

Abstract
Background:
The relative prevalence and clinical importance of monogenic mutations related to familial hypercholesterolemia and of high polygenic score (cumulative impact of many common variants) pathways for early-onset myocardial infarction remain uncertain. Whole genome sequencing enables simultaneous ascertainment of both monogenic mutations and polygenic score for each individual.
Methods:
We performed deep-coverage whole genome sequencing of 2,081 patients from four racial subgroups hospitalized in the U.S. with early-onset myocardial infarction (age ≤ 55 years) recruited using a 2:1 female to male enrollment design. We compared these genomes with 3,761 population-based controls. We first identified individuals with a rare, monogenic mutation related to familial hypercholesterolemia. Second, we calculated a recently developed polygenic score of 6.6 million common DNA variants to quantify the cumulative susceptibility conferred by common variants. We defined high polygenic score as the top 5% of the control distribution, as this cutoff has been previously shown to confer similar risk to familial hypercholesterolemia mutations.
Results:
Mean age of the 2,081 patients presenting with early-onset myocardial infarction was 48 years and 66% were female. A familial hypercholesterolemia mutation was present in 36 (1.7%) of these patients and associated with a 3.8-fold (95%CI 2.1 – 6.8; p < 0.001) increased odds of myocardial infarction. 359 (17.3%) of the patients with early-onset myocardial infarction carried a high polygenic score, associated with a 3.7-fold (95%CI 3.1 – 4.6; p < 0.001) increased odds. Mean estimated untreated LDL cholesterol was 206 mg/dl in those with a familial hypercholesterolemia mutation, 132 mg/dl in those with high polygenic score, and 122 mg/dl in those in the remainder of the population. Although associated with increased risk in all racial groups, high polygenic score demonstrated the strongest association in white participants (p-heterogeneity = 0.008).
Conclusions:
Both familial hypercholesterolemia mutations and high polygenic score are associated with a more than three-fold increased odds of early-onset myocardial infarction. However, high polygenic score cannot be reliably identified on the basis of elevated LDL cholesterol and has a ten-fold higher prevalence among patients presenting with early-onset myocardial infarction.
Clinical Trial Registration:
URL: https://clinicaltrials.gov/ct2/show/NCT00597922; Unique identifier: NCT00597922
Keywords: myocardial infarction, genetics, human, familial hypercholesterolemia, polygenic risk score
Background
An increased risk of early-onset myocardial infarction in those with a parental history was first documented in 1951.1 Subsequent research has identified discrete DNA-based underpinnings of heritable risk. An important example is a molecular defect in the gene encoding the LDL receptor (LDLR), identified as a driver of hypercholesterolemia and risk of myocardial infarction in 1985.2 More recent studies have determined that such familial hypercholesterolemia mutations are present in ~1 in 250 individuals in the population and confer a 3- to 4-fold increased risk of early-onset myocardial infarction.3–6
Proposed clinical applications of genomic screening for risk of myocardial infarction have largely focused on finding carriers of rare monogenic mutations, such as those related to familial hypercholesterolemia.4–6 However, a decade of genome-wide association studies (GWAS) has demonstrated that common DNA variants account for the majority of heritable risk for complex diseases.7–10 Polygenic scores quantify the genetic susceptibility conferred by the cumulative impact of common variants into a single, normally distributed quantitative risk factor. We recently developed and validated a polygenic score for myocardial infarction comprised of a genome-wide set of 6.6 million common DNA variants.11 This score demonstrated substantially better predictive capacity than our previously published score restricted to the 50 most significantly associated variants in previous GWAS analyses.12
Whole genome sequencing captures the complete spectrum of genetic variation – both rare and common – but has never been applied at scale to patients with myocardial infarction. We performed whole genome sequencing in 2,081 patients presenting to U.S. hospital with early-onset myocardial infarction from the multiethnic Variation in Recovery: Role of Gender on Outcomes of Young AMI Patients (VIRGO) study.13 We compare the prevalence and clinical impact of monogenic (single large-effect mutation) and polygenic (cumulative effect of many variants of small effect) risk pathways associated with myocardial infarction.
Methods
Data Availability
Whole genome sequencing data for the VIRGO study and MultiEthnic Study of Atherosclerosis study have been uploaded to the database of Genotypes and Phenotypes (dbGaP) repository under accession numbers phs001259 and phs001416, respectively.
The genome-wide polygenic score for myocardial infarction used in this paper is available for download.14 Python scripts used to extract the polygenic score for each individual from a whole genome sequencing variant call format (.vcf) file and R code to standardize the distribution of this score across racial/ethnic groups are provided in Supplemental Code I-III of the online-only Data Supplement.
Study Populations
Patients with early-onset (age ≤ 55 years) myocardial infarction were derived from the previously described VIRGO study.13 VIRGO investigators enrolled a multiethnic population of patients presenting to one of 103 United States hospitals with a myocardial infarction between August 2008 and January 2012 using a 2:1 female-to-male enrollment design. Eligible participants had elevated cardiac biomarkers (troponin I or T or creatine kinase-myocardial band), with at least one biomarker >99th percentile of the upper reference limit within 24 hours of admission. Additional evidence of acute myocardial ischemia was required, including either symptoms of ischemia or electrocardiogram changes indicative of new ischemia. This report represents the first genetic analysis conducted in the VIRGO study and as such, VIRGO data has not been represented in any earlier genetic studies of myocardial infarction.
Population-based controls were derived from the MESA study.15 MESA is a multiethnic prospective cohort that enrolled individuals in the United States free of cardiovascular disease between 2000 and 2002. Whole genome sequencing was performed in MESA participants who consented to genetic study and had sufficient DNA volume in a central lab repository. For this study, sequenced MESA participants were included as controls if they remained free of incident cardiovascular disease over a median follow-up of 13.2 years through the end of 2014 (mean age at end of follow-up 73 years).
Additional details about the VIRGO and MESA studies are provided in the Supplementary Methods of the online-only Data Supplement. Written informed consent was obtained for all study participants by the VIRGO and MESA investigators. The present analysis was approved by the Partners HealthCare (Boston, MA) Institutional Review Board.
Whole Genome Sequencing
Whole genome sequencing was performed using the Illumina HiSeqX platform at the Broad Institute of Harvard and MIT (Cambridge, MA). Reads were aligned to the human reference genome hg19. Whole genome sequencing was performed on 6,033 individuals, of whom 41 were excluded for sample quality control reasons and an additional 150 were excluded due to relatedness (second-degree or closer) to another sample within the study (Table I of the online-only Data Supplement).
In order to minimize potential confounding from whole genome sequencing performed in separate batches for the VIRGO and MESA cohorts, we assembled a single joint variant call set across all participants in this study. We subsequently confirmed that the overall number of variants per individual was similar between VIRGO patients and MESA controls in a race stratified analysis (Figure I of the online-only Data Supplement). Next, we performed an association study of all observed common (allele frequency ≥ 1%) variants and a gene-based burden test aggregating rare (allele frequency < 1%), confirming no significant inflation of test statistics (Figure II of the online-Only Data Supplement).
Additional sequencing parameters are described in the online-only Data Supplement.
Ascertainment of familial hypercholesterolemia mutations
Heterozygous familial hypercholesterolemia is an autosomal dominant genetic condition caused by mutations in any of three genes – LDLR, APOB (apolipoprotein B), or PCSK9 (proprotein convertase subtilisin/kexin type 9).16 Within each of these three genes, we aggregated three classes of mutations as performed previously.4
First, we identified inactivating mutations in LDLR leading to premature truncation of a protein (nonsense), insertions or deletions of DNA sequence that alter the reading frame (frameshift), point mutations at sites of pre-messenger ribonucleic acid splicing that alter the splicing process (splice-site), or structural variants that perturb the LDLR coding sequence. Structural variation was assessed using a targeted analysis for variants in proximity to the LDLR locus (± 1 megabase) using a recently described method.17 Second, we included mutations in LDLR, APOB, or PCSK9 annotated as ‘pathogenic’ or ‘likely pathogenic’ for familial hypercholesterolemia in the ClinVar database.18 Variants with conflicting annotations or those that had been deemed ‘benign’ or ‘likely benign’ were removed. Third, we identified missense variants in LDLR predicted to be damaging or possibly damaging by each of 5 computer prediction algorithms (LRT score, MutationTaster, PolyPhen-2 HumDiv, PolyPhen-2 HumVar, and Sorting Intolerant From Tolerant [SIFT]), as described previously.3, 4
Ascertainment of polygenic score
We recently developed a genome-wide polygenic score for myocardial infarction comprised of 6,630,150 common (allele frequency >1%) variants.11 This score was based on association statistics for millions of variants derived from a previously published GWAS of up to 60,801 individuals with myocardial infarction and 123,504 controls.9 We used a computational algorithm to optimize a polygenic score that integrates the cumulative impact of all available variants.19 We validated this new score in a population of >400,000 individuals of European ancestry from the UK Biobank.20 In the UK Biobank, our previously published polygenic score comprised of only 50 variants demonstrated that the top 5% of the distribution had 2.1-fold increased odds of coronary artery disease as compared with the remainder of the population.11 By contrast, use of the expanded genome-wide polygenic score noted that the top 5% of the polygenic score distribution had 3.3-fold increased odds of coronary artery disease compared with the remainder of the population, a magnitude of risk comparable to that observed in previous studies of familial hypercholesterolemia mutations.3–6 We thus defined high polygenic score as the top 5% of the distribution.
After application of stringent sequencing quality control parameters, 6,286,512 of 6,630,150 (94.8%) of the variants were available for scoring, and this polygenic score was calculated in each of the early-onset myocardial infarction patients and controls (Supplementary Code I & II of the online-only Data Supplement). Additional details are provided in the online-only Data Supplement.
Statistical analysis
Principal components of ancestry facilitate quantification of genetic ancestry and adjustment for these principal components minimizes genetic association test confounding due to population stratification.21 Given the multiethnic population studied here, we fit a linear regression model using the first four principal components of ancestry to predict the polygenic score within control participants. The residual from this model was used to create an ancestry-corrected reference distribution, with those in the top 5% of this distribution considered to have high polygenic score (Supplementary Code III of the online-only Data Supplement).
The relationship of familial hypercholesterolemia mutations and high polygenic score to early-onset myocardial infarction was assessed using logistic regression models adjusted for the first four principal components of ancestry. In order to estimate untreated values for LDL cholesterol, measured values for those reporting use of statin medications were divided by 0.7 as we and others have performed previously.4, 5 Associations of familial hypercholesterolemia mutations and high polygenic score with LDL cholesterol among early-onset myocardial infarction patients were assessed in linear regression models adjusting for age, sex, and the first four principal components of ancestry.
Analyses were performed using R version 3.2.2 software (The R Foundation).
Results
We analyzed whole genome sequencing data of 2,081 patients presenting to U.S. hospitals with early-onset myocardial infarction (Table 1). Mean age of patients was 48 years, 66% were female, and 28% reported use of statins prior to hospitalization. Patients included 1,537 (75%) white, 336 (16%) black, 168 (8%) Hispanic, and 40 (2%) Asian individuals. Racial designations were based on a combination of patient self-report and inferred genotypic ancestry as assessed by principal compenents (detailed in the online-only Data Supplement). The genomes of these 2,081 patients were compared to 3,761 controls, including 1,544 (41%) white, 962 (26%) black, 751 (20%) Hispanic, and 504 (13%) Asian participants. Principal components analysis confirmed that the patients and controls were well-matched with respect to genetic ancestry (Figure 1).
Table 1.
Baseline characteristics of patient with early-onset myocardial infarction and controls
| Early-Onset MI Patients N = 2081 | Controls N = 3761 | |
|---|---|---|
| Race, N (%) | ||
| White | 1537 (74%) | 1544 (41%) |
| Black | 336 (16%) | 962 (26%) |
| Hispanic | 168 ( 8%) | 751 (20%) |
| Asian | 40 ( 1.9%) | 504 (13%) |
| Male sex, N (%) | 709 (34%) | 1731 (46%) |
| Age, years; Mean (SD) | 47.6 (5.9) | 60.3 (9.7) |
| Hypertension, N (%) | 1345 (65%) | 1467 (39%) |
| Diabetes, N (%) | 735 (36%) | 358 ( 9.5%) |
| Current Smoking, N (%) | 1055 (51%) | 437 (12%) |
| Statin Use, N (%) | 575 (28%) | 527 (14%) |
| Lipid Levels, mg/dl | ||
| LDL Cholesterol; Mean (SD)* | 125 (49) | 123 (34) |
| HDL Cholesterol; Mean (SD) | 40 (14) | 53 (15) |
| Triglycerides; Median (Q1—Q3) | 136 [93, 210] | 110 [77, 159] |
MI – myocardial infarction; N – number; SD – standard deviation; LDL – low density lipoprotein; HDL – high density lipoprotein; Q1 – quartile 1; Q3 – quartile 3
In order to estimate untreated values for low-density lipoprotein cholesterole, measured values for those reporting use of statin medications were divided by 0.7.
Figure 1.
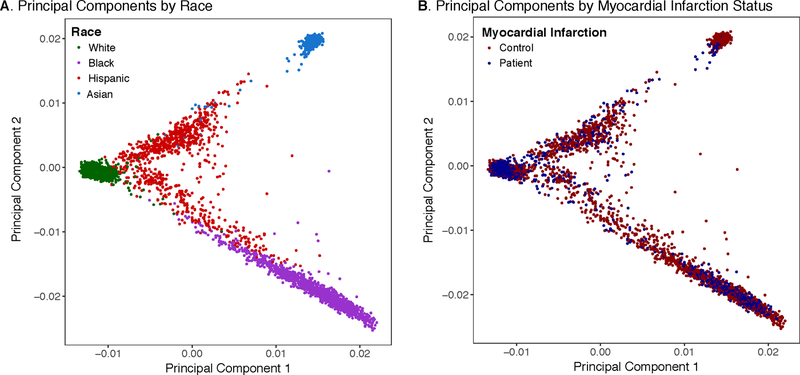
Principal Components of Ancestry According to Race and Myocardial Infarction Status
The first two principal components of ancestry are plotted according to race (A) and patient versus control status (B), confirming that the population were well matched with respect to genetic background. Additional details on principal components calculation are provided in the Supplementary Appendix.
Prevalence and clinical importance of familial hypercholesterolemia mutations
A familial hypercholesterolemia mutation was present in 36 of 2,081 (1.7%) patients with early-onset myocardial infarction. Carriers included 8 individuals with a loss-of-function mutation in LDLR, 12 with a LDLR missense mutation previously annotated as pathogenic in the ClinVar online database, and 16 with a rare LDLR missense mutation predicted to be damaging by each of five computer prediction algorithms. No familial hypercholesterolemia mutations in the APOB or PCSK9 genes were detected in the 2,081 patients.
Of the 8 individuals with a loss-of-function LDLR mutation, one was ascertained only via detailed structural variant analysis – a 7.9 kilobase deletion leading to loss of 4 exons (Figure 2). This mutation was noted in a Hispanic female who presented with a myocardial infarction at age 51. On-statin LDL cholesterol at time of presentation was 193 mg/dl (estimated untreated LDL cholesterol of 276 mg/dl). An advantage of whole genome sequencing is the ability to detect such variants, typically missed in genetic testing based on genotyping array or whole exome sequencing.17
Figure 2. A four-exon deletion of LDLR identified by structural variant analysis of whole genome sequencing.
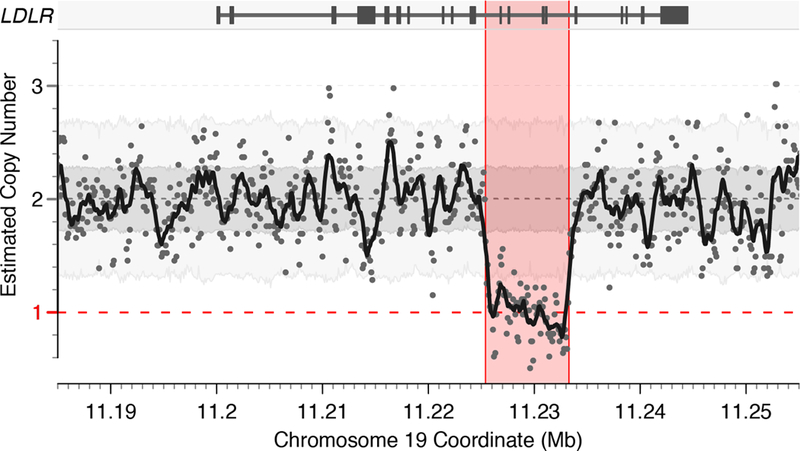
Visualization of copy-number estimates in 100 base pair sequential bins for 5,842 individuals with whole genome sequencing data available. Background shading represents the range of copy-number estimates from whole genome sequencing for the middle 50% and 90% of samples for the darker and lighter shades of gray, respectively. Points represent copy-number estimates per 100 base pair bin in the individual in whom a 7,889 base pair deletion encompassing four exons of LDLR was noted. The solid black line represents the rolling mean copy-number estimate in 1 kb windows. This variant is predicted to result in loss of function of the low-density lipoprotein receptor gene (LDLR) gene resulting in heterozygous familial hypercholesterolemia.
Among early-onset myocardial infarction patients found to be carriers of a familial hypercholesterolemia mutation, 17 of 36 (47%) reported being on a statin prior to presentation. This finding is consistent with previous reports that suggest inadequate recognition and treatment of familial hypercholesterolemia in current clinical practice,22 although it is also conceivable that treated mutation carriers were systematically underrepresented in patients presenting with myocardial infarction. Average estimated untreated LDL cholesterol was 202 mg/dl and 58% had severe hypercholesterolemia (LDL cholesterol ≥ 190 mg/dl). In a model adjusted for age, sex, and principal components of ancestry, LDL cholesterol levels were 82 mg/dl (95%CI 65 –99; p < 0.0001) higher in patients who carried a familial hypercholesterolemia mutation as compared with non-carriers. This effect on increased LDL cholesterol levels was most pronounced in those with a loss-of-function mutation as compared with a LDLR missense mutation (Table II in the online-only Data Supplement).
In aggregate, a familial hypercholesterolemia mutation was present in 36 of 2081 (1.7%) of patients with early-onset myocardial infarction as compared with 23 of 3,761 (0.6%) of controls. Details of observed mutations are provided in Table III of the online-only Data Supplement. In a logistic regression model adjusted for principal components of ancestry, a familial hypercholesterolemia mutation was associated with a 3.76-fold (95%CI 2.12 – 6.82; p < 0.0001) increased odds of early-onset myocardial infarction.
Prevalence and clinical importance high polygenic score
We next calculated a polygenic score comprised of 6.6-million common genetic variants in all participants. As expected given variation in allele frequencies by race, the raw polygenic score distribution varied across the four racial groups. These differences were minimized after correction for principal components of ancestry, as described in Supplemental Code III of the online-only Data Supplement (Figure 3A-B).
Figure 3.

Variation in Polygenic Score Distribution and Clinical Importance According to Race
The distributions of the polygenic score across racial groups within 3,761 control participants are displayed based on raw values (A) and after adjustment for genetic ancestry using the first four principal components (B). Values were scaled to a mean of 0 and standard deviation of 1 to facilitate interpretation. We next determined the relationship between high polygenic score (top 5% of the distribution) and risk of early-onset myocardial infarction (C). Odds ratios were calculated using a logistic regression model adjusted for the first four principal components of ancestry. The clinical importance of high polygenic score varied across racial groupings (p-heterogeneity = 0.008).
MI – myocardial infarction; OR – odds ratio; CI – confidence interval
We noted a significant enrichment of increased polygenic score among patients with early-onset myocardial infarction as compared with controls, with median polygenic score among patients in the 72nd percentile (p < 0.0001); Figure 4. To permit direct comparison to traditional carrier versus non-carrier analyses of monogenic mutations, we analyzed a ‘carrier’ group with high polygenic score (top 5%) versus a reference ‘non-carrier’ group comprised of the remaining 95% of the population. A high polygenic score was present in 359 of 2081 (17.3%) of patients with early-onset myocardial infarction as compared with 188 of 3,761 (5.0%) of controls. In a logistic regression model adjusted for principal components of ancestry, high polygenic score was associated with a 3.73-fold (95%CI 3.06 – 4.56; p < 0.0001) increased odds of early-onset myocardial infarction.
Figure 4.
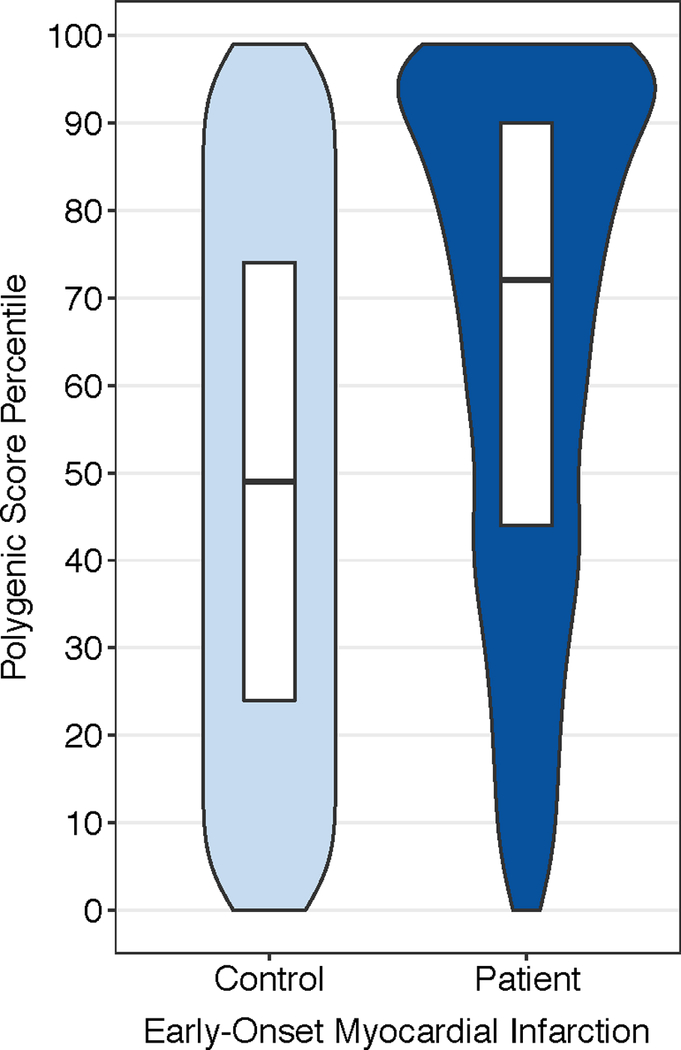
Polygenic Score Percentile Among Early-onset Myocardial Infarction Patients Versus Controls
A reference distribution for polygenic score percentiles adjusted for genetic ancestry was constructed in the control population. Median polygenic score percentile among patients with early-onset myocardial infarction was in the 72nd percentile of the distribution. Violin plots display the polygenic score percentile distribution in patients versus controls. Within the white boxplot insets, the horizontal line in each box indicates the median score, and the top and bottom of the boxes indicate the 75th and 25th percentiles, respectively.
High polygenic score was associated with increased risk within each racial subgroup of patients with early-onset myocardial infarction, but the effect was most pronounced in white participants (p-heterogeneity = 0.008). For example, high polygenic score was associated with a 5.1-fold increased odds among white participants as compared with a 2.0, 3.4, and 3.3-fold increased odds among black, Hispanic, and Asian participants respectively (Figure 3C).
Traditional risk factor assessment would not allow early-onset myocardial infarction patients with high polygenic score to be distinguished from the remainder of the patients (Table IV in the online-only Data Supplement). For example, hypertension was prevalent in 69% of those with high polygenic score versus 64% of the remainder of the distribution (p = 0.13). Diabetes had been diagnosed in 38% of those with high polygenic score as compared with 35% of the remainder of the distribution (p = 0.32). As compared with the remainder of the distribution, individuals with high polygenic score had slightly higher LDL cholesterol (Mean 132 versus 124 mg dl; p = 0.007) and triglycerides (Median 155 versus 133 mg/dl; p = 0.001). The impact of high polygenic score on risk of early-onset myocardial infarction did not vary with respect to age, sex, hypertension, diabetes, current smoking, or circulating lipid levels (p-heterogeneity > 0.05 for each).
Integrated assessment of monogenic and polygenic contributions to early-onset myocardial infarction
We examined the quantitative importance and interplay of monogenic and polygenic risk pathways as they related to inherited risk of myocardial infarction. Among the 2,081 patients with myocardial infarction, 32 (1.5%) carried a familial hypercholesterolemia mutation but did not have high polygenic score, 355 (17.1%) carried high polygenic score but had no familial hypercholesterolemia mutation, and 4 (0.2%) carried both a familial hypercholesterolemia mutation and a high polygenic score.
Assessment of the baseline characteristics of patients with early-onset myocardial infarction across strata of monogenic and polygenic risk was most notable for differences in observed LDL cholesterol levels (Table 2 & Figure 5). Mean estimated untreated values of LDL cholesterol levels were 202 mg/dl in those with only a familial hypercholesterolemia mutation, 130 mg/dl in those with only a high polygenic score, 235 mg/dl in those with both a familial hypercholesterolemia mutation and a high polygenic score, and 122 mg/dl in those with neither.
Table 2.
Baseline Characteristics of Patients with Early-onset Myocardial Infarction According to Genetic Risk Strata
| Neither | Only High Polygenic Score | Only FH Mutation | Both FH Mutation & High Polygenic Score | |
|---|---|---|---|---|
| N | 1690 | 355 | 32 | 4 |
| Race, N (%) | ||||
| White | 1232 (72.9) | 281 (79.2) | 20 (62.5) | 4 (100.0) |
| Black | 296 (17.5) | 35 ( 9.9) | 5 (15.6) | 0 ( 0.0) |
| Hispanic | 129 ( 7.6) | 32 ( 9.0) | 7 (21.9) | 0 ( 0.0) |
| Asian | 33 ( 2.0) | 7 ( 2.0) | 0 ( 0.0) | 0 ( 0.0) |
| Male sex, N (%) | 563 (33.3) | 123 (34.6) | 21 (65.6) | 2 ( 50.0) |
| Age, years; Mean (SD) | 47.6 (5.9) | 47.8 (5.7) | 46.8 (6.5) | 46.3 (10.5) |
| Hypertension, N (%) | 1075 (63.9) | 243 (68.5) | 24 (75.0) | 3 ( 75.0) |
| Diabetes, N (%) | 593 (35.3) | 134 (37.7) | 6 (18.8) | 2 ( 50.0) |
| Current Smoking, N (%) | 848 (50.4) | 190 (53.5) | 14 (43.8) | 3 ( 75.0) |
| Statin Use, N (%) | 445 (26.5) | 113 (31.8) | 15 (46.9) | 2 ( 50.0) |
| Lipid Levels, mg/dl | ||||
| LDL Cholesterol; Mean (SD)* | 122.1 (45.75) | 130.4 (51.0) | 201.5 (82.0) | 235.4 (41.6) |
| HDL Cholesterol; Mean (SD) | 40.7 (13.75) | 38.9 (13.0) | 37.6 (8.1) | 57.8 (24.5) |
| Triglycerides; Median (Q1—Q3) | 133 (91 – 205) | 155 (105 – 222) | 162 (91 – 246) | 102 (82 – 137) |
FH – familial hypercholesterolemia; N – number; SD- standard deviation; LDL – low density lipoprotein; HDL – high density lipoprotein; Q1 – quartile 1; Q3 – quartile 3
In order to estimate untreated values for LDL, measured values for those reporting use of statin medications were divided by 0.7.
Figure 5.
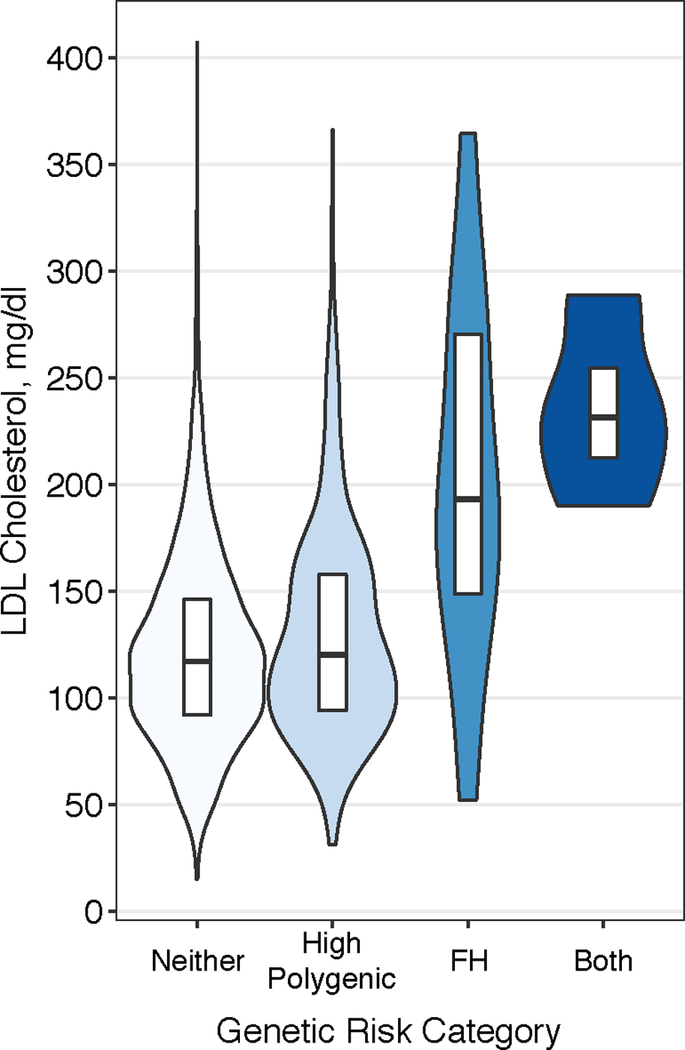
LDL Cholesterol according to Monogenic and Polygenic Risk Strata
Among patients presenting with early-onset myocardial infarction, violin plots display the low-density lipoprotein (LDL) cholesterol distribution according to genetic risk category – only high polygenic score, only familial hypercholesterolemia (FH) mutation, both high polygenic score and familial hypercholesterolemia, or neither. Within the white boxplot insets, the horizontal line in each box indicates the median score, and the top and bottom of the boxes indicate the 75th and 25th percentiles, respectively.
Discussion
We performed whole genome sequencing in 2,081 patients hospitalized for an early-onset myocardial infarction to assess the prevalence and clinical importance of familial hypercholesterolemia mutations and a high polygenic score. We observed a familial hypercholesterolemia mutation in 1.7% of patients and a high polygenic score in 17% of patients, each of which were associated with a more than three-fold increased odds of early-onset myocardial infarction.
These findings may have important clinical implications for the prevention and treatment of early-onset myocardial infarction. High polygenic score was ten times as common as familial hypercholesterolemia mutations among afflicted individuals yet conferred a similar increase in risk. LDL cholesterol levels are available as a biomarker, albeit imperfect, to identify individuals most severely affected by familial hypercholesterolemia mutations without genetic testing. By contrast, no available clinical risk factor or circulating biomarker can reliably identify individuals with a high polygenic score. Moreover, although whole genome sequencing was used to ascertain polygenic scores in the present study, we previously demonstrated that the polygenic scores for a range of diseases can be readily calculated using data from a standard genotyping array – available at a cost of less than 100 U.S dollars.11
If individuals with high polygenic scores were identified prior to suffering a myocardial infarction, the increased risk is modifiable – individuals with high polygenic score derive the greatest absolute risk reduction from adherence to a healthy lifestyle or treatment with statin medications.12, 23, 24 Despite this evidence for increased benefit, only 32% of the patients presenting with an early-onset myocardial infarction and high polygenic score in our study had been prescribed a statin within routine clinical practice prior to hospitalization.
We believe the identification of individuals with high polygenic score at a young age has the potential to facilitate preventive interventions, but several outstanding issues remain. First, polygenic scores are a normally distributed quantitative trait and designation of a threshold for ‘high’ is necessarily arbitrary. For example, if we designated the top 10% of the population distribution as high in our study, high polygenic score would be present in 560 of 2,081 (27%) of patients and associate with a 3.2-fold increase in odds of myocardial infarction. Furthermore, ongoing efforts will seek to seamlessly integrate an individual’s polygenic score with other clinical and lifestyle factors to help guide patient treatment.
Second, additional efforts are needed to optimize polygenic scores in individuals of non-European ancestry.25 We provide a framework for correcting the observed polygenic score for genetic ancestry, effectively creating a uniform distribution across races. Our study additionally demonstrates that high polygenic score has predictive utility across all races (Figure 3), but the prognostic importance remains highest among white individuals. This is a natural consequence of a polygenic score derived from a genome-wide association study performed in primarily white individuals.9 Moving forward, inclusion of a more diverse set of participants in genetic analyses and new computational approaches will enable improved race-specific risk estimates.
Third, we anticipate increased study of how to best disclose polygenic score testing results to healthy individuals. A 2011 landmark paper outlined a framework for genomic medicine and highlighted practical systems for assessment and disclosure of polygenic scores as a key scientific imperative.26 A survey designed to assess interest in the AllofUs Research Program of the Precision Medicine Initiative noted that 90% of people deemed learning about their health as a primary incentive to participate and 74% wanted to receive results from genetic testing.27 However, a robust evidence-base that such disclosure can motivate lifestyle change or facilitate more efficient use of pharmacologic therapies does not exist at present.
Beyond clinical risk stratification, the polygenic score may additionally foster insights into the mechanistic underpinnings of myocardial infarction. This risk associated with a high polygenic score is not the result of a discrete underlying mechanism, but rather a quantitative blend of numerous risk pathways.28 Nevertheless, the relative contributions of gene pathways related to lipid metabolism, inflammation, cellular proliferation, vascular tone, or other as yet undiscovered pathways may provide important insights.10 Moreover, individuals who manifest myocardial infarction despite a favorable polygenic score warrant further study in larger populations. The discordance between the polygenic score and clinical phenotype in these individuals could result from a disproportionate influence of environment, the effect of a rare, large-effect mutation not captured by the polygenic score, or other undetermined factors.
In conclusion, we performed whole genome sequencing in a multiethnic cohort and demonstrate that both familial hypercholesterolemia mutations and a high polygenic score are associated with a 3- to 4-fold increased risk of early-onset myocardial infarction, but a high polygenic score is 10-fold more prevalent.
Supplementary Material
Clinical Perspectives:
What is new?
Whole genome sequencing performed and analyzed in 2,081 patients presenting to a U.S. hospital with early-onset (age ≤ 55 years) myocardial infarction.
A monogenic mutation – a single mutation that significantly increases risk – related to familial hypercholesterolemia identified in 1.7% of the patients and associated with a 3.8-fold increased odds of myocardial infarction.
High polygenic score – reflective of the cumulative impact of many common variants and defined as the top 5% of the control population distribution – was identified in ten times as many patients (17%), and associated with a similar 3.7-fold increased odds of myocardial Infarction.
What are the clinical implications?
A polygenic score comprised of 6.6 million common DNA variants can identify 5% of the population who inherit risk equivalent to that of a familial hypercholesterolemia mutation.
Unlike familial hypercholesterolemia mutation carriers, who typically have high LDL cholesterol levels, ‘carriers’ of a high polygenic score cannot be identified using conventional risk factors or biomarkers.
These findings lay the scientific foundation for the systematic identification of individuals born with a substantially increased risk of myocardial infarction – due to either a familial hypercholesterolemia mutation or high polygenic score – and delivery of a lifestyle or pharmacologic intervention to attenuate inherited risk.
Acknowledgements
We gratefully acknowledge the studies and participants who provided biological samples and data for this analysis. The VIRGO study was facilitated by Mary Geda, Nancy Lorenze, Kelly Strait, and Zhenqiu Lin.
Funding
The VIRGO study was supported by grant R01 HL081153-01A1K from the National Heart, Lung, and Blood Institute. The MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420, UL1-TR-001881, and DK063491.
Whole genome sequencing of the VIRGO cohort was funded by grant 5UM1HG008895-02 from the National Human Genome Research Institute’s Center for Common Disease Genomics. Whole genome sequencing of the MESA cohort was funded through the Trans-Omics for Precision Medicine (TOPMed) Program of the National Heart, Lung, and Blood Institute. General study coordination was provided by the TOPMed Data Coordinating Center (3R01HL-12393-02S1).
Dr. Khera reports funding support from a KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst funded by the National Institutes of Health (TR001100) and a Junior Faculty Research Award from the National Lipid Association. Dr. Aragam is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (T32 HL007208). Dr. Natarajan is supported by 17SDG33680041 from the American Heart Association. Dr. Lander is supported by 5UM1HG008895 from the National Human Genome Research Institute. Dr. Kathiresan is supported by an Ofer and Shelly Nemirovsky Research Scholar Award from Massachusetts General Hospital, RO1 HL127564 from the National Heart, Lung, and Blood Institute, and 5UM1HG008895 from the National Human Genome Research Institute.
Disclosures
A.V.K. and S.K. are listed as co-inventors on a patent application for the use of genetic risk scores to determine risk and guide therapy. S.K. reports grant funding from Bayer AG to study the inherited basis for myocardial infarction.
References
- 1.Gertler MM, Garn SM and White PD. Young candidates for coronary heart disease. J Am Med Assoc 1951;147:621–625. [DOI] [PubMed] [Google Scholar]
- 2.Lehrman MA, Schneider WJ, Sudhof TC, Brown MS, Goldstein JL and Russell DW. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 1985;227:140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, Guella I, Asselta R, Lange LA, Peloso GM, Auer PL, Girelli D, Martinelli N, Farlow DN, DePristo MA, Roberts R, Stewart AF, Saleheen D, Danesh J, Epstein SE, Sivapalaratnam S, Hovingh GK, Kastelein JJ, Samani NJ, Schunkert H, Erdmann J, Shah SH, Kraus WE, Davies R, Nikpay M, Johansen CT, Wang J, Hegele RA, Hechter E, Marz W, Kleber ME, Huang J, Johnson AD, Li M, Burke GL, Gross M, Liu Y, Assimes TL, Heiss G, Lange EM, Folsom AR, Taylor HA, Olivieri O, Hamsten A, Clarke R, Reilly DF, Yin W, Rivas MA, Donnelly P, Rossouw JE, Psaty BM, Herrington DM, Wilson JG, Rich SS, Bamshad MJ, Tracy RP, Cupples LA, Rader DJ, Reilly MP, Spertus JA, Cresci S, Hartiala J, Tang WH, Hazen SL, Allayee H, Reiner AP, Carlson CS, Kooperberg C, Jackson RD, Boerwinkle E, Lander ES, Schwartz SM, Siscovick DS, McPherson R, Tybjaerg-Hansen A, Abecasis GR, Watkins H, Nickerson DA, Ardissino D, Sunyaev SR, O’Donnell CJ, Altshuler D, Gabriel S and Kathiresan S. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015;518:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S and Kathiresan S. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol 2016;67:2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benn M, Watts GF, Tybjaerg-Hansen A and Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J 2016;37:1384–94. [DOI] [PubMed] [Google Scholar]
- 6.Abul-Husn NS, Manickam K, Jones LK, Wright EA, Hartzel DN, Gonzaga-Jauregui C, O’Dushlaine C, Leader JB, Lester Kirchner H, Lindbuchler DM, Barr ML, Giovanni MA, Ritchie MD, Overton JD, Reid JG, Metpally RP, Wardeh AH, Borecki IB, Yancopoulos GD, Baras A, Shuldiner AR, Gottesman O, Ledbetter DH, Carey DJ, Dewey FE and Murray MF. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016;354, pii: aaf7000. [DOI] [PubMed] [Google Scholar]
- 7.Golan D, Lander ES and Rosset S. Measuring missing heritability: inferring the contribution of common variants. Proc Natl Acad Sci U S A 2014;111:E5272–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rare Gibson G. and common variants: twenty arguments. Nat Rev Genet 2012;13:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikainen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, Konig IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimaki T, Loos RJF, Melander O, Metspalu A, Marz W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ and Farrall M. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khera AV and Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet 2017;18:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT and Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, Fuster V, Boerwinkle E, Melander O, Orho-Melander M, Ridker PM and Kathiresan S. Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease. N Engl J Med 2016;375:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lichtman JH, Lorenze NP, D’Onofrio G, Spertus JA, Lindau ST, Morgan TM, Herrin J, Bueno H, Mattera JA, Ridker PM and Krumholz HM. Variation in recovery: Role of gender on outcomes of young AMI patients (VIRGO) study design. Circ Cardiovasc Qual Outcomes 2010;3:684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardiovascular Disease Knowledge Portal URL: http://www.broadcvdi.org/informational/data
- 15.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr., Kronmal R, Liu K, Nelson JC, O’Leary D, Saad MF, Shea S, Szklo M and Tracy RP. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 16.Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes-Virella M, Watts GF, Wierzbicki AS, American Heart Association Atherosclerosis H, Obesity in Young Committee of Council on Cardiovascular Disease in Young CoC, Stroke Nursing CoFG, Translational B, Council on L and Cardiometabolic H. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation 2015;132:2167–2192. [DOI] [PubMed] [Google Scholar]
- 17.Werling DM, Brand H, An JY, Stone MR, Zhu L, Glessner JT, Collins RL, Dong S, Layer RM, Markenscoff-Papadimitriou E, Farrell A, Schwartz GB, Wang HZ, Currall BB, Zhao X, Dea J, Duhn C, Erdman CA, Gilson MC, Yadav R, Handsaker RE, Kashin S, Klei L, Mandell JD, Nowakowski TJ, Liu Y, Pochareddy S, Smith L, Walker MF, Waterman MJ, He X, Kriegstein AR, Rubenstein JL, Sestan N, McCarroll SA, Neale BM, Coon H, Willsey AJ, Buxbaum JD, Daly MJ, State MW, Quinlan AR, Marth GT, Roeder K, Devlin B, Talkowski ME and Sanders SJ. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet 2018;50:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W and Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilhjalmsson BJ, Yang J, Finucane HK, Gusev A, Lindstrom S, Ripke S, Genovese G, Loh PR, Bhatia G, Do R, Hayeck T, Won HH, Kathiresan S, Pato M, Pato C, Tamimi R, Stahl E, Zaitlen N, Pasaniuc B, Belbin G, Kenny EE, Schierup MH, De Jager P, Patsopoulos NA, McCarroll S, Daly M, Purcell S, Chasman D, Neale B, Goddard M, Visscher PM, Kraft P, Patterson N and Price AL. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet 2015;97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T and Collins R. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA and Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 22.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg-Hansen A and European Atherosclerosis Society Consensus P. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–3490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, Nordio F, Hyde C, Cannon CP, Sacks F, Poulter N, Sever P, Ridker PM, Braunwald E, Melander O, Kathiresan S and Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015;385:2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, Sartori S, Fuster V, Reilly DF, Butterworth A, Rader DJ, Ford I, Sattar N and Kathiresan S. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation 2017;135:2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD and Kenny EE. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am J Hum Genet 2017;100:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green ED, Guyer MS and National Human Genome Research I. Charting a course for genomic medicine from base pairs to bedside. Nature 2011;470:204–13. [DOI] [PubMed] [Google Scholar]
- 27.Kaufman DJ, Baker R, Milner LC, Devaney S and Hudson KL. A Survey of U.S Adults’ Opinions about Conduct of a Nationwide Precision Medicine Initiative(R) Cohort Study of Genes and Environment. PLoS One 2016;11:e0160461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khera AV and Kathiresan S. Is Coronary Atherosclerosis One Disease or Many? Setting Realistic Expectations for Precision Medicine. Circulation 2017;135:1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing data for the VIRGO study and MultiEthnic Study of Atherosclerosis study have been uploaded to the database of Genotypes and Phenotypes (dbGaP) repository under accession numbers phs001259 and phs001416, respectively.
The genome-wide polygenic score for myocardial infarction used in this paper is available for download.14 Python scripts used to extract the polygenic score for each individual from a whole genome sequencing variant call format (.vcf) file and R code to standardize the distribution of this score across racial/ethnic groups are provided in Supplemental Code I-III of the online-only Data Supplement.