

Abstract
Background
The phosphatidylinositol 3‐kinase (PI3K) pathway is frequently altered in cancer. This report describes the landscape of PI3K alterations in solid tumors as well as co‐alterations serving as potential resistance/attenuation mechanisms.
Methods
Consecutive samples were analyzed in a commercial Clinical Laboratory Improvement Amendment‐certified laboratory using comprehensive genomic profiling performed by next‐generation sequencing (315 genes). The co‐alterations evaluated included the Erb‐B2 receptor tyrosine kinase 2 (ERBB2), ERBB3, ERBB4, RAS, MET proto‐oncogene tyrosine kinase (MET), and mitogen‐activated protein kinase kinase (MAP2K) genes as well as tumor protein 53 (TP53), estrogen receptor 1 (ESR1), and androgen receptor (AR).
Results
Alterations in any of 18 PI3K‐pathway associated genes were identified in 44% of 60,991 tumors. Although single base and insertions/deletions (indels) were the most frequent alterations, copy number changes and rearrangements were identified in 11% and 0.9% of patients, respectively. Overall, the most frequently altered genes were PIK3 catalytic subunit α (PIK3CA) (13%), phosphatase and tensin homolog (PTEN) (9%), and serine/threonine kinase 11 (STK11) (5%). Tumor types that frequently harbored at least 1 PI3K alteration were uterine (77%), cervical (62%), anal (59%), and breast (58%) cancers. Alterations also were discerned frequently in tumors with carcinosarcoma (89%) and squamous cell carcinoma (62%) histologies. Tumors with a greater likelihood of co‐occurring PI3K pathway and MAPK pathway alterations included colorectal cancers (odds ratio [OR], 1.64; P < .001), mesotheliomas (OR, 2.67; P = .024), anal cancers (OR, 1.98; P = .03), and nonsquamous head and neck cancers (OR, 2.03; P = .019). The co‐occurrence of ESR1 and/or AR alterations with PI3K alterations was statistically significant in bladder, colorectal, uterine, prostate, and unknown primary cancers.
Conclusions
Comprehensive genomic profiling reveals altered PI3K‐related genes in 44% of solid malignancies, including rare disease and histology types. The frequency of alterations and the co‐occurrence of resistance pathways vary by tumor type, directly affecting opportunities for targeted therapy.
Keywords: cancer genome, molecular profile, phosphoinositide 3‐kinase catalytic subunit α (PIK3CA), precision oncology, targeted therapy
Short abstract
Comprehensive genomic profiling of solid tumors reveals frequent genetic alterations in several genes of the phosphatidylinositol 3‐kinase (PI3K) pathway. Data from this analysis suggest that in‐depth characterization of the PI3K pathway along with concomitant resistance alterations in other pathways can provide a genomic background for the development of future treatments.
Introduction
The phosphatidylinositol 3‐kinase (PI3K) pathway is frequently deregulated in human cancers, and the catalytic PI3K subunit P110 is the mediator of the effects of many deregulated extracellular tyrosine kinase receptors. Moreover, other major nodes of this pathway, including AKT serine/threonine kinase (AKT) and mammalian target of rapamycin (mTOR), can be activated though constitutive or redundant intracellular processes.1 Negative regulators of the PI3K pathway, such as phosphatase and tensin homolog (PTEN), also have been well characterized. This important cancer pathway is involved in important fluctuating processes that promote malignant growth and resistance.2, 3
Altered PI3K signaling may be caused by several types of genomic alterations, including mutation, amplification, and methylation.4 Indeed, across various solid tumor types sampled from patients with cancer in a hot‐spot analysis of known regions of PI3K pathway genes, at least 1 PI3K pathway alteration was described in 38%.5 Malignancies frequently associated with PI3K alterations include endometrial, breast, lung, and prostate cancers,6, 7, 8 but almost any tumor type can harbor PI3K genomic alterations in a subset of patients.5 Because of the importance of this pathway for human cancers, numerous small molecules targeted to inhibit different steps of its activation have entered clinical development.4, 6
However, despite extensive drug‐development efforts over many years, few PI3K pathway inhibitors actually have been approved. Approvals were obtained for everolimus, temsirolimus (both of which are mTOR inhibitors), and idelalisib (which blocks the p110‐δ subunit of the PI3K enzyme). Recently, copanlisib, a potent PI3K‐α and PI3K‐δ inhibitor (but with significant inhibitory activity against PI3K‐β and PI3K‐γ) was approved for lymphomas.9 However, many other drugs failed to demonstrate clinical efficacy.10, 11 Overall, PI3K inhibitors are characterized by low activity as monotherapies, an absence of well characterized genomic predictive markers, and redundant mechanisms of resistance.12, 13
Recently, targeted therapies developed under a biomarker‐driven rationale have exhibited greater efficacy and a more successful development pathway compared with agents that were developed for unselected patients with cancer.14, 15, 16, 17 However, biomarker‐driven studies are not always successful.18 The reasons why this type of development has been less successful for PI3K‐directed agents are not clear but could be related to the molecules themselves or to co‐existing resistance pathways. Indeed, it is well known that mitogen‐activated protein kinase kinase (MEK) pathway alterations are more common in patients with PI3K signaling anomalies than in those without such anomalies and that MEK anomalies can mediate resistance.19, 20 An in‐depth characterization of the PI3K genomic landscape, along with a description of concomitant genetic alterations that could lead to resistance to pathway inhibition, is urgently needed. Herein, we characterize the PI3K‐related genomic portfolios of 60,991 patients, including rare disease and histology types not previously well assessed, who underwent clinical‐grade next‐generation sequencing.
Materials and Methods
Tissue Sampling
Consecutive samples submitted by thousands of physicians world‐wide were analyzed using a commercial Clinical Laboratory Improvement Amendment‐certified laboratory (Foundation Medicine, Inc, Cambridge, MA; available at: https://www.foundationmedicine.com). Indications for genomic testing were at the discretion of the ordering physicians. Tissue diagnoses were designated according to the pathology report described by the ordering physicians and further verified by a pathologist at Foundation Medicine. DNA was extracted from formalin‐fixed, paraffin‐embedded tissue, as previously described.21 Patient identification was anonymized for the study. Approval for the Foundation Medicine cohort, including a waiver of informed consent and a Health Insurance Portability and Accountability Act of 1996 waiver of authorization, was obtained from the Western Institutional Review Board (protocol no. 20152817).
Next‐Generation Sequencing
DNA was extracted from formalin‐fixed, paraffin‐embedded sections, and comprehensive genomic profiling was performed on hybridization‐captured, adaptor ligation‐based libraries to a median depth of coverage of >500X.21 The platform simultaneously sequenced the coding regions of 315 cancer‐related genes plus introns from 28 genes that often are rearranged or altered in cancer. Alterations captured by next‐generation sequencing included base‐pair substitutions, insertions/deletions (both short and long), copy‐number alterations, and rearrangements.
Clustering of Genetic Alterations and Tumor Types
Genomic alterations were classified as activators of the PI3K pathway (18 genes) or mediators of PI3K resistance (Supporting Table 1). An analysis of frequencies was performed according to disease ontologies (clustered according to the American Joint Committee on Cancer’s AJCC Cancer Staging Handbook, seventh edition)22 and also according to tumor histologies (according to the ordering physician’s pathology report).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, La Jolla, CA), Python 2.7 (Python Software Foundation, Beaverton, OR), and Anaconda (version 4‐4.3.21; Anaconda Inc, Austin, TX). A co‐occurrence analysis was performing matching genomic alterations in the PI3K pathway with 3 different subsets of genomic alterations (the mitogen‐activated kinase [MAPK] pathway, the tumor protein 53 [TP53] pathway, and the estrogen receptor 1 [ESR1] and/or androgen receptor [AR] [hormone receptor] pathway).
Results
Alterations in any gene of the PI3K pathway were identified in 44% of the 60,991 tumors analyzed (Fig. 1). The most frequently altered gene was PIK3 catalytic subunit α (PIK3CA) (13.3%), followed by PTEN (9.4%), serine/threonine kinase 11 (STK11) (4.8%), and F‐box and WD repeat domain containing 7 (FBXW7) (3.4%) (Fig. 1C,F). Alterations in some genes were rare in our population, including regulatory‐associated protein of mTOR/independent companion of mTOR complex 2 (RICTOR) (0.02%), PIK3 regulatory subunit 2 (PIK3R2) (0.36%), and inositol polyphosphatate‐4‐phosphatase type IIB (INPP4B) (0.43%). Important variations in the frequency of PI3K alterations were detected according to histology and tumor type.
Figure 1.
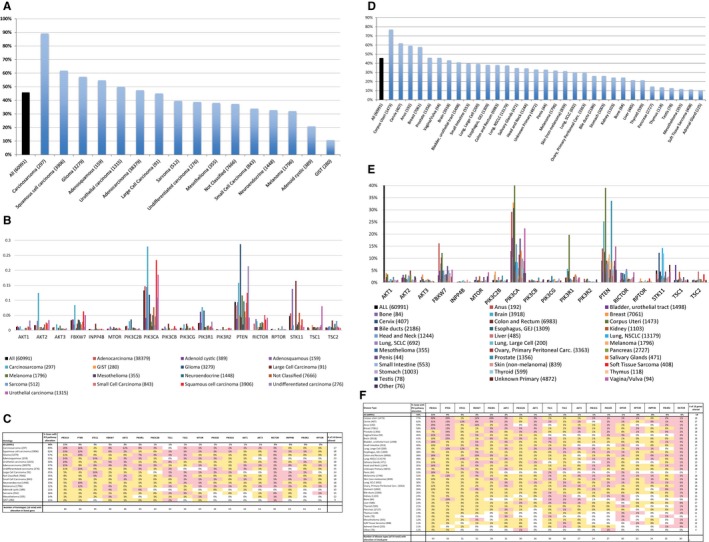
Genetic alterations in the phosphoinositide 3‐kinase (PI3K) pathway are illustrated in patients with cancer. The percentages of patients who had alterations are indicated on the y‐axis. The analysis of alteration frequency (%) was calculated based on at least 1 alteration per patient. Numbers in parentheses indicate the numbers of patients. (A) Results from an analysis of overall alterations are illustrated according to histology. “All” represents all samples, regardless of histology. GIST indicates gastrointestinal stromal tumor. (B) Specific gene alteration frequencies are illustrated according to histology. (C) The percentages of gene alterations are illustrated according to histology corresponding to A and B. The far right column lists the number of genes that were altered for each associated histologic type, and the bottom row indicates the number of histologies (from a total of 16) that had alterations in the listed genes (columns). (D) Results from an analysis of the overall percentages of alterations are illustrated according to disease type. Carc. indicates carcinoma; GEJ, gastroesophageal junction; NSCLC, nonsmall cell lung cancer; SCLC, small cell lung cancer. (E) Percentages of specific gene alterations are illustrated according to disease type. Other types include parathyroid carcinoma, placenta choriocarcinoma, spine ependymoma, soft tissue paraganglioma, spine glioma, and eye lacrimal duct carcinoma. (F) The percentages of gene alterations are illustrated according to disease type corresponding to D and E. The far right column lists the number of genes that were altered in for each associated disease type, and the bottom row indicates the number of disease types (from a total of 34) that had alterations in the listed genes (columns). On charts C and F, pink shading denotes the percentage above the median, yellow shading denotes the percentage below the median; 0% without shading indicates values of 0%, and 0% with shading indicates values between 0.001% and 0.5%. AKT1 indicates AKT serine/threonine kinase 1; AKT2, AKT serine/threonine kinase 2; AKT3, AKT serine/threonine kinase 3; FBXW7, F‐box and WD repeat domain containing 7; INPP4B, inositol polyphosphatate‐4‐phosphatase type IIB; MTOR, mammalian target of rapamycin; PIK3C2B, PIK3 catalytic subunit 2β; PIK3CA, PIK3 catalytic subunit α; PIK3CB, PIK3 catalytic subunit β; PIK3CG, PIK3 catalytic subunit γ; PIK3R1, PIK3 regulatory subunit 1; PIK3R2, PIK3 regulatory subunit 2; PTEN, phosphatase and tensin homolog; RICTOR, regulatory‐associated protein of mTOR/independent companion of mTOR complex 2; RPTOR, regulatory‐associated protein of mTOR complex 1; STK11, serine/threonine kinase 11; TSC1, tuberous sclerosis complex subunit 1; TSC2, tuberous sclerosis complex subunit 2.
Analysis by Histology
The distribution of alterations of 18 genes in the PI3K pathway was analyzed according to histology. Sixteen histologies were evaluated, and all had at least 1 PI3K pathway alteration (Fig. 1A‐C). There was significant variation in the patterns of PI3K alterations between histologies. Carcinosarcomas (89% had PI3K pathway alterations) and squamous cell carcinomas (62% had PI3K pathway alterations) were the most altered histologies, whereas sarcomas (16% had PI3K pathway alterations), mesotheliomas (14% had PI3K pathway alterations), and gastrointestinal stromal tumors (GISTs) (11% had PI3K pathway alterations) were the least common.
PIK3CA was the most frequently altered gene across the 16 histologies (13% overall) and was altered in every histology evaluated (Fig. 1B). Carcinosarcomas had the highest incidence of PIK3CA alteration (28% had PIK3CA alterations), followed by squamous cell carcinomas (23% had PIK3CA alterations), urothelial carcinomas (19% had PIK3CA alterations), adenocarcinomas (15% had PIK3CA alterations), and adenosquamous carcinomas (14% had PIK3CA alterations). Although PIK3CA was altered most frequently across histologies, at a 1.4 times higher rate than PTEN (13% vs 9% had PIK3CA alterations), a subset of histologies had significantly higher rates (P < .0001) of PTEN alterations than PIK3CA alterations: these included gliomas (29% vs 12%), melanomas (12% vs 3%), neuroendocrine tumors (10% vs 5%), and GISTs (5% vs 0%). Although the difference was not significant, PTEN also was altered at a higher rate than PIK3CA in undifferentiated carcinomas (21% vs 11%; P = .0755).
Other alteration rates to note by histology included RICTOR, which was altered at a very low rate overall (0.02% of patients) but was altered in all histologies except adenoid cystic. Urothelial cancers had a significantly higher rate of tuberous sclerosis complex subunit 1 (TSC1) alterations than all other histologies (8% vs 0%‐2%), whereas carcinosarcomas had a significantly higher rate of AKT serine/threonine kinase 2 (AKT2) alterations than other histologies (12% vs 1%‐3%). STK11 was altered at a 5% rate across histologies but was aberrant in 14% and 16% of patients with adenosquamous and large‐cell carcinomas, respectively. Although PIK3R2 and regulatory‐associated protein of mTOR complex 1 (RPTOR) were altered at a slightly higher rate than RICTOR (0.36% and 0.44%, respectively), they were the least frequently altered genes according to histology and were altered in 11 of the 16 histologies at a rate of 1% or less.
Analysis by Disease Type
Six of the 34 disease types had higher rates of PI3K pathway alterations than the overall average of 44%, including 4 of 5 “female” cancers (ie, cancers of the uterus [77% of patients had an alteration], cervix [62%], breast [58%], and vagina/vulva [46%]) (Fig. 1D‐F). The exception was ovarian cancers, which were altered in 30% of patients. Lung cancers, including large cell carcinomas and nonsmall cell lung cancers (NSCLCs), had alterations in 39% and 38% of patients, respectively; whereas small cell lung cancers were altered in 30% of patients. Soft tissue sarcomas and adrenal gland cancers were the least frequently altered (in 11% of patients), whereas testis cancers had the lowest number of altered PI3K pathway genes (only 6 of 18 genes) (Fig. 1E).
PIK3CA was altered most frequently across disease types (13% of patients had an alteration) and was the only gene altered in every disease type. PTEN was altered in 9% of patients and in all but 1 disease type (adrenal gland). Notably, in addition to the female cancers listed above, anus cancer also had a higher than average number of alterations in both PIK3CA and PTEN. Similar to histologic differences, several cancers had significantly higher rates (P < .0001) of PTEN alterations than PIK3CA alterations, including melanoma (12%), brain cancer (25%), prostate cancer (34%), and bone cancer (10%; P = .03). STK11 was altered at a significantly higher rate in cervical cancer, lung large cell carcinoma, and NSCLC, whereas altered FBXW7 was notably more frequent in corpus uteri cancer, anal cancer, colorectal cancer, and melanoma. TSC2 was altered in 5% of liver cancers, compared with 1% overall. Bladder (7%) and kidney (4%) cancers had significant more mutations in TSC1 compared with other disease types (1%).
Uterine cancers
Because uterine cancers had the highest overall rate of PI3K pathway alterations (77% of patients with uterine cancer had alterations vs 62% of those with cervical cancer; P < .0001) and had a significantly higher percentage of PIK3R1 alterations than the next highest disease type (20%; P < .0001), we performed an additional, detailed analysis of the top PI3K pathway genes that were altered in this disease (see Fig. 1E,F). PIK3CA was altered in 40% of patients, followed closely by PTEN. In 2% of patients, PIK3R1 was the only PI3K gene altered, whereas PIK3CA was the only PI3K gene altered in 10% of the uterine cancer cohort (with an overall uterine cohort alteration rate of 40%).
Rare cancers
Rare cancers for which, to our knowledge, comprehensive genomic profiles have not been previously published by The Cancer Genome Atlas (TCGA) (although some have been reported outside of TCGA),23, 24, 25, 26 include adenoid cystic, anus, neuroendocrine, nonmelanoma skin, salivary gland, small intestine, thyroid, vaginal and vulvar, and unknown primaries (Fig. 2A‐I). Unknown primaries differ in their genomic alterations, as might be suspected, based on the histologic differences described above. Thyroid cancers are primarily altered in PIK3CA and PTEN, including a higher proportion of PIK3CA alterations in the anaplastic subtype (12%) compared with well differentiated thyroid cancers (5%). Higher proportions of genomic alterations in FBXW7, MTOR, PIK3CA, and STK11 were detected in vulvar cancers compared with vaginal cancers. Adenoid cystic cancers had very few alterations in the PI3K pathway, but PIK3R1 alterations (6%) were detected more frequently compared with other cancers in general (2%). In anal cancers, higher frequencies of alterations were detected in FBXW7, PIK3CA, and PTEN, which were observed almost exclusively in tumors that had squamous histology compared with basaloid carcinomas. Neuroendocrine cancers had low rates of PIK3CA alterations compared with other cancers (<10%). It is important to note the high frequency of PTEN alterations in prostate small cell carcinomas (35%), similar to prostate cancers overall (34%).
Figure 2.
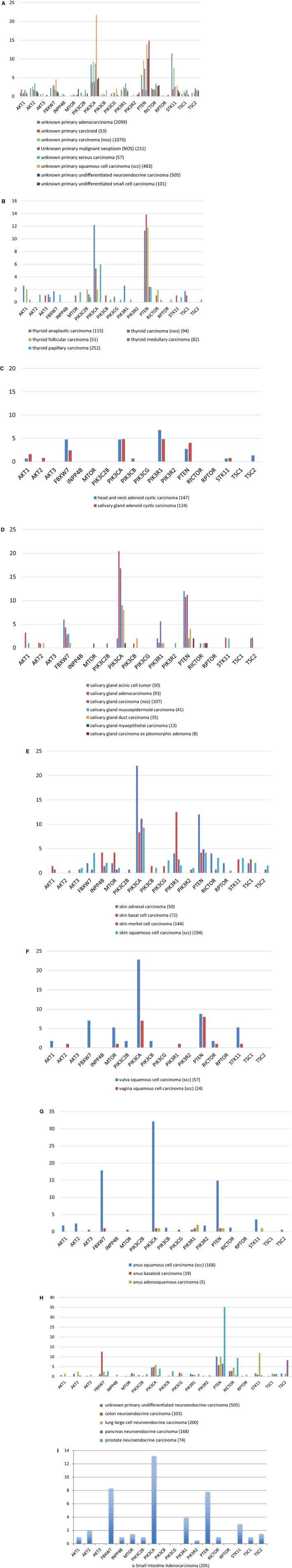
Phosphoinositide 3‐kinase (PI3K) pathway alterations are illustrated in rare or uncommon cancer types. The percentages of patients who had alterations are indicated. Numbers in parentheses represent the numbers of patients. The cancer types illustrated include: (A) unknown primaries, (B) thyroid, (C) adenoid cystic, (D) salivary gland, (E) skin (nonmelanoma), (F) vaginal and vulvar, (G) anus, (H) neuroendocrine tumors, and (I) small intestine. AKT1 indicates AKT serine/threonine kinase 1; AKT2, AKT serine/threonine kinase 2; AKT3, AKT serine/threonine kinase 3; FBXW7, F‐box and WD repeat domain containing 7; INPP4B, inositol polyphosphatate‐4‐phosphatase type IIB; MTOR, mammalian target of rapamycin; NOS, not otherwise specified; PIK3C2B, PIK3 catalytic subunit 2β; PIK3CA, PIK3 catalytic subunit α; PIK3CB, PIK3 catalytic subunit β; PIK3CG, PIK3 catalytic subunit γ; PIK3R1, PIK3 regulatory subunit 1; PIK3R2, PIK3 regulatory subunit 2; PTEN, phosphatase and tensin homolog; RICTOR, regulatory‐associated protein of mTOR/independent companion of mTOR complex 2; RPTOR, regulatory‐associated protein of mTOR complex 1; STK11, serine/threonine kinase 11; TSC1, tuberous sclerosis complex subunit 1; TSC2, tuberous sclerosis complex subunit 2.
Co‐Occurrence of PI3K Alterations and MAPK, TP53, and Hormone Receptor Pathway Alterations
The likelihood of a co‐occurrence of an alteration in the PI3K pathway, in either the MAPK pathway or the hormone receptor pathway, or in TP53 was analyzed by disease type. Some tumors revealed a higher likelihood of a co‐occurrence of alterations in both pathways compared with an isolated alteration, especially for colorectal cancers (odds ratio [OR], 1.64; P < .001), mesotheliomas (OR, 2.67; P = .024), anal cancers (OR, 1.98; P = .03), and nonsquamous head and neck cancers (OR, 2.03; P = .019) (Fig. 3A, Table 1). In contrast, for liver cholangiocarcinomas, endometrial endometrioid tumors, lung and unknown primary squamous cell carcinomas, lobular breast carcinomas, and glioblastomas, alterations in the PI3K or MAPK pathway most likely occurred as isolated events.
Figure 3.
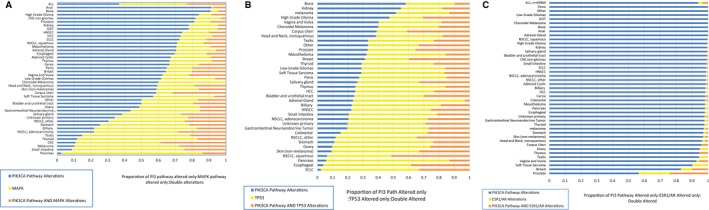
Charts illustrate the co‐occurrence of pathway alterations in the phosphoinositide 3‐kinase (PI3K) pathway and the (A) mitogen‐activated kinase (MAPK) pathway, (B) tumor protein 53 (TP53), and (C) estrogen receptor 1 (ESR1) and/or androgen receptor (AR) (hormone receptor) pathways. (A) The co‐occurrence of alterations in the MAPK pathway and the PI3K pathway is illustrated. The ratio of alterations in the PI3K pathway only, in the MAPK pathway only, and in both pathways is depicted for all disease types, and a significant association is noted between the 2 pathways (P ≤ .05 for co‐occurrence; in total, 14 disease types had a significant association). (B) The co‐occurrence of pathway alterations in the TP53 pathway and the PI3K pathway is illustrated. The ratio of alterations in the PI3K pathway only, in the TP53 pathway only, and in both pathways is depicted for all disease types, and a significant association is noted between the 2 pathways (P ≤ .05 for co‐occurrence; in total, 21 disease types had a significant association). (C) The co‐occurrence of alterations in the hormone receptor pathways and the PI3K pathway is illustrated. The ratio of alterations in the PI3K pathway only, in the ESR1 and/or AR pathway only, or in both pathways is depicted for all disease types, and a significant association is noted between the 2 pathways (P ≤ .05 for co‐occurrence; in total, 33 disease types had a significant association). No alterations in either ESR1 or AR were observed in kidney clear cell carcinoma, cervix adenocarcinoma, bladder urothelial (transitional cell) carcinoma, small intestine adenocarcinoma, breast metaplastic carcinoma, kidney urothelial carcinoma, duodenum adenocarcinoma, cervix squamous cell carcinoma (SCC), lung large cell neuroendocrine carcinoma, salivary gland carcinoma (not otherwise specified [NOS]), head and neck adenoid cystic carcinoma, unknown primary undifferentiated small cell carcinoma, ovary carcinosarcoma, uterus endometrial adenocarcinoma (NOS), uterus endometrial adenocarcinoma endometrioid, thymus carcinoma (NOS), brain ependymoma, or diffuse type stomach adenocarcinoma. CNS indicates central nervous system; CRC, colorectal cancer; GIST, gastrointestinal stromal tumor; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma; NSCLC, nonsmall cell lung cancer; SCLC, small cell lung cancer.
Table 1.
Co‐Occurrence of Alterations Between the Phosphatidylinositol 3‐Kinase Pathway and the Mitogen‐Activated Kinase Pathwaya
| Disease Type | PI3K Pathway Exclusive Alteration | MAPK Pathway Exclusive Alteration | Double Mutant | Neither Alteration | OR for Altered PI3K in MAPK Mutant vs Wild Type | OR for MAPK Mutant in PI3K Altered vs Wild Type | P |
|---|---|---|---|---|---|---|---|
| All tumors | 16,004 | 17,574 | 9932 | 26,450 | 0.96 | 0.96 | .0001 |
| Anal | 107 | 1 | 9 | 93 | 1.68 | 7.29 | .0252 |
| Biliary | 333 | 924 | 261 | 964 | 0.86 | 0.90 | .0341 |
| Cervix | 233 | 61 | 56 | 129 | 0.74 | 0.60 | .0022 |
| CRC | 665 | 3050 | 2351 | 1842 | 1.64 | 1.25 | .0001 |
| Gastrointestinal NET | 169 | 160 | 59 | 666 | 1.33 | 1.34 | .0343 |
| Gliomas, high grade | 1294 | 115 | 130 | 825 | 0.87 | 0.75 | .0159 |
| Gliomas, low grade | 329 | 171 | 34 | 887 | 0.61 | 0.58 | .0013 |
| Head and neck, nonsquamous | 41 | 15 | 13 | 138 | 2.03 | 2.46 | .0185 |
| Melanoma | 160 | 965 | 431 | 444 | 1.17 | 1.06 | .0485 |
| Mesothelioma | 46 | 13 | 6 | 343 | 2.67 | 3.16 | .0235 |
| NSCLC, adenocarcinoma | 1587 | 3633 | 2060 | 3281 | 1.11 | 1.07 | .0001 |
| NSCLC, squamous | 865 | 191 | 160 | 718 | 0.83 | 0.74 | .0025 |
| Pancreas | 61 | 2170 | 353 | 240 | 0.69 | 0.95 | .0056 |
| Salivary gland | 80 | 72 | 55 | 185 | 1.43 | 1.45 | .0125 |
| SCLC | 276 | 61 | 44 | 617 | 1.36 | 1.53 | .0268 |
Abbreviations: CRC: colorectal carcinoma; MAPK, mitogen‐activated kinase; NET: neuroendocrine tumor; NSCLC: nonsmall cell lung cancer; OR, odds ratio; PI3K, phosphatidylinositol 3‐kinase; SCLC: small cell lung cancer.
Only tumors with a statistically significant association are included in this table.
When all tumors were analyzed, there were no significant co‐occurrences between PI3K and TP53 pathway alterations (Fig. 3B, Table 2). Nonetheless, among other tumor types, positive co‐occurrences were detected for some gastrointestinal tumors, including colorectal cancer (OR, 1.55; P < .001), hepatocellular carcinoma (OR, 2.32; P < .001), and gastric cancer (OR, 2.87; P = .006).
Table 2.
Co‐Occurrence of Alterations Between the Phosphatidylinositol 3‐Kinase Pathway and Tumor Protein 53a
| Disease Type | PI3K Pathway Exclusive Alteration | TP53 Alteration | Double Mutant | Neither Alteration | OR for Altered PI3K in TP53 Mutant vs Wild Type | OR for Mutant TP53 in PI3K Altered vs Wild Type | P |
|---|---|---|---|---|---|---|---|
| All tumors | 11,227 | 24,670 | 14,709 | 19,354 | 1.02 | 1.01 | .0835 |
| Adrenal gland | 23 | 72 | 3 | 150 | 0.30 | 0.33 | .0399 |
| Colorectal | 1076 | 4024 | 1940 | 868 | 1.55 | 1.66 | .0001 |
| Corpus uteri | 644 | 329 | 679 | 115 | 0.59 | 0.78 | .0001 |
| Gastrointestinal NET | 96 | 262 | 132 | 564 | 0.98 | 1.00 | .0001 |
| Gliomas, low grade | 234 | 444 | 129 | 614 | 1.82 | 2.02 | .035 |
| HCC | 63 | 131 | 58 | 298 | 2.32 | 2.50 | .0005 |
| HNSCC | 176 | 412 | 197 | 234 | 0.94 | 0.92 | .0007 |
| Kidney | 234 | 116 | 76 | 842 | 0.75 | 0.83 | .0001 |
| Mesothelioma | 35 | 56 | 17 | 300 | 0.91 | 0.91 | .0058 |
| NSCLC, other | 253 | 1121 | 725 | 564 | 0.99 | 0.99 | .0001 |
| NSCLC, squamous | 114 | 751 | 911 | 158 | 1.27 | 1.11 | < .001 |
| Prostate | 360 | 342 | 385 | 543 | 0.43 | 0.54 | .0001 |
| Salivary gland | 60 | 93 | 75 | 164 | 1.33 | 1.34 | .0003 |
| SCLC | 21 | 583 | 299 | 95 | 1.67 | 1.54 | .0005 |
| Skin (nonmelanoma) | 37 | 227 | 130 | 134 | 1.87 | 1.09 | .0007 |
| Soft tissue sarcoma | 35 | 66 | 20 | 397 | 1.10 | 1.07 | .0002 |
| Stomach | 206 | 1201 | 442 | 421 | 2.87 | 2.55 | .0058 |
| Testis | 10 | 13 | 7 | 70 | 0.82 | 0.92 | .0397 |
| Thyroid | 72 | 117 | 58 | 437 | 1.89 | 1.69 | .0001 |
| Unknown primary | 673 | 1730 | 972 | 1391 | 2.34 | 2.11 | .0165 |
| Vagina and vulva | 37 | 38 | 17 | 25 | 1.10 | 1.07 | .0027 |
Abbreviations: HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma; NET, neuroendocrine tumor; OR, odds ratio; PI3K, phosphatidylinositol 3‐kinase; SCC, squamous cell carcinoma; SCLC, small cell lung cancer; TP53, tumor protein 53.
Only tumors with a statistically significant association are included in this table.
A significant co‐occurrence ratio was present between PI3K pathway alterations and hormone receptor alterations in ESR1 and AR (OR, 1.53; P < .01) (Fig. 3C, Table 3). However, this positive correlation was restricted to a few tumor types (bladder, colorectal, corpus uteri, prostate, and unknown primary cancers).
Table 3.
Co‐Occurrence of Alterations Between the Phosphatidylinositol 3‐Kinase Pathway and Estrogen Receptor 1/Androgen Receptora
| Disease Type | PI3K Pathway Exclusive Alteration | ESR1/AR Alteration | Double Mutant | Neither Alteration | OR for Altered PI3K in ESR1/AR Mutant vs Wild Type | OR for Mutant ESR1/AR in PI3K Altered vs Wild Type | P |
|---|---|---|---|---|---|---|---|
| All tumors | 25,025 | 718 | 911 | 43,306 | 1.53 | 2.15 | .0001 |
| Bladder and urothelial tract | 680 | 0 | 6 | 998 | 2.47 | —b | .0045 |
| Colorectal | 2984 | 23 | 32 | 4869 | 1.53 | 2.26 | .0031 |
| Corpus uteri | 1272 | 3 | 51 | 441 | 1.27 | 5.71 | .0003 |
| Prostate | 523 | 183 | 222 | 702 | 1.28 | 1.44 | .0001 |
| Unknown primary | 1627 | 16 | 18 | 3105 | 1.54 | 2.13 | .0292 |
Abbreviations: AR, androgen receptor; ESR1, estrogen receptor 1; OR, odds ratio; PI3K, phosphatidylinositol 3‐kinase pathway; SCLC: small cell lung cancer.
Only tumors with a statistically significant association are included in this table.
Because there were no ESR1/AR alterations, a value could not be computed.
Types of Genetic Alterations
Different types of alterations were identified in the 18 PI3K pathway genes (Supporting Table 2). Only alterations that were considered pathogenic or likely pathogenic were reported. Single nucleotide changes were the predominant genetic alterations in 15 of the18 genes (83%), whereas copy number changes were predominant in AKT2 (1.6%), AKT3 (0.9%), and PIK3 catalytic subunit 2β (PIK3C2B) (1.2%). Although copy number changes and rearrangements were identified infrequently (11% and 0.9%, respectively), they still represent approximately 7300 patients for whom these alterations might inform potential treatments. Of the single nucleotide changes reported, 63% were missense, 29% were nonsense, and 8% were splice variants (Supporting Fig. 1).
Discussion
PI3K pathway alterations are observed frequently in diverse solid tumors, making this pathway an attractive target for cancer therapies. Historically, monotherapy against this pathway has had mixed efficacy. An analysis of specific alterations in the key genes of the pathway and of the co‐occurrence of complementary, activated resistance signals may inform nuanced treatment options and combination strategies that were not previously considered. To our knowledge, this is the largest analysis to date in terms of both the total number of patients evaluated (n = 60,991) and the number of PI3K pathway genes interrogated (18 genes), and it includes data for multiple, rare cancers that were not previously selected by TCGA.
Overall, we report that at least1 PI3K pathway alteration occurred in 44% of tumors. For comparison, our previous analysis included 19,784 solid tumors and reported ≥1 alteration in 38% of patients.5 In that previous study, alterations were restricted to PIK3CA, PTEN, and AKT1 but also included an analysis of PTEN loss by immunohistochemistry (not reported here), which was present in 30% of samples. Several other studies were cataloged by the cBioPortal,27, 28 including an extensive analysis by TCGA and the Memorial Sloan Kettering Impact project.29 This combined analysis included approximately 33,000 patients with various types of solid tumors and indicated that 37% had alterations in ≥1 of the analyzed PI3K pathway genes.
We also focused on reporting PI3K pathway alterations in rare tumor types, for which few or no studies are available.23, 24, 25, 26 Overall, PIK3CA continues to be the most frequently altered gene, predominantly in squamous cell histologies, including anal (32%), vulvar (23%), and unknown primary (22%) sites. Neuroendocrine carcinomas in general were silent for PI3K alterations. Relevant exceptions occurred, 12% of tumors at colon neuroendocrine sites had alterations in FBXW7, and 35% of tumors at prostate neuroendocrine sites had PTEN mutations, mirroring the general tumor site frequency rather than the histology itself.30
Carcinosarcoma was the histologic type with the most frequent number of PI3K pathway alterations (89%). We included 297 samples (predominantly uterine [n = 178] and ovarian [n = 99] in origin), which represent the largest series of molecular‐profiled carcinosarcomas to date. These tumors are rare and usually have a worse prognosis compared with carcinomas of the same anatomic site, and scarce therapeutic options are available.31 The cBioPortal catalog included 78 patients with uterine carcinosarcomas and reported PI3K pathway alterations in 71% of samples.27 Here, we describe a lower frequency of alterations in individual genes compared with previous small series that reported mutations in PIK3CA (41%), PTEN (41%), and PIK3R1 (14%).32 A significantly greater number of samples and a higher proportion of carcinosarcomas of ovarian origin in our series may explain these differences. It is interesting to note our data suggesting that the PI3K landscape of alterations is more similar to that of uterine carcinomas than general sarcomas, reinforcing the biologic relatedness of both.31 An important difference was the greater number of AKT2 alterations in carcinosarcomas (12% vs 3% in uterine cancers in general vs 2% in solid tumors). AKT2 has been implicated in the epithelial‐to‐mesenchymal transition,33 for which carcinosarcomas represent 1 of the best examples in human cancers.
Several reviews have provided a good background describing the difficulties in developing efficacious PI3K pathway inhibitors as well as the paucity of successful clinical trials that resulted in drug approval targeted to specific alterations in this pathway.4, 34 Descriptions of novel genomic alterations in the pathway that predict sensitivity to targeted therapies are emerging.35 It is important to note that the presence of concomitant alterations in alternative pathways may lead to resistance to PI3K inhibitors.19, 36 Conversely, the presence of PI3K alterations can cause resistance to other treatments, especially hormone therapies.37, 38 In the current study, we demonstrated that the co‐occurrence of PI3K pathway alterations and alterations in the MAPK, TP53, and hormone pathways varied according to tumor types, as expected. A higher co‐occurrence rate of PI3K and MAPK alterations was observed in some gastrointestinal tumors, including anal and colorectal cancers, which could significantly affect the activity of PI3K pathway inhibitors.12 ESR1 mutations are being implicated as an evolutionary mechanism of acquired resistance to endocrine manipulation, especially in patients with metastatic, previously treated breast cancer.39 It is also noteworthy that, for the first time, we report the absence of a significant co‐occurrence of ESR1 mutations with PI3K pathway alterations for breast cancer (OR, 1.01; P = .85). Nonetheless, a significant correlation was detected for uterine cancers, suggesting a distinct biologic pathway for the development of resistance between both sites.
AKT1 is 1 of the PI3K pathway genes activated by mutations, including the most frequent glutamic acid‐to‐lysine (E17K) hotspot mutation.40 AKT inhibitor monotherapy has been tested in this setting with initial promising results.41 In this study, we reported AKT1 alterations in 1% of samples. Alterations in AKT1 are identified more frequently in uterine cancers, breast cancers, and undifferentiated carcinomas. We also analyzed genetic alterations in STK11, which recently has been implicated as a resistance mechanism to programmed cell death 1 (PD‐1)/PD‐1 inhibitors.42 Although the overall frequency of this alteration was 5%, it is interesting to note that some tumor types had significantly higher frequencies, including NSCLC (14%), large cell lung cancer (12%), and cervical cancer (12%). These results may be important for the future selection of patients for checkpoint inhibitors, especially in these tumor types.
Our current analysis also highlights the need to use a comprehensive genomic profiling approach to identify the full spectrum of alteration types that might be identified in a gene. Hot‐spot panels and other sequencing methods do not interrogate across all 4 classes of genomic alterations (ie, these panels detect single nucleotide variants but do not identify copy numbers, large insertions and deletions, or rearrangements, which occur in an important minority of patients) (Supporting Table 2). Single nucleotide alterations that are not located in common or known regions of the genes also would be missed using hot‐spot technology.
In conclusion, comprehensive genomic profiling of solid tumors has revealed frequent genetic alterations in several genes of the PI3K pathway. Gynecologic, breast, and prostate cancers, along with carcinosarcoma and squamous cell carcinomas of different sites, more frequently harbor PI3K alterations. Our data also suggest that there are different frequencies of alterations and co‐occurrence patterns of resistance pathways according to tumor types, which can directly affect targeted therapeutic opportunities and clinical trial design.
Funding Support
This study was funded in part by National Cancer Institute grant P30 CA023100 and by the Joan and Irwin Jacobs Fund Philanthropic Fund (all funds were received by Razelle Kurzrock).
Conflict of Interest Disclosures
Denis L. Jardim reports personal fees from Roche outside the submitted work. Razelle Kurzrock reports research funding from Incyte, Genentech, Merck, Serono, Pfizer, Sequenom, Foundation Medicine, and Guardant outside the submitted work; personal fees from X Biotech, Loxo, and Actuate Therapeutics, and Roche outside the submitted work; and has an ownership interest in Curematch Inc. Vincent A. Miller reports personal fees from Revolutions Medicines outside the submitted work. Siraj M. Ali serves on the Incysus Therapeutics Scientific Advisory Board. Sherri Z. Millis, Lee Albacher, Jeffrey S. Ross, Vincent A. Miller, and Siraj M. Ali are employees of Foundation Medicine Inc.
Author Contributions
Sherri Z. Millis: Conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, writing–original draft, and writing–review and editing. Denis L. Jardim: Conceptualization, formal analysis, investigation, methodology, project administration, writing–original draft, and writing–review and editing. Lee Albacker: Funding acquisition, resources, software, and writing–review and editing. Jeffrey S. Ross: Funding acquisition, resources, software, and writing–review and editing. Vincent A. Miller: Funding acquisition, resources, software, and writing–review and editing. Siraj M. Ali: Funding acquisition, resources, software, and writing–review and editing. Razelle Kurzrock: Conceptualization, data curation, funding acquisition, investigation, methodology, project administration, resources, supervision, visualization, writing–original draft, and writing–review and editing.
Supporting information
Contributor Information
Denis L. Jardim, Email: jardimde@gmail.com.
Razelle Kurzrock, Email: kurzrock@ucsd.edu.
References
- 1. Engelman JA. Targeting PI3K signaling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550‐562. [DOI] [PubMed] [Google Scholar]
- 2. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34:3803‐3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol‐3‐kinase pathway alterations across 19,784 diverse solid tumors. JAMA Oncol. 2016;2:1565‐1573. [DOI] [PubMed] [Google Scholar]
- 6. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu G, Xing M, Mambo E, et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7:R609‐R616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. US Food and Drug Administration (FDA) . Drug Approvals and Database. Available at: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm576098.htm. Accessed February 15, 2018.
- 10. Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015;146:53‐60. [DOI] [PubMed] [Google Scholar]
- 11. Cheah CY, Fowler NH. Idelalisib in the management of lymphoma. Blood. 2016;128:331‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janku F, Wheler JJ, Naing A, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early phase clinical trials. Cancer Res. 2013;73:276‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wheler JJ, Molder SL, Naing A, et al. Anastrozole and everolimus in advanced gynecologic and breast malignancies: activity and molecular alterations in the PI3K/AKT/mTOR pathway. Oncotarget. 2014;5:3029‐3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jardim DL, Schwaederle M, Hong DS, Kurzrock R. An appraisal of drug development timelines in the era of precision oncology. Oncotarget. 2016;7:53037‐53046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwaederle M, Zhao M, Lee JJ, et al. Impact of precision medicine in diverse cancers: a meta‐analysis of phase 2 clinical trials. J Clin Oncol. 2015;33:3817‐3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jardim DL, Schwaederle M, Wei C, et al. Impact of a biomarker‐based strategy on oncology drug development: a meta‐analysis of clinical trials leading to FDA approval [serial online]. J Natl Cancer Inst. 2015;107:djv423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 2016;76:3690‐3701. [DOI] [PubMed] [Google Scholar]
- 18. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumor molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicenter, open‐label, proof‐of‐concept, randomized, controlled phase 2 trial. Lancet Oncol. 2015;16:1324‐1334. [DOI] [PubMed] [Google Scholar]
- 19. Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early‐phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsimberidou AM, Hong DS, Ye Y, et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): an MD Anderson precision medicine study. JCO Precis Oncol. 2017;1:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edge S, Byrd DR, Compton CC, Fritz AG, Greene F, Trotti A, eds. AJCC Cancer Staging Handbook. From the AJCC Cancer Staging Manual, 7th edn New York: Springer‐Verlag; 2010. [Google Scholar]
- 23. Ross JS, Wang K, Rand JV, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next‐generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol. 2014;38:235‐238. [DOI] [PubMed] [Google Scholar]
- 24. Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965‐2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris LG, Chandramohan R, West L, et al. The molecular landscape of recurrent and metastatic head and neck cancers: insights from a precision oncology sequencing platform. JAMA Oncol. 2017;3:244‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Groisberg R, Hong DS, Roszik J, et al. Clinical next‐generation sequencing for precision oncology in rare cancers. Mol Cancer Ther. 2018;17:1595‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mohanty SK, Kim SA, DeLair DF, et al. Comparison of metastatic neuroendocrine neoplasms to the breast and primary invasive mammary carcinomas with neuroendocrine differentiation. Mod Pathol. 2016;29:788‐798. [DOI] [PubMed] [Google Scholar]
- 31. Pang A, Carbini M, Moreira AL, Mark RG. Carcinosarcomas and related cancers: tumors caught in the act of epithelial‐mesenchymal transition. J Clin Oncol. 2018;36:210‐216. [DOI] [PubMed] [Google Scholar]
- 32. Jones S, Stransky N, McCord CL, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes [serial online]. Nat Commun. 2014;5:5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lan A, Qi Y, Du J. Akt2 mediates TGF‐β1‐induced epithelial to mesenchymal transition by deactivating GSK3β/snail signaling pathway in renal tubular epithelial cells. Cell Physiol Biochem. 2014;34:368‐382. [DOI] [PubMed] [Google Scholar]
- 34. Massacesi C, Di Tomaso E, Urban P, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Croessmann S, Sheehan JH, Lee KM, et al. PIK3CA C2 domain deletions hyperactivate phosphoinositide 3‐kinase (PI3K), generate oncogene dependence and are exquisitely sensitive to PI3Kα inhibitors. Clin Cancer Res. 2018;24:1426‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858‐2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beltran H, Antonarakis ES, Morris MJ, Attard G. Emerging molecular biomarkers in advanced prostate cancer: translation to the clinic. Am Soc Clin Oncol Educ Book. 2016;35:131‐141. [DOI] [PubMed] [Google Scholar]
- 38. Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross‐talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10(1 pt 2):331S‐336S. [DOI] [PubMed] [Google Scholar]
- 39. Jeselsohn R. Are we ready to use ESR1 mutations in clinical practice? Breast Care (Basel). 2017;12:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bleeker FE, Felicioni L, Buttitta F, et al. AKT1(E17K) in human solid tumors. Oncogene. 2008;27:5648‐5650. [DOI] [PubMed] [Google Scholar]
- 41. Hyman DM, Smyth LM, Donoghue MTA, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35:2251‐2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skoulidis F, Hellmann MD, Awad MM, et al. STK11/LKB1 co‐mutations to predict for de novo resistance to PD‐1/PD‐L1 axis blockade in KRAS‐mutant lung adenocarcinoma [abstract]. J Clin Oncol. 2017;35(15 suppl):9016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials