these data are consistent with plasma PK measurements of released TEL well under 400 ng/mL (Fig. S19 – S22). Altogether, we conclude that TEL-2-BASP has an excellent safety profile and is well tolerated in both rodents and dogs.
TEL-2-BASP Shows Sustained Release and Reduces Fibrosis.
The in vivo activity of TEL-2-BASP was first assessed in the STAM metabolic mouse model of nonalcoholic steatohepatitis (NASH) where the liver disease is driven by a combination of diabetes and a high-calorie diet.33,34 Specifically, NASH was induced in male mice (C57BL/6) by injection of a streptozotocin (STZ, 200 μg) solution 2 days after birth, followed by feeding a high fat diet (HFD) starting at 4 weeks of age. The animals (n = 7 per group) were maintained on HFD for 9 weeks and administered two doses of TEL-2-BASP (300 mg/kg i.v.) at the beginning of week 6 and week 7 of the HFD. Animals were then sacrificed and assessed for liver fibrosis by PicroSirius Red (PSR) staining (Fig. S23a). While the TEL-2-BASP treatment showed a statistically significant (P < 0.05) reduction of fibrosis compared to that of the untreated animals (Fig. S23b), the STAM model displayed only a mild fibrosis phenotype consistent with this being a diet-induced model of nonalcoholic fatty liver disease. Hence, we next turned our attention to the effects of TEL-2-BASP in a chemically induced carbon tetrachloride (CCl4) mouse model, which is characterized by a pronounced fibrosis phenotype as evident from our biomarker analyses (vide infra, Fig. 5). Specifically, three groups of mice (n = 10 to 12 per group) were challenged with 1 mL/kg of CCl4 (i.p. twice weekly for 6 weeks) to induce liver fibrosis. Fibrosis was confirmed by week 4 (Fig. 6 and Figs. S24 – S31) based on histology (H&E and PSR), immunohistochemistry, and digital immunofluorescent quantitative analyses (α-smooth muscle actin, nestin, and collagen-1). During the last two weeks of the continuous CCl4 administration, diseased animals were treated with equivalent amounts of TEL over a 2 week period: either 14 doses of generic TEL (10 mg/kg daily perorally, Fig. 5a: grey arrows) or two total doses of TEL-2-BASP (700 mg/kg i.v; 10% TEL loading w/w, Fig. 5a: orange arrows) leveraging the sustained release of TEL from TEL-2-BASP observed in Fig. 2b. It should be noted that a 700 mg/kg TEL-2-BASP dosage is feasible for human translation based on allometric scaling.35 With our dosing regimen, all animals received, on average, the same amount of TEL over the 2 week period (140 mg/kg). Notably, in the CCl4 liver fibrosis model, TEL-2-BASP showed efficacy even during ongoing CCl4 intoxication based on blood biochemistry (Fig. S32), liver function, histology, and biomarker endpoints (Fig. 6 and Figs. S24 – S31). Moreover, both generic TEL and TEL-2-BASP-treated groups showed no significant body weight change compared to healthy or untreated diseased animals over the course of the 14 days treatment regimen (Fig. 5a). Terminal liver tissue PK analysis of released and bound TEL revealed that more than 10% of the systemically injected dose of TEL-2-BASP was present in the diseased liver 7 days post last dosing (Fig. 5b). Moreover, the concentration of polymer-conjugated TEL (221,689 ng/g) was ~170-fold higher than the concentration of generic TEL (6720 ± 619 ng/g) and the amount of released drug from TEL-2-BASP (1843 ± 118 ng/g) was within 3-fold of generic TEL; the large concentration of TEL prodrug in the diseased liver acts as a depot to provide a constant therapeutic concentration of released TEL long after the generic TEL dosage is cleared.
Figure 5. Design and efficacy of TEL-2-BASP and TEL in a chemically induced CCl4 mouse model.
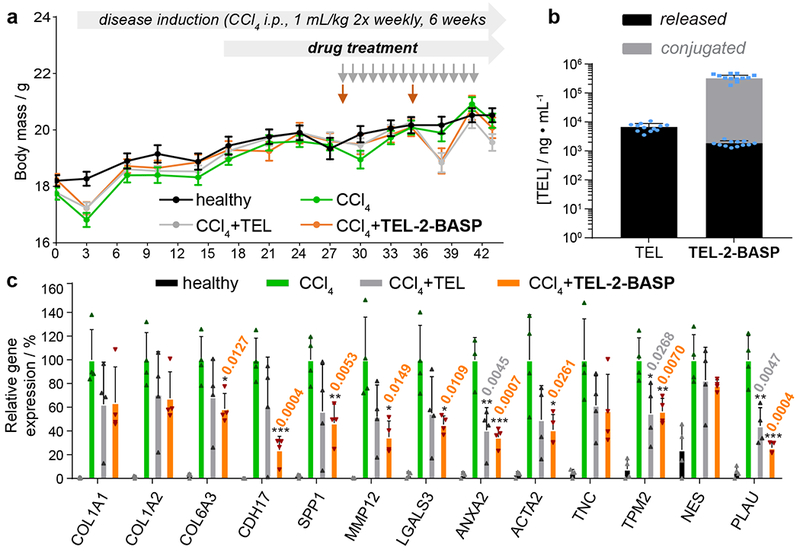
a. Experimental study design for the CCl4-induced mouse liver fibrosis model showing body weight measurement for all treatment groups (healthy, untreated, and test agents: TEL (10 mg/kg, p.o. q.d.; total of 14 doses) and TEL-2-BASP (700 mg/kg i.v. q.w.; total of two doses) during 6 weeks of CCl4 intoxication; data reported as mean ± SEM. b. Terminal liver PK measurements (week 6) for TEL and TEL-2-BASP (released and conjugated TEL); data reported as mean ± SEM. c. Gene expression analysis from a repeat CCl4 efficacy study comparing two doses of TEL-2-BASP (300 mg/kg i.v. q.w.) with daily administration of TEL (10 mg/kg p.o. q.d.; total of 14 doses). Relative expression levels of 13 genes that become over expressed in fibrotic tissue and are suppressed by TEL-2-BASP are shown; data reported as mean ± SD, statistical analyses performed with two-tailed Student’s t test relative to levels of CCl4 group (* = P < 0.05, ** = P < 0.01, *** = P < 0.001). Individual P values are noted above each bar when statistical differences are present.
Figure 6. Assessing fibrosis reversal with PicroSirus Red histological images.
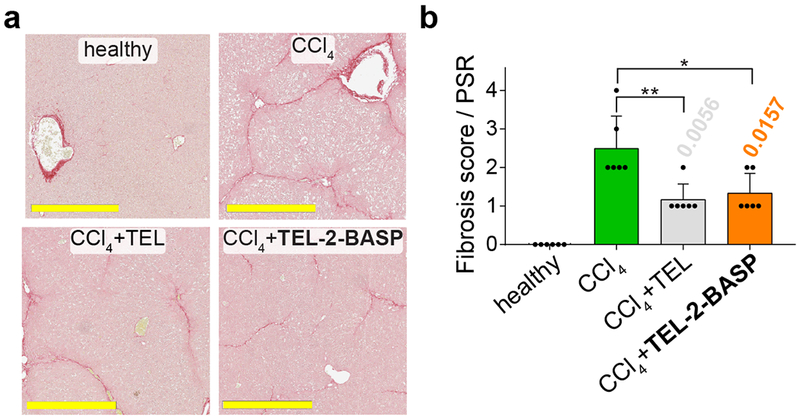
a. PicroSirus Red (PSR) stained images (from a representative animal challenged with CCl4) reveals an overall reduction of fibrosis upon treatment of TEL-2-BASP. For full sets of PSR stained histological images from three separate groups of animals (out of 6 random animals that were analyzed per group), see Figures S24 – S26 (full liver images) and Figures S27 – S29 (magnified liver images). Scale bars = 500 μm. b. Visualization of fibrosis scores (PSR) of healthy, diseased (CCl4), and treated (TEL or TEL-2-BASP) animals (n = 6 random animals analyzed & scored per group). Data reported as mean ± SD, statistical analyses performed with two-tailed Student’s t test (* = P < 0.05, ** = P < 0.01). Individual P values are noted above each bar when statistical differences are present.
Importantly, consistent with the superior biodistribution to diseased liver tissue, TEL-2-BASP exhibited similar-to-improved efficacy compared to generic TEL for all endpoints examined. At the 6-week endpoint, liver enzyme levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and total bilirubin (TB) were significantly decreased in TEL-2-BASP-treated animals compared to untreated or those treated with generic TEL (Fig. S32). Analyses of intact tissue protein biomarkers based on immunohistochemistry and quantitative digital imaging of immunofluorescence, as well as histopathology, confirmed that TEL-2-BASP exerted a significantly superior protective effect against periportal hepatocyte necrosis (Fig. 6 and Figs. S24 – S31). It also was more efficacious in restoring glycogen content (Fig. S33) compared to generic TEL. Histopathology scoring for necrosis and inflammation (Figs. S27 – S29 and S34) and independent apoptosis scoring through cleaved caspase-3 quantification (Fig. S30a) both showed more than a 4-fold reduction in the liver cell death score for TEL-2-BASP-treated animals compared to tissue from the untreated groups. Moreover, stage-specific embryonic antigen 1 (SSEA1) levels were significantly decreased for TEL-2-BASP-treated animals compared to those treated with generic TEL (Fig. S30b). Along with significant hepatocyte protection by TEL-2-BASP treatment, images stained for α-smooth muscle actin, nestin, and collagen-1 (Fig. S31), as well as scoring of PSR histological images by a blinded pathologist (Figs. 6 and S24 – S29), strongly support the notion that TEL-2-BASP efficacy is due at least in part to reversal of fibrosis. In a repeat CCl4 efficacy study, we found that TEL-2-BASP reduced mRNA expression from 13 genes representative of inflammation and extracellular matrix synthesis and turn-over in fibrotic tissues, lending further credence to an anti-fibrotic mechanism of action via microenvironment modulation (Fig. 5c). From these data collectively, we conclude that despite an infrequent dosing paradigm (i.e., weekly), the efficacy of TEL-2-BASP significantly exceeds that of generic TEL administered daily in this mouse model of liver fibrosis.
Outlook.
We have shown that the optimization of the design and synthesis of macromolecular telmisartan prodrugs can enable superior biodistribution and release of the active drug in fibrotic diseased tissues, concomitant with a low release of the active drug systemically. The superior efficacy and unique pharmacokinetic profile of TEL-BASP, with a depot-like release and long half-life of active released TEL in the diseased tissue, suggests that an attractive infrequent dosing schedule might be feasible in the clinic. Overall, TEL-BASPs may enable tissue-microenvironment-targeted therapies for chronic inflammatory and fibrotic conditions. Moreover, the modularity and scalability of BASP synthesis demonstrated here make BASPs attractive for a range of additional applications in nanomedicine.
Methods
Detailed methods are described in the Supplementary Information.
General Procedure for Synthesis of TEL-MM.
A 20-mL scintillation vial was charged with one of three TEL-N3 compounds (TEL-1-N3, TEL-2-N3, or TEL-3-N3) or Cy7.5-N3, PEG-Alkyne-MM, and a stir bar. In a nitrogen filled glovebox, the reagents were dissolved in CH2Cl2 (1 mL CH2Cl2 per 100 mg PEG-alkyne-MM) to which was added approximately 3 equiv of copper (I) acetate. The reaction was stirred until consumption of PEG-alkyne-MM (ca. 1 h) was observed by LC-MS analysis. As the relative extinction coefficients of the TEL-N3 and PEG-alkyne-MM are vastly different (the former being much larger), it is important that a small amount of TEL-N3 is visible by LC-MS. It is difficult to remove unreacted PEG-Alkyne-MM and its presence is detrimental to the subsequent ROMP processes. The crude product was purified by prep-HPLC or prep-GPC. For material purified by prep-HPLC, fractions containing product were concentrated by rotary evaporation and the resulting residue was redissolved in CH2Cl2 and dried over sodium sulfate. After concentration by rotary evaporation, the solid polymer was washed with cold diethyl ether by centrifugation of solids and decantation of ether. This wash step was repeated three times to afford the TEL-MM or Cy7.5-MM as white and green solids, respectively. For material purified by prep-GPC, the crude reaction mixture was first concentrated and taken up in minimal CH2Cl2. The solution was passed thru a short column of neutral alumina (~10% MeOH/CH2Cl2) and the eluent was concentrated by rotary evaporation. Then after purification by prep-GPC, fractions containing product were concentrated by rotary evaporation. The resulting solid was washed with cold diethyl ether three times (as above) to afford TEL-MM as a white powder.
General Procedure for Synthesis of TEL-BASP.
TEL-2-MM (2.00 g, 9.9 equiv) and Cy7.5-MM (20.1 mg, 0.1 equiv) were added to a 40 mL vial charged with a Teflon-coated stir bar and septum. In a nitrogen-filled glovebox, the following solutions were prepared: G3 (49.8 mg in 2.49 mL 1,4-dioxane = 0.02 g/mL) and AXL (304 mg in 5.24 mL 1,4-dioxane = 0.1 M). To a stirred solution of MM (3.07 mL 1,4-dioxane) was added an aliquot of G3 (1.75 mL, 1 equiv) as a single stream. The final volume in the reaction vial (3.07 mL + 1.75 mL = 4.82 mL) affords a MM concentration of 0.1 M. Upon initiating ROMP, the reaction was allowed to proceed for 15 – 20 minutes before removing from the glovebox and transferring immediately to a Schlenk line under positive N2 pressure. A 10 mL syringe fitted with a stainless steel needle was charged with the solution of AXL solution (5.24 mL, 0.1 M), and the assembly was placed into a syringe pump (KD Scientific, Legato 100). The tip of the stainless steel needle was carefully placed just under the level of the ROMP reaction medium, and AXL solution (4.34 mL, 9 equiv) was directly infused at a rate of 1 equiv/min. Upon completion of AXL addition, the crosslinking reaction was allowed to proceed for approximately 120 minutes before quenching with ethyl vinyl ether (0.300 mL). The material was diluted with nanopure H2O (ca. 9.5 mL, 1:1 dilution) before transferring to dialysis tubing (RC, 8 kDa MWCO) and dialyzing against nanopure H2O (15 L, 3 × 2 – 3 h cycles). The contents of the dialysis tubing were then transferred to clean 20 mL vials and lyophilized (at least 24 h) to afford a dry powder (2.1 g, 90%).
In vitro TEL Release Assay.
Approximately 5 mg of a given MM (TEL-1-MM, TEL-2-MM, TEL-3-MM) was weighed into a clean 4 mL vial. Separately, a stock solution of porcine liver esterase (17 units/mg solid) was made from 24 mg of solid esterase powder and 4 mL of PBS (pH 7.6) such that the final activity of esterase in solution was approximately 100 units/mL. Each vial containing 5 mg of a given MM was dissolved in 1 mL of the esterase stock solution. The polymer solutions were briefly vortexed and immediately portioned into 1 mL vials with Teflon-coated caps. All vials were sealed and placed in a 37 °C static incubation oven. At each time point, a vial for a given MM was removed from the incubation oven and cooled to room temperature. To each vial was added 200 μL DMSO, and the resulting solution was briefly vortexed before filtering through a 0.45 μm Nylon syringe filter. Analysis by LC provided insight to the amount of TEL-MM remaining at a given time point.
BD and PK Analysis in vivo.
The lyophilized BASP product was reconstituted in PBS (40 mg/mL), sterile filtered, and administered intravenously (i.v., 200 mg/kg) to BALB/c mice (n = 15) with blood and liver tissue samples collected for longitudinal LC-MS/MS measurements of both free (“unconjugated”) and polymer conjugated TEL. The presence of polymer could be quantified in liver homogenate by Cy7.5 imaging after resolution on an SDS-PAGE gel following a higher dose of TEL-2-BASP (300 mg/kg). Similarly, PEG clearance was measured by anti-PEG ELISA (Fig. 4e and Fig. S17). For the ELISA assay, liver tissues were homogenized and lysates prepared in T-PER reagent (Tissue protein extraction reagent, ThermoFisher Scientific) and normalized for protein concentration. Anti-PEG ELISA was performed on the liver homogenates following standard procedures (PEGylated protein ELISA kit, ab133065, Abcam). Briefly, test samples were added to 96-well plate coated with monoclonal antibody specific to PEGylated protein. This is followed by addition of biotinylated PEG protein and incubation at room temperature for 30 min. The wells are washed to remove excess unbound protein and a solution of streptavidin-HRP conjugate is then added. Following incubation, excess reagents are washed and TMB substrate is added and further incubated at room temperature. Stop solution is then added and O.D. absorbance was measured at 450 nm using a SpectraMax plate reader.
CCl4 Mouse Model.
Female mice (BALB/c) were ordered from Beijing Vital River Laboratory Animal Co. Ltd and hosted at PharmaLegacy Laboratories in a separate room required due to the toxicity of CCl4. The animals were specific pathogen free and approximately 6 – 7 weeks old upon arrival at PharmaLegacy Laboratories vivarium where they were housed in clear polycarbonate plastic cages (260 mm × 160 mm × 127 mm); 5 animals per cage. All procedures were performed in accordance with PharmaLegacy regulations and were approved by PharmaLegacy Laboratories IACUC. A health inspection was performed on each animal to include evaluation of the coat, extremities and orifices. Each animal was also examined for any abnormal signs in posture or movement. The period of acclimatization was 7 days. Animals had ad libitum access to rodent food (irradiated, Shanghai SLAC Laboratory Animal Co. Ltd., China). The bedding material was autoclaved corn-cob bedding (Shanghai MaoSheng Biologic Science & Technology Development Co., Ltd., China) that was changed twice per week. The room was supplied with HEPA filtered air at the rate of 15 – 25 air changes per hour. The temperature was maintained at 20 – 26 °C (68 – 79 °F) with a relative humidity of 40 – 70%. Temperature and humidity were continuously monitored and recorded. Illumination was fluorescent light for 12-hour light (08:00 – 20:00) and 12-hour dark (Cat# MO1-F). Animals were assigned to treatment groups (n = 10 – 12 animals per group) by randomization in BioBook software to achieve similar group mean weight, which provided for control of bias. Additionally, disease animals (CCl4 group) were assigned to the different groups according to their ALT/AST levels first and body weight second after 4 weeks of CCl4 treatment. Animals were injected intraperitoneally with CCl4 (i.p.) 1 mL/kg (4 mL/kg of 25% CCl4 in olive oil), twice per week for a total period of 6 weeks with test agents administrated during the last two weeks of study. Induction of fibrosis was assessed first in both male and female BALB/c mice by PicroSirius Red staining of harvested liver tissue. The aggressive, bi-weekly, intraperitoneal CCl4 dosing regimen led to a more consistent level of fibrosis in female mice compared to male mice. The male mice displayed higher variability and mortality rates during the 6 week continued CCl4 induction. Hence, female mice were chosen so there would be consistency across treatment groups when comparing effect sizes. Telmisartan (TEL) was formulated from dry powder in 0.5% hydroxyl propyl methyl cellulose (HPMC) with 0.2% Tween-80 solution and the test agent was administrated by daily oral gavage. TEL-2-BASP (lyophilized material) was reconstituted in PBS on the day of dosing (70 mg/mL solution), sterile filter (0.22 μM) and dosed intravenously according to body weight (i.e. 200 μL per 20 g animal; total of two doses). At the conclusion of the study, the whole liver tissue was flushed quickly with ice-cold PBS, blotted briefly on paper towel, and weighed. Liver tissue was dissected into pieces: the right lobe was fixed in 10% neutral formalin; the left lobe was shock frozen in liquid nitrogen for tissue PK analysis. For tissue fixation, tissues were placed into 10% neutral buffered formalin (NBF; 20X volume) and incubated overnight (at least 12 hours) at 4 °C. The following day the tissue was moved to room temperature for 4-5 hours, before being washed twice with PBS and stored at 4 °C for subsequent paraffin embedding and IHC processing. Animal blood samples (non-fasting) were collected at sacrifice for terminal PK analysis (whole blood in EDTA tubes) and plasma was processed prepared by refrigerated centrifugation within 30 min of collection (2000 × g for 10 minutes at 4 °C) and the plasma levels of alanine transferase (ALT), aspartate transaminase (AST) and total bilirubin (TB) were measured using an automatic biochemistry analyzer (HITACHI 7020).
Supplementary Material
Acknowledgements
Funding was provided by XTuit Pharmaceuticals. J.A.J. acknowledges the National Institutes of Health (1R01CA220468-01) for support of this work. M.R.G. acknowledges the National Institutes of Health for a postdoctoral fellowship (1F32EB023101). H.V.-T. N. thanks the National Science Foundation for a Graduate Research Fellowship. We thank Dr. R. Bronson for assistance with histopathology analysis.
Footnotes
Reporting Summary. Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability. The authors declare that all data supporting the findings of this study are available within the paper and its Supporting Information.
Competing interests
J.L., J.N.A, M.V.S, S.J.H, B.V., K.D.E, A.M.N., J.C.A., J.B., S.P., S.W.B., E.J.H., J.K.S-S., P.W.K., D.E.C., and P.B.-J. are former employees and shareholders of XTuit Pharmaceuticals. P.B.-J. is President and Founder of Acrivon Therapeutics. J.A.J. is a Co-Founder of Acrivon Therapeutics.
Supplementary Information is available for this paper at ________.
References
- 1.Mokdad AA et al. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Medicine 12, 145, doi: 10.1186/s12916-014-0145-y (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL, Sheppard D, Duffield JS & Violette S Therapy for Fibrotic Diseases: Nearing the Starting Line. Sci. Transl. Med 5, 167sr161, doi: 10.1126/scitranslmed.3004700 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Fleming KM, Aithal GP, Card TR & West J All-cause mortality in people with cirrhosis compared with the general population: a population-based cohort study. Liver International 32, 79–84, doi: 10.1111/j.1478-3231.2011.02517.x (2012). [DOI] [PubMed] [Google Scholar]
- 4.Trautwein C, Friedman SL, Schuppan D & Pinzani M Hepatic fibrosis: Concept to treatment. J. Hepatol 62, S15–S24, doi: 10.1016/j.jhep.2015.02.039 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Lee YA, Wallace MC & Friedman SL Pathobiology of liver fibrosis: a translational success story. Gut 64, 830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wynn TA Cellular and molecular mechanisms of fibrosis. J. Pathol 214, 199–210, doi: 10.1002/path.2277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bataller R & Brenner DA Liver fibrosis. J. Clin. Investig 115, 209–218, doi: 10.1172/jci24282 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgescu EF, Ionescu R, Niculescu M, Mogoanta L & Vancica L Angiotensin-receptor blockers as therapy for mild- to-moderate hypertension-associated non-alcoholic steatohepatitis. World J. Gastroenterol 15, 942–954, doi: 10.3748/wjg.15.942 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boehringer-Ingelheim. Micardis [package insert]. Ridgefield, CT: (2014). [Google Scholar]
- 10.Chauhan VP et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun 4, 2516, doi: 10.1038/ncomms3516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge PS & Runyon BA Treatment of Patients with Cirrhosis. N. Engl. J. Med 375, 767–777, doi: 10.1056/NEJMra1504367 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Hamidreza NL a. Polymeric Conjugates for drug delivery. Chem. Mater 24, 840–853, doi: 10.1021/cm2031569.Polymeric (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinnear C, Moore TL, Rodriguez-lorenzo L, Rothen-rutishauser B & Petri-fink A Form Follows Function : Nanoparticle Shape and Its Implications for Nanomedicine. Chem. Rev 117, 11476–11521, doi: 10.1021/acs.chemrev.7b00194 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Blanco E, Shen H & Ferrari M Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol 33, 941–951, doi: 10.1038/nbt.3330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giannitrapani L, Soresi M, Bondì ML, Montalto G & Cervello M Nanotechnology applications for the therapy of liver fibrosis. World J. Gastroenterol 20, 7242–7251, doi: 10.3748/wjg.v20.i23.7242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conner SD & Schmid SL Regulated portals of entry into the cell. Nature 422, 37–44, doi: 10.1038/nature01451 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Bartneck M, Warzecha KT & Tacke F Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg. Nutr 3, 364–376, doi: 10.3978/j.issn.2304-3881.2014.11.02 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmad Z, Shah A, Siddiq M & Kraatz HB Polymeric micelles as drug delivery vehicles. RSC Adv 4, 17028–17028, doi: 10.1039/c3ra47370h (2014). [DOI] [Google Scholar]
- 19.Leroux J-C Drug Delivery: Too Much Complexity, Not Enough Reproducibility? Angew. Chem. Int. Ed 56, Early View, doi: 10.1002/anie.201709002 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Bobo D, Robinson KJ, Islam J, Thurecht KJ & Corrie SR Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res 33, 2373–2387, doi: 10.1007/s11095-016-1958-5 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Johnson JA et al. Drug-Loaded, Bivalent-Bottle-Brush Polymers by Graft-through ROMP. Macromolecules 43, 10326–10335, doi: 10.1021/ma1021506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JA et al. Core-Clickable PEG-Branch-Azide Bivalent-Bottle-Brush Polymers by ROMP: Grafting-Through and Clicking-To. J. Am. Chem. Soc 133, 559–566, doi: 10.1021/ja108441d (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J et al. “Brush-first” method for the parallel synthesis of photocleavable, nitroxide-labeled poly(ethylene glycol) star polymers. J. Am. Chem. Soc 134, 16337–16344, doi: 10.1021/ja3067176 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Liao L et al. A convergent synthetic platform for single-nanoparticle combination cancer therapy: Ratiometric loading and controlled release of cisplatin, doxorubicin, and camptothecin. J. Am. Chem. Soc 136, 5896–5899, doi: 10.1021/ja502011g (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao AX, Liao L & Johnson JA Synthesis of acid-labile PEG and PEG-doxorubicin-conjugate nanoparticles via Brush-First ROMP. ACS Macro Lett 3, 854–857, doi: 10.1021/mz5004097 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnes JC et al. Using an RNAi Signature Assay to Guide the Design of Three-Drug-Conjugated Nanoparticles with Validated Mechanisms, in Vivo Efficacy, and Low Toxicity. J. Am. Chem. Soc 138, 12494–12501, doi: 10.1021/jacs.6b06321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liederer BM & Borchardt RT Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci 95, 1177–1195, doi: 10.1002/jps.20542 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Cabral H et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotech 6, 815–823, doi: 10.1038/nnano.2011.166 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Jain Rakesh K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 26, 605–622, doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumann A, Tuerck D, Prabhu S, Dickmann L & Sims J Pharmacokinetics, metabolism and distribution of PEGs and PEGylated proteins: Quo vadis? Drug Discov. Today 19, 1623–1631, doi: 10.1016/j.drudis.2014.06.002 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Ivens IA et al. PEGylated Biopharmaceuticals. Toxicol. Pathol 43, 959–983, doi: 10.1177/0192623315591171 (2015). [DOI] [PubMed] [Google Scholar]
- 32.M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals <https://www.fda.gov/downloads/drugs/guidances/ucm073246.pdf> (2010). [PubMed]
- 33.Fujii M et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol 46, 141–152, doi: 10.1007/s00795-013-0016-1 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Takaura K et al. Characterization of Non-Alcoholic Steatohepatitis-derived Hepatocellular Carcinoma as a Human Stratification Model in Mice. Anticancer Res 34, 4849–4855 (2014). [PubMed] [Google Scholar]
- 35.Nair AB & Jacob S A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharma 7, 27–31, doi: 10.4103/0976-0105.177703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.