

Abstract
The family of adhesion G protein-coupled receptors (aGPCRs) consists of 33 members in humans. Although the majority continue to be orphan receptors with unknown functions, many reports have demonstrated critical functions for some members of this family in organogenesis, neurodevelopment, myelination, angiogenesis, and cancer progression. Importantly, mutations in several aGPCRs have been linked to human diseases. The crystal structure of a shared protein domain, the GPCR autoproteolysis inducing (GAIN) domain, has enabled the discovery of a common signaling mechanism—a tethered agonist—for this class of receptors. A series of recent reports has shed new light on their biological functions and disease relevance. This review will focus on these recent advances in our understanding of aGPCR biology in the nervous system and the untapped potential of aGPCRs as novel therapeutic targets for neurological disease.
Keywords: Adhesion G protein-coupled receptors, aGPCRs, GPCR Autoproteolysis INducing (GAIN) domain, neurological disease, neurodevelopment
A brief overview
The superfamily of G protein-coupled receptors (GPCRs) is comprised of the largest family of cell membrane receptors. These seven transmembrane (7TM) proteins inwardly transmit external signals by interactions between their N-terminal extracellular domains (ECDs) and diverse stimuli, including hormones, peptides and proteins, small molecules, metabolites, ions, and light. G protein-dependent and -independent pathways transduce these myriad external signals and initiate context-dependent downstream signaling, thereby providing cells with rapid means of evaluating their environments. Owing to their diversity, specificity, and involvement in human disease, GPCRs have been a central focus of pharmaceutical drug discovery and development efforts. Indeed, nearly one in three clinical agents targets GPCRs [1].
The adhesion GPCR (aGPCR) family, the second largest subfamily of GPCRs, contains 33 human orthologs. They are subdivided into nine subfamilies by N-terminal domain architecture and phylogenetic relationships: ADGRL (latrophilins; LPHLs), ADGRE (EMRs), ADGRA, ADGRC (CELSRs), ADGRD, ADGRF, ADGRB (BAIs), ADGRG, and ADGRV (GPR98) (Figure 1) [2]. They share many commonalities that unite GPCRs: (1) they possess 7TM domains; (2) they are known to couple to G proteins [3]; and (3) there exist hints that similar desensitization and internalization pathways might terminate aGPCR signaling [4, 5]. Two features shared by all aGPCR family members, however, distinguishes them from their phylogenetic ancestors. These include their large, multi-domain N-termini and their unique juxtamembrane GPCR Autoproteolysis INducing (GAIN) domain (see Glossary) [4, 6].
Figure 1: Phylogenetic organization, domain structure, and G protein coupling of human adhesion GPCR subfamilies I-IX.
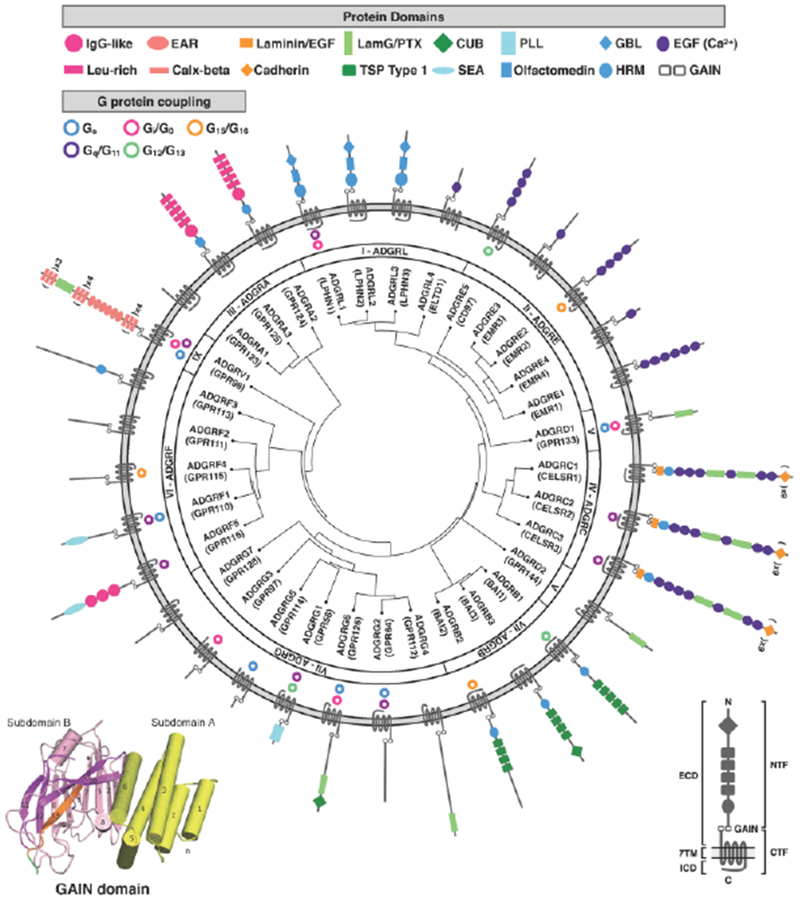
The left inset shows the GAIN domain crystal structure of rat latrophilin with subdomain A (yellow) and subdomain B (magenta), modified from [111]. The right inset identifies general domains and features of aGPCRs. All human aGPCRs are phylogenetically clustered by homology in the center ring, with gene names, aliases, and aGPCR families listed in the middle ring. G protein-coupling, when known, is illustrated in the outer ring, with the corresponding aGPCR structure and protein domains (not to scale). Receptor homology and phylogenetic clustering was performed by GPCRdb (accessed 3/19/2018). Receptor domains were determined by the NCBI Conserved Domain calculator (accessed 3/19/2018), the UniProt database (accessed 3/19/2018), and published reports. G protein coupling was determined by the Guide To Pharmacology (IUPHAR/BPS; accessed 3/19/2018) and published reports [81, 112–114]. Abbreviations used: ECD: extracellular domain; 7TM: seven transmembrane domain; ICD: intracellular domain; NTF: N-terminal fragment; GAIN: GPCR autoproteolytic-inducing domain; CTF: C-terminal domain; IgG: immunoglobulin G; Leu-rich: leucine-rich; EAR: epilepsy-associated repeat; Calx: calcium exchanger; EGF: epidermal growth factor-like domain; LamG/PTX: laminin G/pentraxin; TSP: thrombospondin repeat; CUB: complement C1r/C1s, Uegf, Bmp1; SEA: sperm protein/enterokinase/agrin; PLL: pentraxin (PTX)/laminin/neurexin/sex-hormone-binding globulin (LNS)-like; GBL: galactose-binding lectin; HRM: hormone motif; EGF (Ca2+): calcium-binding EGF domain.
GAIN domain crystal structure and signaling mechanism
The GAIN domain structure consists of ~320 amino acid residues and is found in all members of the aGPCR family, except ADGRA1 (GPR123), and in five human polycystic kidney disease proteins (Figure 1, inset) [6]. The GPCR proteolysis site (GPS) motif is part of the GAIN domain that cannot function by itself, but rather needs to be within the context of the GAIN domain to mediate autoproteolysis.
GAIN domain autoproteolysis during protein maturation cleaves the receptor into two fragments that remain noncovalently associated at the cell surface: a N-terminal fragment (NTF), containing its adhesion domains and most of its GAIN domain, and a C-terminal fragment (CTF), containing its conserved Stachel (a cryptic tethered agonist), 7TM, and intracellular domains (Figure 1, inset). The activity of a number of aGPCRs is significantly enhanced after separation of their NTF and CTF domains by truncation or chemical dissociation [3, 4, 7–10]. This observation has led to three current models to describe aGPCR activity modulation (Figure 2).
Figure 2: Three proposed models of aGPCR activation.
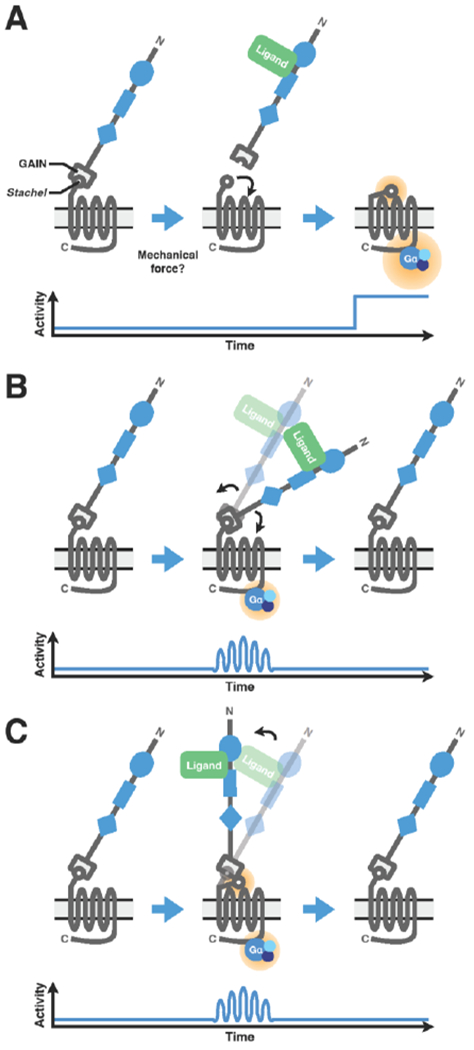
(A) In the first model, NTF and N-terminal GAIN domains conceal the conserved, cryptic tethered agonist Stachel structure in the CTF. Binding of extracellular ligand, possibly combined with mechanical force, removes the NTF, thereby exposing the Stachel structure, which interacts with extracellular loops of the 7TM domain and initiates conformational changes leading to constitutive downstream G protein signaling. (B) In the second model, ligand binding induces conformational changes independent of Stachel structure exposure, leading to transient and reversible downstream G protein signaling. (C) In the third model, ligand binding induced conformational changes that result in exposure of the Stachel structure and Stachel-dependent, potentially transient downstream signaling mediated through the 7TM.
In the first, binding of an extracellular ligand causes shedding of the NTF, possibly through mechanical force (reviewed in [3, 4]), thereby exposing its tethered agonist, facilitating direct interaction with its 7TM domain, and subsequent initiation of downstream signaling via heterotrimeric G proteins [7–10] (Figure 2A). In the second, direct and transient interaction between the extracellular and 7TM domains—presumably in response to ligand-induced conformational changes and possibly in response to mechanical or vibrational stimulations—ultimately alters downstream signaling in a Stachel-independent manner [11, 12] (Figure 2B). In the third, ligand binding induces a conformation change that exposes the Stachel sequence and leads to receptor activation without cleavage and/or removal of the NTF [13] (Figure 2C). These models are unlikely to be mutually exclusive and could in principle occur in the same receptor. Owing to the buried nature of the Stachel sequence and the requirement for NTF-CTF dissociation, receptor activation in the first model is likely to be irreversible [7, 8]. By contrast, aGPCR activation in the second and third models is predicted to be transient and reversible, a form of fine-tuning [3]. While compelling evidence exists to support these models, it remains to be seen how universal each model applies to the broader family of aGPCRs, as well as how receptor activity is regulated in complex cellular and tissue contexts in vivo.
While many aGPCRs signal via coupling to various G proteins [3, 4] (Figure 1), some aGPCRs are capable of transducing signals through non-canonical, G protein-independent pathways. The most heavily studied aGPCRs in this regard are ADGRB1-3 (BAI1-3) and ADGRA2 (GPR124). ADGRB1 contains a long C-terminal tail that interacts with a number of PDZ domain-containing synaptic scaffold proteins [14, 15]. Some of these interactions can regulate ADGRB1 signaling through G proteins [15], while others, such as ADGRB1 coupling to the Rac-GEF Tiam1, can mediate G protein-independent signaling [14]. ADGRB1 has also been shown to associate with ELMO/Dock180 to activate Rac during phagocytosis of apoptotic cells [16] and Gram-negative bacteria [17, 18], as well as during myoblast fusion [19]. ADGRA2 plays an important role during brain angiogenesis and functions as a WNT7A/WNT7B-specific co-activator of beta-catenin signaling in the brain endothelium [20–22]. Recently, two groups independently demonstrated that ADGRA2 is specifically required to deliver Reck-bound Wnt7 in the assembling of Reck/ADGRA2/Frizzled/Lrp5/6 complexes [23, 24].
Domain-specific functions
Distinct functions of NTF and CTF.
The release of NTF regions from their cognate CTF structures has been demonstrated in some cases to enable distinct NTF- and CTF-independent functions. For example, loss of lat-1 in Caenorhabditis elegans confers lethality and infertility. Interestingly, wildtype lat-1 can fully rescue both lethality and infertility phenotypes in lat-1 null animals, whereas the CTF-deficient mutant constructs can only complement the fertilization defect but not the lethality phenotype, suggesting a CTF-independent function of lat-1 in C. elegans [25]. Loss of ADGRG6 (GPR126) causes both cardiac hypotrabeculation, leading to impaired contraction and energy metabolism in the heart, and hypomyelination of the peripheral nervous system (PNS) in zebrafish and mice, producing a grave motor defect [26–29]. Ectopic expression of ADGRG6-NTF in zebrafish can only rescue the trabeculation phenotype in the heart but not the myelination phenotype in the PNS [29].
In regards to Schwann cell-mediated myelination of the peripheral nervous system, ADGRG6 exerts distinct NTF- and CTF-dependent function [30]. While the NTF of ADGRG6 is both necessary and sufficient to maintain Schwann cells in an immature state and for radial sorting, a necessary first step in the myelination of peripheral axons, the CTF of GPR126 promotes Schwann cell maturation and ensheathment of peripheral axons by promoting the generation of the second messenger cyclic AMP and the expression of myelinogenic transcriptional programs (Figure 3). These domain-specific, independent processes depend on interaction with extracellular Laminin-211 derived from the basal lamina. These findings are the basis of a signaling concept that differentiates between a trans (NTF-mediated) and a cis (CTF-mediated) signal induced by aGPCRs.
Figure 3: Domain-specific functions of ADGRG6.
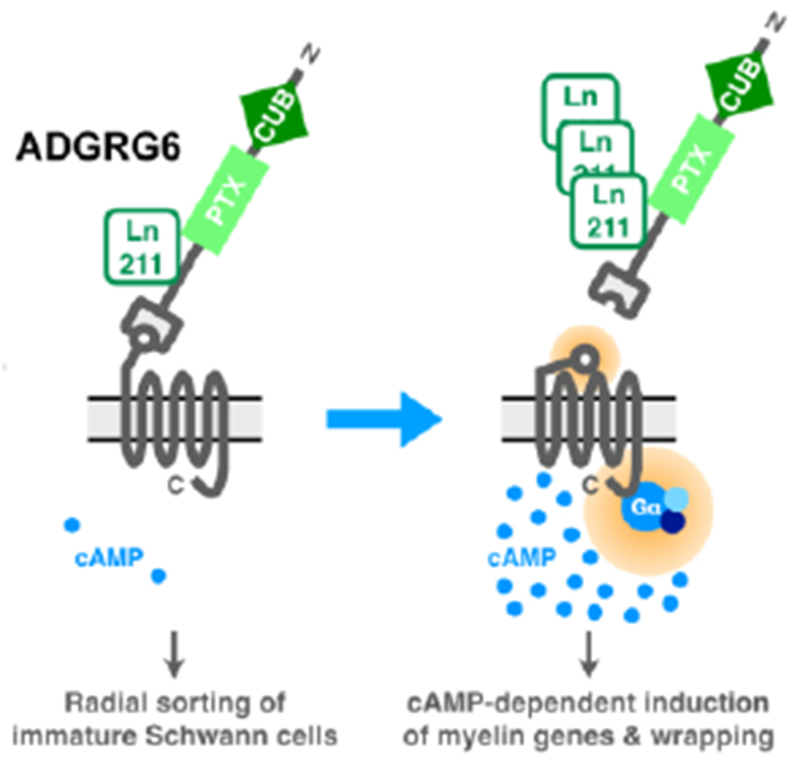
The NTF attachment of ADGRG6 to its CTF suppresses receptor activity, thereby maintaining Schwann cells in an immature state and allowing for radial sorting of cells to axons. Increased accumulation and binding of its ligand laminin-211 removes the NTF, thereby allowing for Stachel-dependent activation of ADGRG6, association with Gαs, accumulation of cyclic AMP (cAMP), and the initiation of cAMP-dependent myelinogenic gene programs and the myelination of peripheral axons. Adapted from [30].
Splicing isoforms.
A parallel question about domain-specific functions stems from the widespread abundance of aGPCR isoforms. Splicing isoforms have been predicted or detected for all aGPCRs except ADGRA1 (Table 1). A well-studied example of aGPCR isoform-specific functions is ADGRG1 (GPR56), an evolutionarily conserved aGPCR required for normal brain development [31]. There are multiple alternative splicing sites in the noncoding first exon of ADGRG1 that produce alternatively spliced isoforms with diverse expression patterns in both the fetal and adult brain [32]. These splicing isoforms are highly variable between mice and humans. A 15-base pair deletion in this region was shown to selectively disrupt human cortical development in the peri-Sylvian region bilaterally by abolishing regional ADGRG1 expression, resulting in a specific human brain malformation [33].
Table 1:
Human and mouse adhesion GPCR isoforms.
| Subclass | aGPCR | Human | Mouse | ||||
|---|---|---|---|---|---|---|---|
| Reported splicing isoform(s) | PubMed ID | Predicted splicing isoform(s) | Reported splicing isoform(s) | PubMed ID | Predicted splicing isoform(s) | ||
| I | ADGRL1 (LPHN1) | 2 | 9261169; 9920906 | 3 | 2 | 16141072 | 5 |
| I | ADGRL2 (LPHN2) | 7 | 14681372; 10030676; 17456239 | 4 | 2 | 16141072 | 15 |
| I | ADGRL3 (LPHN3) | 4 | 14681372 | 13 | 6 | 16141072; 15489334 | 18 |
| I | ADGRL4 (ELTD1) | 1 | 11050079 | 2 | 1 | 11050079 | 1 |
| II | ADGRE1 (EMR1) | 5 | 14702039; 15489334 | 1 | 1 | 15883173 | 0 |
| II | ADGRE2 (EMR2) | 6 | 14702039; 10903844 | 3 | 0 | - | 0 |
| II | ADGRE3 (EMR3) | 3 | 12975309; 11279179 | 3 | 0 | - | 0 |
| II | ADGRE4 (EMR4) | 3 | 16753812; 12731063 | 0 | 1 | 12023293 | 0 |
| II | ADGRE5 (CD97) | 3 | 8955192; 7636245 | 3 | 3 | 10540231; 10744645 | 5 |
| III | ADGRA1 (GPR123) | 3 | 12565841; 11347906 | 0 | 1 | 17212699 | 0 |
| III | ADGRA2 (GPR124) | 3 | 14702039; 15489334; 11347906; 21421844 | 1 | 1 | 25558062 | 3 |
| III | ADGRA3 (GPR125) | 4 | 15489334; 12975309; 12565841 | 2 | 1 | 17882221 | 0 |
| IV | ADGRC1 (CELSR1) | 2 | 15489334; 20353824 | 1 | 1 | 9858697 | 2 |
| IV | ADGRC2 (CELSR2) | 1 | 10907856 | 0 | 2 | 15489334 | 2 |
| IV | ADGRC3 (CELSR3) | 2 | 9693030 | 0 | 1 | 11677057 | 3 |
| V | ADGRD1 (GPR133) | 4 | 14702039; 15489334; 15203201 | 3 | 1 | 22025619 | 1 |
| V | ADGRD2 (GPR144) | 1 | 12679517 | 3 | 0 | - | 0 |
| VI | ADGRF1 (GPR110) | 4 | 20149256 | 3 | 1 | 22837050 | 0 |
| VI | ADGRF2 (GPR111) | 2 | 17056209 | 1 | 1 | 22837050 | 0 |
| VI | ADGRF3 (GPR113) | 3 | 12975309 | 2 | 1 | 15780750 | 1 |
| VI | ADGRF4 (GPR115) | 1 | 14702039; 23840300 | 1 | 1 | 22837050 | 2 |
| VI | ADGRF5 (GPR116) | 3 | 17974005 | 0 | 1 | 28570277 | 4 |
| VII | ADGRB1 (BAI1) | 1 | 9393972 | 2 | 1 | 11875720 | 6 |
| VII | ADGRB2 (BAI2) | 4 | 15489334; 9533023 | 6 | 3 | 16141072; 15489334 | 4 |
| VII | ADGRB3 (BAI3) | 2 | 14702039 | 3 | 1 | 15225653 | 2 |
| VIII | ADGRG1 (GPR56) | 4 | 19572147 | 47 | 4 | 27657451 | 10 |
| VIII | ADGRG2 (GPR64) | 10 | 12420295 | 0 | 5 | 12420295 | 3 |
| VIII | ADGRG3 (GPR97) | 1 | 16141072; 12435584 | 3 | 1 | 24113187 | 1 |
| VIII | ADGRG4 (GPR112) | 3 | 12435584 | 0 | 2 | 14681372 | 4 |
| VIII | ADGRG5 (GPR114) | 1 | 12679517 | 1 | 2 | 14681372; 26499266 | 2 |
| VIII | ADGRG6 (GPR126) | 4 | 15489334; 15189448; 17974005; 15225624 | 4 | 1 | 21124978 | 1 |
| VIII | ADGRG7 (GPR128) | 1 | 12565841; 19797732 | 1 | 1 | 15203201; 24574718 | 0 |
| IX | ADGRV1 (GPR98) | 5 | 15207856; 11606593; 10976914 | 14 | 5 | 11545713; 15606908 | 6 |
Alternative splicing of exons 2, 3, and 10 of ADGRG1 is predicted to generate four isoforms: isoforms 1 and 2 are six and one amino acid shorter than full-length ADGRG1, respectively, whereas isoforms 3 and 4 lack 170 and 175 amino acids from their N-termini, respectively (Figure 4A) [32]. The fourth isoform (S4) contains a 5’ deletion in exon 2, thus generating a new frameshifted transcriptional start site [32]. The internal start site of S4 lies between two large domains—the N-terminal pentraxin (PTX)/laminin/neurexin/sex-hormone-binding globulin (LNS)-like (PLL) domain and its GAIN domain [34]. As a consequence, the S4 isoform of ADGRG1 lacks the PLL domain, which mediates interactions with its two known extracellular ligands, collagen III and tissue transglutaminase 2 (TG2)[34–37] (Figure 4B-D). Although the ligand and biological function of S4 isoform remain unknown, its basal activity in a luciferase-based downstream reporter assay is more pronounced than the full length ADGRG1 [34].
Figure 4: Splice variants of ADGRG1.
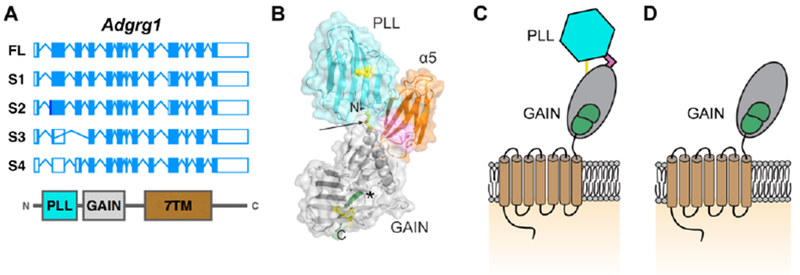
(A) Gene structure of human full-length (FL) ADGRG1 and related isoforms S1-S4. The corresponding protein domains are shown below. Adapted from [32]. (B). Crystal structure of the extracellular domain of ADGRG1 in complex with α5 (orange). Disulfide bonds in yellow, linker in pink. PLL and GAIN domain in cyan and grey, respectively. Modified from [34]. (C). Schematic of ADGRG1 with same color-coding as C. The single yellow line indicating two disulfide bonds between PLL and GAIN domains. (D). Schematic of S4 isoform of ADGRG1.
Alternative splicing is an important regulatory mechanism for diverse protein functions. In ADGRLs (latrophilins), alternative splicing of a five-residue insertion located in the connecting linker region regulates binding to teneurins TEN2 and TEN4 [38]. Conversely, a splice variant of TEN2 with the insertion of the 7-amino acid extension in a β-propeller domain renders it incapable of mediating interactions with ADGRL1 (LPHN1) [39, 40]. With the continued advancement in RNA sequencing technology, studies may soon provide the opportunity to assess the importance of splicing variants of aGPCRs systematically across species and in both healthy and pathological human tissues.
aGPCRs in neurological diseases
Given their widespread cellular and tissue distribution, it is not surprising that the expression and activity of aGPCRs have been implicated in a number of human conditions and diseases. Mutations in many aGPCRs have been shown to contribute to or cause human diseases, and we will discuss some of these findings in the nervous system below.
ADGRL3 (LPHN3).
A loss-of-function (LOF) variant of ADGRL3 has been shown to confer susceptibility to attention deficit hyperactivity disorder (ADHD) [41–44]. Consistent with these findings in humans, studies in mice have revealed that targeted deletion of ADGRL3 results in significant changes in the relative strengths of connections between different layers of the neocortex [45]. ADGRL3 is expressed in ADHD-related human brain regions, including the amygdala, caudate nucleus, cerebellum and cerebral cortex [41, 46, 47]. Ablation of ADGRL3 in mice and zebrafish results in abnormal dopaminergic brain wiring and causes an ADHD-like hyperactive/impulsive behavior [48, 49]. This behavioral phenotype can be rescued with the ADHD treatments methylphenidate and atomoxetine [49]. Thus, ADGRL3 plays a key role in modulating nervous system development and function, and the human genetic data implicate this receptor in psychiatric and neurological disorders. From a drug development perspective, this available data suggest that enhanced ADGRL3 signaling might ameliorate an ADHD phenotype. Further understanding of ADGRL3 signaling pathways or regulation of ADGRL3 expression would be needed to realize this potential.
ADGRC1-3 (CELSR1-3).
Mutations in ADGRC1 (CELSR1) significantly enhance the risk of neurodevelopmental disorders, such as neural tube defects [50, 51] and spina bifida [52]. ADGRC1-3 are the vertebrate orthologs of the Drosophila aGPCR flamingo, which has been intensively studied as a key determinant of planar cell polarity (PCP) [53]. Similar to flamingo, ADGRC1-3 strongly regulates PCP, and thus knockout of these genes has striking effects on neural development as well as the development of other systems. Ablation of ADGRC1 causes neural tube closure defects in a substantial fraction of the knockout mice [54], consistent with reports that ADGRC1 mutations greatly enhance the risk of neural tube defects [50, 51] and spina bifida [52] in humans and suggesting that the human variants confer LOF. Knockout of ADGRC1 also regulates hair patterning and the development of other organs, as would be expected from deletion of a receptor that plays a key role in PCP [55, 56]. Deletion of ADGRC2 or ADGRC3 result in hydrocephalus, due at least in part to defective ciliogenesis in ependymal cells in the brains of knockout mice [57]. Additionally, genetic ablation of ADGRC3 results in major perturbations in axonal pathfinding in vivo in both the central [58–62] and peripheral [63] nervous systems.
Excitingly, two reports using whole exome sequencing in more than 800 Tourette disorder patient-and-parent trios identified an excess of de novo sequence variants in ADGRC3 in probands of simplex but not multiplex families [64, 65]. These variants were predicted to be damaging missense mutations suggesting LOF, and their frequency was sufficient to categorize ADGRC3 as a Tourette disorder risk gene. Sequence variants and copy number variants (CNVs) were also detected in other genes related to cell polarity, consistent with a conserved underlying pathophysiology, which remains to be deciphered. Importantly, the identified sequence variants overlapped with those implicated in obsessive-compulsive disorder, while the CNVs overlapped with those associated to autism spectrum disorder indicating that the genetic clues provided by ADGRC3 sequence variants and other cell-polarity genes may carry widespread significance for neurodevelopmental disorders. It now remains to be determined whether the cell-polarity function, a downstream function, or a completely distinct function of these related gene products are responsible for the disease associations.
ADGRB1-3 (BAI1-3).
Knockout studies on ADGRB1-3 have identified important roles for these receptors in synaptogenesis, as well as a potential role in neurological pathologies. Ablation of ADGRB1 in mice results in structural, post-synaptic alterations in various brain regions with concomitant defects in synaptic plasticity and learning [66]. ADGRB2 knockout mice have enhanced neural stem cell proliferation in the hippocampus and improved performance in rodent models of depression, suggesting a role for ADGRB2 in regulating the wiring of brain areas related to mood [67]. The overall implication of these findings is that enhanced ADGRB2 activity might be deleterious in the context of mood disorders. Consistent with this possibility, an activating ADGRB2 variant causes spastic paraparesis [68], although the mechanism involved altered G-protein coupling. Compounds that selectively inhibit ADGRB2 activity could provide a novel approach to treating the depressive aspect of mood disorders. Conversely, deletion of ADGRB3 results in perturbations to dendritic structure in the cerebellum [69]. Thus, the three members of the ADGRB subfamily all have distinct roles in vivo, although their actions appear to control synaptic development.
ADGRG1 (GPR56).
The most intensively studied example of an aGPCR associated with a human genetic disease in this regard is ADGRG1, LOF mutations of which cause a devastating brain malformation called bilateral frontoparietal polymicrogyria (BFPP) [31]. MRI analyses of BFPP brains show signal changes in the white matter, suggesting a CNS myelination defect. Work in mice and zebrafish has further established the connection between loss of ADGRG1 and defective CNS myelination [70, 71]. These studies demonstrated that signaling of ADGRG1 via RhoA activation promotes the proliferation of oligodendrocyte precursor cells (OPCs). A recent report demonstrated that the ligand of OPC ADGRG1 is tissue transglutaminase (TG2, gene symbol Tgm2), produced by microglia. Deleting Tgm2 phenocopies CNS myelination defects associated with ablation of ADGRG1 in OPCs [72]. Importantly, loss of either OPC ADGRG1 or microglial TG2 leads to impairment of myelin repair following demyelinating injury [72]. These findings indicate that selectively enhancing ADGRG1 activity in OPCs might lead to improved remyelination in demyelinating disorders such as multiple sclerosis [72]. In addition to its function in CNS myelination, ADGRG1 also plays a role in Schwann cell functions in the peripheral nervous system (PNS) and regulates PNS myelin thickness and maintenance [73].
ADGRG6 (GPR126).
LOF mutations in ADGRG6 cause a severe form of arthrogryposis multiplex congenita (AMC) that is perinatally lethal [74]. Disease-causing mutations in ADGRG6 impair cleavage of the receptor’s GAIN domain, underscoring the necessity of GAIN domain autoproteolysis for the receptor’s function in vivo. Similar to ADGRG1, ADGRG6 has been shown to be a key regulator of myelination, albeit in the PNS rather than CNS. Mutational analyses in zebrafish revealed a critical role of ADGRG6 in initiating myelination in Schwann cells [26]. Subsequent work revealed that knockout mice lacking ADGRG6 exhibit severe hypomyelination in the PNS [27, 28], in addition to deficits in cardiovascular [29, 75] and skeletal [76] development. As mentioned above, ADGRG6 mutations cause a severe form of AMC, and myelination defects associated with LOF mutations to ADGRG6 may plausibly contribute to this disorder. However, the connections that have been reported between certain variants of ADGRG6 and adolescent-onset idiopathic scoliosis, the most common skeletal disease in children, are most likely due to action of ADGRG6 in cartilage rather than peripheral nerves [4, 77].
ADGRV1 (GPR98).
Mutations to ADGRV1 cause Usher syndrome type 2C, a genetic disorder typified by deafness and blindness [78]. ADGRV1 is highly expressed in the stereocilia of the cochlea and the ciliary membrane of photoreceptors [79, 80], presumably regulating aspects of ciliary function. ADGRV1 is the largest aGPCR in terms of total amino acids, and many distinct disease-causing mutations have been identified on the receptor’s massive (>5,000 amino acid) N-terminus [78]. Some of the disease causing mutations introduce stop codons and result in a truncated protein that lacks its CTF [78], suggesting underlying receptor LOF. Furthermore, one disease-associated mutation is found on the receptor’s cytoplasmic C-terminus and has been shown to modulate the G protein coupling of ADGRV1 [81], consistent with the possibility that altered G protein coupling can confer a LOF phenotype. Taken together, both the NTF and CTF of ADGRV1 appear to be required for cochlear and photoreceptor development. Visual impairment in Usher’s 2C becomes evident late in life often between ages 50 and 70, indicating a role for ADGRV1 both in development and maintenance of photoreceptors. The disorder is autosomal recessive with LOF mutations, suggesting a general role in photoreceptor health and underscoring the value of a drug target capable of enhancing ADGRV1 pathway signaling.
aGPCR variants as risk factors.
Whole genome sequencing and deep sequencing has allowed for the rapid identification of disease-associated mutations and small nucleotide polymorphisms (SNPs). Although most members of aGPCR family appear in GWAS studies of neurological disorders with a p-value cutoff of p<1E−4, three aGPCRs—ADGRB3, ADGRG1, and ADGRC1—have SNPs that are more relevant to human neurological disease with a p-value of p<1E−6 (Table 2).
Table 2.
GWAS analysis identifying adhesion GPCRs in neurological diseases.
| aGPCR | CNS disease | SNP ID | Chrom osome | Change | MAF | p-value | PubMed ID |
|---|---|---|---|---|---|---|---|
| ADGRB3 (BAI3) | Late-onset Alzheimer’s | rs10485435 | 6q | G>T; intronic | 0.331 | 6.1E−7 | 21390209 |
| Autism | rs13208283 | 6q | A>C; intronic | 0.042 | 1.1E−6 | 20663923 | |
| Autism | rs16900553 | 6q | A>C; intronic | 0.141 | 5.2E−6 | 20663923 | |
| Schizophrenia | rs3011917 | 6q | A>C; intronic | 0.115 | 5.1E−6 | 21688384 | |
| ADGRG1 (GPR56) | Late-onset Alzheimer’s | rs1466134 | 16q | G>A; intronic | 0.319 | 8.0E−6 | 21379329 |
| Multiple Sclerosis | rs1376041 | 16q | 996T>C | 0.241 | 7.3E−6 | 19525953 | |
| ADGRC1 (CELSR1) | Myopia | rs3827410 | 22q | C>A; intronic | 0.430 | 9.9E−6 | 21640322; 23406873 |
SNP data was collected using the NIH GRASP database, applying a p-value cutoff of p<1E−6 and within neurological disorders. MAF: minor allelic frequency.
Enrichment of SNPs within ADGRB3 have been identified in patients with late-onset Alzheimer’s disease [82] and with schizophrenia [83–86], underscoring its reported involvement in synaptic development, maintenance, and function. Interestingly, SNPs in ADGRB3 have also been correlated with neurodevelopmental delays and symptoms of autism [87]. ADGRG3 lies in a cohort of genes and alleles on Chromosome 6q whose deletion has been linked to autism spectrum symptoms [88, 89]. It will be important to define the causal relationship of ADGRB3 in autism-associated phenotypes using murine models of neurodevelopmental diseases and patient-derived models [90, 91].
SNPs of ADGRG1 have been indicated by GWAS studies of late-onset Alzheimer’s disease and multiple sclerosis. Given the wide expression of ADGRG1 in neuronal precursor cells, glia, the CNS-resident immune cells, microglia [92, 93], it is interesting to speculate if ADGRG1 participates in the modulation of neuroinflammation, a feature shared by both of these diseases. Moreover, in light of our laboratory’s findings that ADGRG1 regulates the generation of myelin from oligodendrocytes during CNS development and in response to demyelinating injuries, as in multiple sclerosis [70–72], it will be important in future studies to determine if disease-associated SNPs impact the myelinogenic ability of ADGRG1.
Lastly, a SNP in ADGRC1 has been linked to myopia in the Han Chinese population [94, 95]. Although the implications of these findings and causality require subsequent investigation, it is interesting to note that ADGRC1 is expressed in the developing vertebrate and invertebrate retina [96] where it regulates axonal guidance and wiring [97].
Currently, the impact of those reported SNPs on their associated gene function is yet to be characterized. Unexpectedly, six of the seven SNPs are located in intronic regions (Table 2). It is interesting to speculate if allelic variation in the noncoding regions of aGPCRs alters splicing events, as has been observed in a large number of other genes [98]. Given the abundance of reported and predicted alternatively spliced aGPCR genes (Table 1), and that alternative splicing of at least one aGPCR (ADGRG1) has been linked to human developmental malformations [33], it will be critical in future studies to determine the contributions of these SNPs to splicing events using deep-sequencing and other more sensitive sequencing approaches.
It is worth noting that the reported statistically significant SNP changes and human disease associations involve mainly CNS and limited PNS abnormalities. There is no report in the enteric nervous system. Furthermore, none of those SNPS, based on our knowledge, are used in clinical testing for their associated disease conditions. With recent advances in the scale and depth of sequencing, aGPCR SNPs are likely to be identified in future studies.
Approaches in the therapeutic targeting of aGPCRs
With the great diversity of human diseases and conditions in which they have been implicated, aGPCRs offer phenomenal opportunities for therapeutic targeting. The complex domain structures and protein interactions of aGPCRs present a potentially multimodal approach to modulating their activity, including strategies to disrupt NTD interactions with ligands, small molecule- and peptide-based modulators of CTD activity, and the exploitation of disease state- and mutation-specific downstream signaling partners. The existence of diverse means of targeting aGPCRs is an attractive feature given limitations of specific therapeutic modalities (e.g., the difficulty in generating blood-brain barrier-permeant biologics and peptides). In this final section we will discuss recent discoveries that will be critical in the advancement and development of aGPCR therapeutics.
As with their GPCR relatives, the activity of aGPCRs can be modulated by small molecules. This was first demonstrated with ADGRG3 (GPR97), for which the steroid beclomethasone dipropionate was identified as an agonist in a chemical screen [99]. More recently, Stoveken and colleagues identified the rotenone-derivative dihydromunduletone as a small molecule antagonist of ADGRG1, as well as an antagonist of structurally related ADGRG5 (GPR114), through a high-throughput activity screen [100]. In a subsequent study, the gedunin-derivative 3-α-acetoxydihydrodeoxygedunin and related small molecules were identified as partial agonists for both ADGRG1 and ADGRG5 [101]. While current small molecule approaches target the 7TM domains of ADGRG1 and ADGRG5, it will be important in future studies to identify small molecules that preferentially and specifically bind to the CTF or NTF and modulate aGPCR function. Indeed, recent advances in directed evolution of chemical small molecule libraries and in DNA-encoded libraries in particular are paving the way to new pharmacological avenues [102]. Although the therapeutic efficacy of small molecule regulators in specific disease contexts remains unknown, these findings underscore the amenability of screening aGPCR small molecules through traditional cell-based or biochemical high-throughput screens.
The tethered agonist signaling mechanism of aGPCRs additionally lends itself to peptide-based pharmacological modulation. The possible utility of peptide-based regulation was first suggested with the identification of the aGPCR Stachel-mediated signaling. Purified synthetic Stachel peptides of ADGRG1, ADGRG2, ADGRG6, ADGRD1, ADGRF1, ADGRF5, ADGRL1 and its C. elegans homolog lat-1 activated their respective receptors in aGPCR reconstitution assays and in transfected cells [7–9, 103, 104]. Cross-activation by receptor-specific peptides was observed for some aGPCR-peptide combinations and not observed for others, suggesting that—despite functional and structural similarities in Stachel structures [7, 8, 101] —the development of receptor-specific therapeutics is possible although quite challenging. The therapeutic implications of these findings are further emphasized by experiments performed by Liebscher and colleagues using zebrafish carrying LOF mutations in ADGRG6, which show impaired myelination in the peripheral nervous system. Impressively, the addition of recombinant ADGRG6 Stachel peptide to developing larvae partially rescued this dysmyelination phenotype in an ADGRG6-dependent manner [7]. It will be important to test in future studies the specificity and generalizability of Stachel-derived in modulating the activity of targeted aGPCRs or aGPCR subfamilies, in particular for disease and injury models in which these receptors have been implicated. Moreover, given the high concentration of peptide required for activity, peptidomimetic compounds and chemically-modified peptide variants might improve potency and efficacy.
Using ADGRG1 as a target, Salzman and colleagues recently developed a panel of monobodies (synthetic binding proteins are constructed using non-antibody molecular scaffolds such as fibronectin domain III [Fn3]) raised against specific domains of the receptor [11]. These monobodies behave as synthetic ligands, and either enhance or suppress ADGRG1 activity with nanomolar efficacy in vitro. Inverse agonist monobodies appear to bind both PLL and GAIN domains, potentially restricting the conformational changes needed to effect Stachel-independent signaling, whereas others targeting the PLL domain—to which ADGRG1 ligands collagen III and TG2 are known to bind—and enhance basal activity. This toolset was designed to target ADGRG1 NTF domains of both human and mouse origin, and identifies both cross-species and species-specific monobodies that will be important in any future translational and pre-clinical studies. The efficacy of these ADGRG1-specific monobodies—or monobodies to other aGPCRs—in a disease-relevant in vivo setting remains to be demonstrated. However, recent work has highlighted the utility of monobody technology in a murine leukemia model and provides a proof-of-concept framework for future therapeutic studies [105]. In total, this work signals an exciting approach to the therapeutic targeting of aGPCRs.
Another plausible approach involves the pharmacological targeting of disease- or mutation-specific signaling downstream (biased signaling) of aGPCRs. Two recent reports have highlighted differences in signaling pathways downstream of disease-relevant mutations when compared to wildtype receptors [68, 106]. Two mutations in ADGRG1 that have been linked to BFPP (R565W and L640R) disrupt signaling through Gα12/13 to activate a serum response element (SRE) luciferase reporter, while maintaining parallel signaling to activate a nuclear factor of activated T cells (NFAT) luciferase reporter [106]. Similarly, a disease-causing mutation in ADGRB2 alters G protein coupling: while wild-type ADGRB2 couples primarily with Gαz, ADGRB2-R1465W preferentially couples with Gi. In addition to alterations in signaling, ADGRB2-R1465W suppresses interactions with the endocytic trafficking protein endophilin A1 [68]. Although signaling downstream of these receptors is likely to be cell type- and tissue-specific, it is interesting to speculate on the feasibility of small molecule-based targeting of mutation-specific signaling pathways. Similar approaches to generate biased signaling modulators have been used with aminergic GPCRs [107, 108], including the mu opioid [109] and 5-HT2B receptors [110], highlighting the potential applicability in targeting aGPCRs, their phylogenetic cousins.
Finally, pharmacologically targeting aGPCR ligands could be another potential approach in aGPCR drug discovery. Given the widespread expression of a number of aGPCRs, it may prove more therapeutically tractable to target the tissue- and cell-specific ligands of an aGPCR instead of modulating the activity of the aGPCR itself. For example, our laboratory recently reported that TG2 promotes CNS myelin formation and repair by functioning as the ligand of ADGRG1 in oligodendrocytes. This activity requires the crosslinking enzymatic activity of TG2 and the presence of the ECM protein laminin. It is conceivable to selectively enhance TG2 crosslinking enzymatic activity in a site- and content-dependent manner to promote CNS myelin repair.
Concluding remarks and future perspectives
With their multi-domain structures, widespread cellular and tissue distribution, and amenability to targeted therapies, adhesion GPCRs have demonstrated in the last two decades their great potential in advancing our understanding of both developmental biology and disease. An explosion of recent work has characterized and de-orphanized formerly understudied receptors, as well as provided novel structural insight into their complex N-terminal domains and developed new tools to target individual receptors or subfamilies of aGPCRs. Ongoing and future studies will shed additional light on the universality of Stachel-dependent and Stachel-independent signaling mechanisms and will better define domain-specific functions and context-dependent interacting proteins under native conditions, as well as the importance of alternatively sliced isoforms (see Outstanding Questions). Finally, given the diversity of pathophysiological conditions and diseases in which aGPCRs have been implicated, there now exists fertile ground to develop novel targeted therapies and to test their therapeutic potential in cellular and pre-clinical human disease models.
Outstanding Questions:
How does G protein coupling and subsequent downstream signaling differ for aGPCRs in different cells/tissue, during development, and under pathophysiological conditions?
How can receptor ligands be defined in a context-dependent, physiological manner?
What are the expression, function, and interactome of aGPCR isoforms?
How universal is Stachel-dependent aGPCR activation?
What differences in signaling events lie downstream of Stachel-dependent and Stachel-independent receptor activation models?
Does pharmacological modulation of aGPCR activity (by small molecules, peptides, peptidomimetic drugs) provide therapeutic benefit in models of human diseases?
Highlights.
The class of adhesion GPCRs plays critical functions in various developmental processes, such as organogenesis, neurodevelopment, myelination, and angiogenesis.
Mutations in adhesion GPCRs link to various human diseases.
GAIN domain-mediated receptor autoproteolysis is required for most aGPCR function and enables tethered agonist signaling mechanism.
With the new understanding of aGPCR signaling mechanism and their diverse implications in human diseases, adhesion GPCRs offer phenomenal opportunities for therapeutic targeting.
Resources:
GPCRdb: http://gpcrdb.org
NCBI Conserved Domain database: https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
IUPHAR/BPS Guide To Pharmacology: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=17
UniProt Database for protein isoforms: https://www.uniprot.org/
NIH GRASP for SNPs: https://grasp.nhlbi.nih.gov/Overview.aspx
Acknowledgements:
This work was supported by grants of the US National Institutes of Health (NS085201, NS094164, NS108446, and NS108312 to X.P.), and the National Multiple Sclerosis Society (RG-1501-02577 to X.P. and FG 2063-A1/2 to S.G.). Figure 4B was reprinted from Neuron, 2016. 91(6): p. 1292-1304, Salzman, G.S., et al., Structural Basis for Regulation of GPR56/ADGRG1 by Its Alternatively Spliced Extracellular Domains; and the lat-1 crystal structure in Figure 1 was reprinted from Trends Pharmacol Sci, 2013. 34(8): p. 470-8, Promel, S., T. Langenhan, and D. Arac, Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins; with permission from Elsevier.
Glossary:
- C-terminal fragment (CTF)
the membrane-spanning C-terminal fragment generated by the autoproteolytic cleavage of the GAIN domain.
- GPCR Autoproteolysis INducing (GAIN) domain
an evolutionarily conserved juxtamembrane domain present in the extracellular domains of all adhesion GPCRs, except ADGRA1, and polycystic kidney disease proteins.
- GPCR proteolysis site (GPS)
a highly conserved juxtamembrane motif within the GAIN domain and site of autoproteolytic cleavage.
- N-terminal fragment (NTF)
the extracellular N-terminal fragment generated by the autoproteolytic cleavage of the GAIN domain.
- Small nucleotide polymorphisms (SNPs)
a single nucleotide change within a gene and present with a certain allelic frequency in a population.
- Splicing isoform
alternative protein isoforms generated by alternative splicing, in which select introns and exons are excised prior to translation, or by frameshifted open reading frames.
- Stachel sequence
a short peptide that is exposed in the GAIN domain following autoproteolytic cleavage, which in some adhesion GPCRs can initiate receptor activation; also known as a tethered agonist.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
None
References
- 1.Hauser AS, et al. , Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov, 2017. 16(12): p. 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjarnadottir TK, et al. , The human and mouse repertoire of the adhesion family of G-protein-coupled receptors. Genomics, 2004. 84(1): p. 23–33. [DOI] [PubMed] [Google Scholar]
- 3.Purcell RH and Hall RA, Adhesion G Protein-Coupled Receptors as Drug Targets. Annu Rev Pharmacol Toxicol, 2018. 58: p. 429–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamann J, et al. , International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev, 2015. 67(2): p. 338–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monk KR, et al. , Adhesion G Protein-Coupled Receptors: From In Vitro Pharmacology to In Vivo Mechanisms. Mol Pharmacol, 2015. 88(3): p. 617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arac D, et al. , A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J, 2012. 31(6): p. 1364–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liebscher I, et al. , A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep, 2014. 9(6): p. 2018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoveken HM, et al. , Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc Natl Acad Sci U S A, 2015. 112(19): p. 6194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demberg LM, et al. , Activation of Adhesion G Protein-coupled Receptors: AGONIST SPECIFICITY OF STACHEL SEQUENCE-DERIVED PEPTIDES. J Biol Chem, 2017. 292(11): p. 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller A, et al. , Oriented Cell Division in the C. elegans Embryo Is Coordinated by G-Protein Signaling Dependent on the Adhesion GPCR LAT-1. PLoS Genet, 2015. 11(10): p. e1005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salzman GS, et al. , Stachel-independent modulation of GPR56/ADGRG1 signaling by synthetic ligands directed to its extracellular region. Proc Natl Acad Sci U S A, 2017. 114(38): p. 10095–10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishore A, et al. , Stalk-dependent and Stalk-independent Signaling by the Adhesion G Protein-coupled Receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J Biol Chem, 2016. 291(7): p. 3385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scholz N, et al. , Mechano-dependent signaling by Latrophilin/CIRL quenches cAMP in proprioceptive neurons. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duman JG, et al. , The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J Neurosci, 2013. 33(16): p. 6964–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephenson JR, et al. , Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J Biol Chem, 2013. 288(31): p. 22248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park D, et al. , BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature, 2007. 450(7168): p. 430–4. [DOI] [PubMed] [Google Scholar]
- 17.Das S, et al. , Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc Natl Acad Sci U S A, 2011. 108(5): p. 2136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, et al. , Brain angiogenesis inhibitor 1 is expressed by gastric phagocytes during infection with Helicobacter pylori and mediates the recognition and engulfment of human apoptotic gastric epithelial cells. FASEB J, 2014. 28(5): p. 2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochreiter-Hufford AE, et al. , Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature, 2013. 497(7448): p. 263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y and Nathans J, Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev Cell, 2014. 31(2): p. 248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posokhova E, et al. , GPR124 functions as a WNT7-specific coactivator of canonical beta-catenin signaling. Cell Rep, 2015. 10(2): p. 123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhollebeke B, et al. , Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vallon M, et al. , A RECK-WNT7 Receptor-Ligand Interaction Enables Isoform-Specific Regulation of Wnt Bioavailability. Cell Rep, 2018. 25(2): p. 339–349 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eubelen M, et al. , A molecular mechanism for Wnt ligand-specific signaling. Science, 2018. 361(6403). [DOI] [PubMed] [Google Scholar]
- 25.Promel S, et al. , The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep, 2012. 2(2): p. 321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monk KR, et al. , A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science, 2009. 325(5946): p. 1402–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monk KR, et al. , Gpr126 is essential for peripheral nerve development and myelination in mammals. Development, 2011. 138(13): p. 2673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogha A, et al. , Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J Neurosci, 2013. 33(46): p. 17976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patra C, et al. , Organ-specific function of adhesion G protein-coupled receptor GPR126 is domain-dependent. Proc Natl Acad Sci U S A, 2013. 110(42): p. 16898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen SC, et al. , The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron, 2015. 85(4): p. 755–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piao X, et al. , G protein-coupled receptor-dependent development of human frontal cortex. Science, 2004. 303(5666): p. 2033–6. [DOI] [PubMed] [Google Scholar]
- 32.Kim JE, et al. , Splicing variants of the orphan G-protein-coupled receptor GPR56 regulate the activity of transcription factors associated with tumorigenesis. J Cancer Res Clin Oncol, 2010. 136(1): p. 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae BI, et al. , Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science, 2014. 343(6172): p. 764–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salzman GS, et al. , Structural Basis for Regulation of GPR56/ADGRG1 by Its Alternatively Spliced Extracellular Domains. Neuron, 2016. 91(6): p. 1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo R, et al. , Disease-associated mutations prevent GPR56-collagen III interaction. PLoS One, 2012. 7(1): p. e29818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, et al. , GPR56 Regulates VEGF production and angiogenesis during melanoma progression. Cancer Res, 2011. 71(16): p. 5558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, et al. , GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci U S A, 2006. 103(24): p. 9023–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boucard AA, Maxeiner S, and Sudhof TC, Latrophilins function as heterophilic cell-adhesion molecules by binding to teneurins: regulation by alternative splicing. J Biol Chem, 2014. 289(1): p. 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, et al. , Structural Basis for Teneurin Function in Circuit-Wiring: A Toxin Motif at the Synapse. Cell, 2018. 173(3): p. 735–748.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson VA, et al. , Structures of Teneurin adhesion receptors reveal an ancient fold for cell-cell interaction. Nat Commun, 2018. 9(1): p. 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arcos-Burgos M, et al. , A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol Psychiatry, 2010. 15(11): p. 1053–66. [DOI] [PubMed] [Google Scholar]
- 42.Acosta MT, et al. , A two-locus genetic interaction between LPHN3 and 11q predicts ADHD severity and long-term outcome. Transl Psychiatry, 2011. 1: p. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribases M, et al. , Contribution of LPHN3 to the genetic susceptibility to ADHD in adulthood: a replication study. Genes Brain Behav, 2011. 10(2): p. 149–57. [DOI] [PubMed] [Google Scholar]
- 44.Jain M, et al. , A cooperative interaction between LPHN3 and 11q doubles the risk for ADHD. Mol Psychiatry, 2012. 17(7): p. 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Sullivan ML, et al. , LPHN3, a presynaptic adhesion-GPCR implicated in ADHD, regulates the strength of neocortical layer 2/3 synaptic input to layer 5. Neural Dev, 2014. 9: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plessen KJ, et al. , Hippocampus and amygdala morphology in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry, 2006. 63(7): p. 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krain AL and Castellanos FX, Brain development and ADHD. Clin Psychol Rev, 2006. 26(4): p. 433–44. [DOI] [PubMed] [Google Scholar]
- 48.Wallis D, et al. , Initial characterization of mice null for Lphn3, a gene implicated in ADHD and addiction. Brain Res, 2012. 1463: p. 85–92. [DOI] [PubMed] [Google Scholar]
- 49.Lange M, et al. , The ADHD-susceptibility gene lphn3.1 modulates dopaminergic neuron formation and locomotor activity during zebrafish development. Mol Psychiatry, 2012. 17(9): p. 946–54. [DOI] [PubMed] [Google Scholar]
- 50.Allache R, et al. , Role of the planar cell polarity gene CELSR1 in neural tube defects and caudal agenesis. Birth Defects Res A Clin Mol Teratol, 2012. 94(3): p. 176–81. [DOI] [PubMed] [Google Scholar]
- 51.Robinson A, et al. , Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum Mutat, 2012. 33(2): p. 440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lei Y, et al. , Identification of novel CELSR1 mutations in spina bifida. PLoS One, 2014. 9(3): p. e92207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keeler AB, Molumby MJ, and Weiner JA, Protocadherins branch out: Multiple roles in dendrite development. Cell Adh Migr, 2015. 9(3): p. 214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curtin JA, et al. , Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol, 2003. 13(13): p. 1129–33. [DOI] [PubMed] [Google Scholar]
- 55.Devenport D and Fuchs E, Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol, 2008. 10(11): p. 1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi D, et al. , Celsr1 is required for the generation of polarity at multiple levels of the mouse oviduct. Development, 2014. 141(23): p. 4558–68. [DOI] [PubMed] [Google Scholar]
- 57.Tissir F, et al. , Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat Neurosci, 2010. 13(6): p. 700–7. [DOI] [PubMed] [Google Scholar]
- 58.Tissir F, et al. , Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci, 2005. 8(4): p. 451–7. [DOI] [PubMed] [Google Scholar]
- 59.Zhou L, et al. , Early forebrain wiring: genetic dissection using conditional Celsr3 mutant mice. Science, 2008. 320(5878): p. 946–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou L, et al. , Role of the atypical cadherin Celsr3 during development of the internal capsule. Cereb Cortex, 2009. 19 Suppl 1: p. i114–9. [DOI] [PubMed] [Google Scholar]
- 61.Feng J, et al. , A role for atypical cadherin Celsr3 in hippocampal maturation and connectivity. J Neurosci, 2012. 32(40): p. 13729–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qu Y, et al. , Genetic evidence that Celsr3 and Celsr2, together with Fzd3, regulate forebrain wiring in a Vangl-independent manner. Proc Natl Acad Sci U S A, 2014. 111(29): p. E2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chai G, et al. , Celsr3 is required in motor neurons to steer their axons in the hindlimb. Nat Neurosci, 2014. 17(9): p. 1171–9. [DOI] [PubMed] [Google Scholar]
- 64.Wang S, et al. , De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell Rep, 2018. 24(13): p. 3441–3454 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willsey AJ, et al. , De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron, 2017. 94(3): p. 486–499 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu D, et al. , BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J Clin Invest, 2015. 125(4): p. 1497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okajima D, Kudo G, and Yokota H, Antidepressant-like behavior in brain-specific angiogenesis inhibitor 2-deficient mice. J Physiol Sci, 2011. 61(1): p. 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Purcell RH, et al. , A disease-associated mutation in the adhesion GPCR BAI2 (ADGRB2) increases receptor signaling activity. Hum Mutat, 2017. 38(12): p. 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lanoue V, et al. , The adhesion-GPCR BAI3, a gene linked to psychiatric disorders, regulates dendrite morphogenesis in neurons. Mol Psychiatry, 2013. 18(8): p. 943–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giera S, et al. , The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat Commun, 2015. 6: p. 6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ackerman SD, et al. , The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Galpha12/13 and RhoA. Nat Commun, 2015. 6: p. 6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giera S, et al. , Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ackerman SD, et al. , GPR56/ADGRG1 regulates development and maintenance of peripheral myelin. J Exp Med, 2018. 215(3): p. 941–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ravenscroft G, et al. , Mutations of GPR126 Are Responsible for Severe Arthrogryposis Multiplex Congenita. Am J Hum Genet, 2015. 96(6): p. 955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waller-Evans H, et al. , The orphan adhesion-GPCR GPR126 is required for embryonic development in the mouse. PLoS One, 2010. 5(11): p. e14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karner CM, et al. , Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum Mol Genet, 2015. 24(15): p. 4365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kou I, et al. , Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat Genet, 2013. 45(6): p. 676–9. [DOI] [PubMed] [Google Scholar]
- 78.Weston MD, et al. , Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet, 2004. 74(2): p. 357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Wijk E, et al. , The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet, 2006. 15(5): p. 751–65. [DOI] [PubMed] [Google Scholar]
- 80.McGee J, et al. , The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci, 2006. 26(24): p. 6543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu QX, et al. , Constitutive Galphai coupling activity of very large G protein-coupled receptor 1 (VLGR1) and its regulation by PDZD7 protein. J Biol Chem, 2014. 289(35): p. 24215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu VW, Addington A, and Hyman A, Novel autism subtype-dependent genetic variants are revealed by quantitative trait and subphenotype association analyses of published GWAS data. PLoS One, 2011. 6(4): p. e19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DeRosse P, et al. , The genetics of symptom-based phenotypes: toward a molecular classification of schizophrenia. Schizophr Bull, 2008. 34(6): p. 1047–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao HM, et al. , Identification and characterization of three inherited genomic copy number variations associated with familial schizophrenia. Schizophr Res, 2012. 139(1–3): p. 229–36. [DOI] [PubMed] [Google Scholar]
- 85.Lips ES, et al. , Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry, 2012. 17(10): p. 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang KS, et al. , Genome-wide association analysis of age at onset in schizophrenia in a European-American sample. Am J Med Genet B Neuropsychiatr Genet, 2011. 156B(6): p. 671–80. [DOI] [PubMed] [Google Scholar]
- 87.Anney R, et al. , A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet, 2010. 19(20): p. 4072–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Engwerda A, et al. , The phenotypic spectrum of proximal 6q deletions based on a large cohort derived from social media and literature reports. Eur J Hum Genet, 2018. 26(10): p. 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vlckova M, et al. , Mechanism and genotype-phenotype correlation of two proximal 6q deletions characterized using mBAND, FISH, array CGH, and DNA sequencing. Cytogenet Genome Res, 2012. 136(1): p. 15–20. [DOI] [PubMed] [Google Scholar]
- 90.Beltrao-Braga PC and Muotri AR, Modeling autism spectrum disorders with human neurons. Brain Res, 2017. 1656: p. 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sztainberg Y and Zoghbi HY, Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci, 2016. 19(11): p. 1408–1417. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, et al. , An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 2014. 34(36): p. 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nowakowski TJ, et al. , Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science, 2017. 358(6368): p. 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi Y, et al. , A genome-wide meta-analysis identifies two novel loci associated with high myopia in the Han Chinese population. Hum Mol Genet, 2013. 22(11): p. 2325–33. [DOI] [PubMed] [Google Scholar]
- 95.Shi Y, et al. , Genetic variants at 13q12.12 are associated with high myopia in the Han Chinese population. Am J Hum Genet, 2011. 88(6): p. 805–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feng J, Han Q, and Zhou L, Planar cell polarity genes, Celsr1–3, in neural development. Neurosci Bull, 2012. 28(3): p. 309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Das G, Reynolds-Kenneally J, and Mlodzik M, The atypical cadherin Flamingo links Frizzled and Notch signaling in planar polarity establishment in the Drosophila eye. Dev Cell, 2002. 2(5): p. 655–66. [DOI] [PubMed] [Google Scholar]
- 98.Xiong HY, et al. , RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science, 2015. 347(6218): p. 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gupte J, et al. , Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett, 2012. 586(8): p. 1214–9. [DOI] [PubMed] [Google Scholar]
- 100.Stoveken HM, et al. , Dihydromunduletone Is a Small-Molecule Selective Adhesion G Protein-Coupled Receptor Antagonist. Mol Pharmacol, 2016. 90(3): p. 214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stoveken HM, et al. , Gedunin- and Khivorin-Derivatives Are Small-Molecule Partial Agonists for Adhesion G Protein-Coupled Receptors GPR56/ADGRG1 and GPR114/ADGRG5. Mol Pharmacol, 2018. 93(5): p. 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goodnow RA Jr., Dumelin CE, and Keefe AD, DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat Rev Drug Discov, 2017. 16(2): p. 131–147. [DOI] [PubMed] [Google Scholar]
- 103.Demberg LM, et al. , Identification of the tethered peptide agonist of the adhesion G protein-coupled receptor GPR64/ADGRG2. Biochem Biophys Res Commun, 2015. 464(3): p. 743–7. [DOI] [PubMed] [Google Scholar]
- 104.Nazarko O, et al. , A Comprehensive Mutagenesis Screen of the Adhesion GPCR Latrophilin-1/ADGRL1. iScience, 2018. 3: p. 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta A, et al. , Facile target validation in an animal model with intracellularly expressed monobodies. Nat Chem Biol, 2018. 14(9): p. 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kishore A and Hall RA, Disease-associated extracellular loop mutations in the adhesion G protein-coupled receptor G1 (ADGRG1; GPR56) differentially regulate downstream signaling. J Biol Chem, 2017. 292(23): p. 9711–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McCorvy JD, et al. , Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol, 2018. 14(2): p. 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wacker D, Stevens RC, and Roth BL, How Ligands Illuminate GPCR Molecular Pharmacology. Cell, 2017. 170(3): p. 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manglik A, et al. , Structure-based discovery of opioid analgesics with reduced side effects. Nature, 2016. 537(7619): p. 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCorvy JD, et al. , Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol, 2018. 25(9): p. 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Promel S, Langenhan T, and Arac D, Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol Sci, 2013. 34(8): p. 470–8. [DOI] [PubMed] [Google Scholar]
- 112.Tang X, et al. , GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Galphaq-p63RhoGEF-Rho GTPase pathway. Cancer Res, 2013. 73(20): p. 6206–18. [DOI] [PubMed] [Google Scholar]
- 113.Shin D, et al. , Very large G protein-coupled receptor 1 regulates myelin-associated glycoprotein via Galphas/Galphaq-mediated protein kinases A/C. Proc Natl Acad Sci U S A, 2013. 110(47): p. 19101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee JW, et al. , Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat Commun, 2016. 7: p. 13123. [DOI] [PMC free article] [PubMed] [Google Scholar]