

Abstract
Liver X receptors α and β (LXRα and LXRβ) are nuclear receptors with pivotal roles in the transcriptional control of lipid metabolism. Transcriptional activity of LXRs is induced in response to elevated cellular levels of cholesterol. LXRs bind to and regulate the expression of genes that encode proteins involved in cholesterol absorption, transport, efflux, excretion and conversion to bile acids. The coordinated, tissue-specific actions of the LXR pathway maintain systemic cholesterol homeostasis and regulate immune and inflammatory responses. LXRs also regulate fatty acid metabolism by controlling the lipogenic transcription factor sterol regulatory element-binding protein 1c and regulate genes that encode proteins involved in fatty acid elongation and desaturation. LXRs exert important effects on the metabolism of phospholipids, which, along with cholesterol, are major constituents of cellular membranes. LXR activation preferentially drives the incorporation of polyunsaturated fatty acids into phospholipids by inducing transcription of the remodelling enzyme lysophosphatidylcholine acyltransferase 3. The ability of the LXR pathway to couple cellular sterol levels with the saturation of fatty acids in membrane phospholipids has implications for several physiological processes, including lipoprotein production, dietary lipid absorption and intestinal stem cell proliferation. Understanding how LXRs regulate membrane composition and function might provide new therapeutic insight into diseases associated with dysregulated lipid metabolism, including atherosclerosis, diabetes mellitus and cancer.
The maintenance of cellular and systemic lipid levels is essential for physiological homeostasis. Perturbations of lipid metabolism are associated with several prevalent diseases, such as diabetes mellitus, atherosclerosis, cancer and neurodegenerative disease1,2. Control of lipid levels involves a carefully orchestrated balance between their endogenous biosynthesis, dietary uptake, metabolism and elimination from the body. Nuclear receptors are a class of ligand-activated transcription factors that have important roles in physiology3. Several nuclear receptors, including liver X receptors (LXRs; also known as oxysterols receptor LXR), farnesoid X receptors (FXRs) and peroxisome proliferator-activated receptors (PPARs), respond to changes in cellular levels of endogenous lipid ligands by regulating the expression of genes that encode proteins involved in lipid metabolism1,4.
First cloned in the 1990s from mice, the LXR family consists of two isotypes, LXRα (encoded by Nr1h3) and LXRβ (encoded by Nr1h2)5,6. LXRα and LXRβ share extensive sequence homology but have distinct tissue distributions. LXRα is highly expressed in metabolically active tissues and cell types, including liver, intestine, adipose tissue and macrophages, whereas LXRβ is expressed ubiquitously7,8. LXRs form obligate heterodimers with the retinoid X receptor-α (RXRα; also known as retinoic acid receptor RXRα) and bind to a specific DNA recognition sequence known as an LXR response element (LXRE) (FIG. 1). LXREs contain variations of the repeated sequence AGGTCA, separated by four nucleotides6. In the absence of ligands, LXR–RXR complexes are believed to bind co-repressors, such as silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) or nuclear receptor co-repressor (NCoR), and repress target gene expression9,10. Ligands binding to LXRs induce a conformational change to the LXR–RXR complexes; this change causes the release of co-repressors and the recruitment of co-activators such as histone acetyltransferase p300 (also known as E1A-associated protein p300) and activating signal co-integrator 2 (ASC2; also known as NCOA6), resulting in the increased transcription of target genes11. A limited number of studies have suggested that LXRs act as monomers; however, these findings have not been widely replicated12,13. Further studies are needed to demonstrate that LXR regulates transcription by acting as a monomer under physiological conditions in vivo. LXRs are activated by endogenous ligands, including cholesterol derivatives such as oxysterols and 24(S),25-epoxycholesterol14–16, and by intermediate precursors in the cholesterol biosynthetic pathway, such as desmosterol17. Synthetic agonists, such as GW3965 and T0901317, have also been widely used as chemical tools to study LXR signalling pathways both in vitro and in vivo4.
Fig. 1 |. LXRs are lipid-responsive transcription factors.
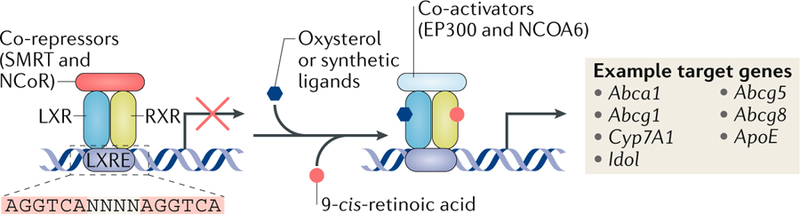
Liver X receptors (LXRs) and retinoid X receptors (RXRs) form heterodimers and bind to LXR response elements (LXREs) in the regulatory regions of their target genes. The target regions consist of variations of the repeated sequence AGGTCA, separated by four nucleotides (N). In the absence of ligands, the LXR–RXR complex binds co-repressors and suppresses gene expression. When LXRs are activated by oxysterols or synthetic ligands and/or RXR is activated by its ligands, such as 9-cis-retinoic acid, the co-repressors are replaced by co-activators, thereby activating the expression of target genes involved in lipid metabolism. ASC2, activating signal co-integrator 2; EP300, histone acetyltransferase p300; NCoR, nuclear receptor co-repressor; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor.
This Review summarizes the roles of LXRs in cholesterol, fatty acid and phospholipid metabolism and their links to diseases related to lipid metabolism. We also discuss emerging strategies for targeting LXRs and their downstream pathways in the setting of metabolic diseases.
LXRs in cholesterol metabolism
Cholesterol excretion from the liver.
The discovery that LXRs were activated by oxysterols was the first clue that they might be involved in maintaining cholesterol homeostasis14. This idea was confirmed through the generation and analysis of Lxrα-knockout mice. Mice lacking LXRα accumulate massive amounts of cholesterol in the liver when fed a high-cholesterol diet18. This early study also identified the gene encoding cytochrome P450 7A1 (Cyp7a1), the rate-limiting enzyme in the bile acid synthesis pathway, as the first direct target of LXRs (FIG. 2). The study showed that loss of LXRα in mice impairs the conversion of cholesterol into bile acids. Subsequent studies revealed that LXRs also stimulate biliary cholesterol excretion directly through their target genes Abcg5 and Abcg8 (REF.19). Mice lacking Abcg5 and Abcg8 fail to increase biliary cholesterol content in response to T0901317 treatment, suggesting that the expression of Abcg5 and Abcg8 is required for LXR-dependent biliary sterol trafficking. The importance of LXRs, especially liver LXRα, in whole-body cholesterol homeostasis was reinforced by studies of hepatic-specific Lxrα-knockout mice, which have impaired reverse cholesterol transport (RCT), cholesterol catabolism and cholesterol excretion20.
Fig. 2 |. LXR signalling pathways in cholesterol and fatty acid metabolism.
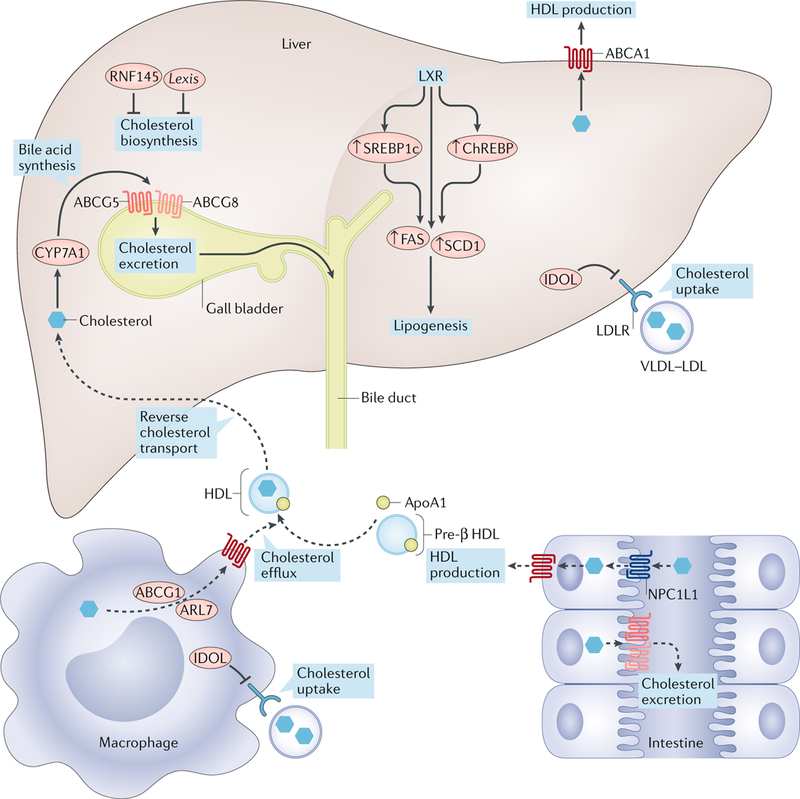
Liver X receptor (LXR) activation modulates cholesterol and fatty acid metabolism in a tissue-specific manner. LXR targets are shown in red. In the liver, LXR agonism promotes the conversion of cholesterol into bile acids via cytochrome P450 7A1 (CYP7A1) and biliary cholesterol excretion through ATP-binding cassette subfamily G members 5 and 8 (ABCG5 and ABCG8, respectively). LXR inhibits cholesterol uptake in the liver and macrophages through inducing the expression of inducible degrader of the LDL receptor (IDOL) and the degradation of LDL receptor (LDLR). LXR activation suppresses cholesterol biosynthesis in liver by inducing the transcription of non-coding RNA LXR-induced sequence (Lexis) and E3 ubiquitin protein ligase RING finger protein 145 (RNF145). LXR activation promotes fatty acid biosynthesis by inducing the expression of sterol regulatory element-binding protein 1c (SREBP1c), carbohydrate-response element-binding protein (ChREBP) and their targets, fatty acid synthase (FASN) and stearoyl-coenzyme A desaturase 1 (SCD1). In peripheral cells such as macrophages, LXRs increase expression of ATP-binding cassette subfamily A member 1 (ABCA1), ABCG1 and ADP-ribosylation factor-like protein 7 (ARL7), thereby promoting cholesterol movement to the plasma membrane and cholesterol efflux to apolipoprotein A1 (ApoA1) or pre-β HDL. In the intestine, LXR activation increases HDL formation via basolateral ABCA1 and promotes cholesterol efflux and trans-intestinal cholesterol excretion through ABCG5 and ABCG8. LXR also inhibits cholesterol absorption by indirectly inhibiting Niemann–Pick C1-like protein 1 (NPC1L1). The dashed arrows indicate reduced cholesterol uptake.
Reverse cholesterol transport.
Another important role of LXRs in maintaining cholesterol homeostasis is to promote RCT, a process by which excess cholesterol in peripheral tissues is transferred to HDL and transported to the liver for bile acid synthesis and excretion. The initial step in RCT is cellular cholesterol efflux, which involves the transfer of free cholesterol to apolipoprotein A1 (ApoA1) or pre-β HDL. Cellular cholesterol efflux is mediated primarily by the ATP-binding cassette transporters ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1), two of the earliest identified LXR-responsive factors21,22. Mutations in ABCA1 in humans cause Tangier disease, a disorder characterized by severe reductions in plasma levels of HDL and increased cholesterol accumulation in peripheral tissues23,24. Structural analysis of membrane-bound ABCA1 revealed that two extracellular domains form a hydrophobic tunnel that probably facilitates the export of cholesterol25. ABCG1 was shown to localize to endosomes and to enable the transport of intracellular sterols away from the endoplasmic reticulum (ER)26. Several other LXR targets are also involved in the RCT pathway. For instance, ADP-ribosylation factor-like protein 7 (ARL7; also known as ARL4C), another LXR target, was shown to transport cholesterol between the perinuclear compartment and the plasma membrane27,28. Additionally, LXRs induce the expression of apolipoproteins (including ApoE, ApoC1, ApoC2, ApoC4 (REFS29,30) and ApoD31), as well as genes that encode proteins involved in lipoprotein remodelling (such as phospholipid transfer protein (PLTP)32, cholesteryl ester transfer protein (CETP)33 and lipoprotein lipase (LPL))34. The net effect of LXR activation — the promotion of RCT from the peripheral tissues to the liver — probably results from the coordinated action of many, if not all, these factors.
Cholesterol biosynthesis.
LXRs work hand in hand with the sterol regulatory element-binding protein 2 (SREBP2) pathway to maintain cellular and systemic sterol levels. Whereas LXRs facilitate elimination of excess cholesterol in response to high cellular levels of cholesterol, SREBP2 promotes cholesterol biosynthesis and uptake in response to low cholesterol levels in cells. Research over the past decade has revealed that these two key pathways engage in crosstalk on several levels. SREBP2 was shown to activate the transcription of ABCA1 by generating oxysterol ligands for LXRs35. Conversely, LXR activation in the liver inhibits cholesterol biosynthesis through the induction of the non-coding RNA LXR-induced sequence (Lexis)36 and the E3 ubiquitin protein ligase RING finger protein 145 (RNF145)37. Lexis interacts with the ribonucleoprotein RNA-binding protein Raly (RALY), which acts as a cofactor in the transcription of genes that encode proteins involved in cholesterol biosynthesis. RNF145 is an ER membrane protein that ubiquitylates sterol regulatory element-binding protein cleavage-activating protein (SCAP) and inhibits its transport to the Golgi and the subsequent proteolytic processing of SREBP2 (REF.37).
Cholesterol uptake.
A considerable body of evidence has demonstrated that high levels of LDL cholesterol are a dominant risk factor for atherosclerosis. The LDL receptor (LDLR), a cell surface protein, is essential for the uptake of LDL cholesterol and the maintenance of plasma levels of cholesterol38. Mutations in LDLR in humans or mice cause hypercholesterolaemia and accelerated atherosclerosis owing to reduced LDL clearance39–41. LDLR expression is regulated at both the transcriptional and post-translational levels. The primary transcription factor for LDLR is SREBP2. In response to low cholesterol levels, SREBP2 drives the transcription of genes encoding proteins involved in cholesterol biosynthesis and LDL cholesterol uptake42. In a counter-regulatory mechanism, LXRs induce the expression of inducible degrader of the LDLR (IDOL; also known as MYLIP), an E3 ubiquitin protein ligase that ubiquitylates and degrades LDLR and related proteins VLDL receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2; also known as LRP8), through clathrin-independent endocytosis and multivesicular-body-mediated lysosomal degradation43–45. Crystallography studies characterized the interactions between the ubiquitin-conjugated enzyme E2 D (UBE2D) family (UBE2D1–4) of E2 ligases and IDOL that drive ubiquitylation of LDLR46. Subsequent studies showed that the four-point-one, ezrin, radixin, moesin (FERM) domain of IDOL binds to a recognition sequence in the cytoplasmic tails of lipoprotein receptors and mediates their degradation47. Interestingly, the LXR–IDOL pathway appears to exert species-specific influences on LDLR expression and plasma levels of cholesterol48. In mice, LXR agonists or loss of IDOL has little effect on hepatic levels of LDLR protein and plasma levels of cholesterol. By contrast, LXR agonists reduce hepatic LDLR expression and raise plasma levels of LDL cholesterol in primates and humans, probably through an IDOL-dependent mechanism48,49. Genome-wide association studies identified an Asn342Ser polymorphism in the gene encoding IDOL (MYLIP) that is associated with plasma lipid levels in a Mexican population50. Individuals carrying the Ser342 allele had higher cholesterol and LDL levels than those carrying the Asn342 allele. Variation at the MYLIP locus is also linked with response to statin treatment51. These findings suggest that IDOL inhibition could be an alternative approach to lowering LDL cholesterol in humans.
Sterol metabolism in the intestine.
As the gateway for cholesterol entry into the body, the intestine has a crucial role in maintaining whole-body cholesterol homeostasis. Dietary cholesterol absorption from the intestinal lumen is mediated by apical membrane Niemann–Pick C1-like protein 1 (NPC1L1; also known as NPC1-like intracellular cholesterol transporter 1)52. Cholesterol efflux from enterocytes to the circulation is mediated by basolateral membrane ABCA1 (REFS53–55). Cholesterol efflux from enterocytes into the lumen56,57 and transintestinal cholesterol excretion are mediated by heterodimers of apical membrane ATP-binding cassette subfamily G members 5 and 8 (ABCG5 and ABCG8, respectively)58. LXRs regulate multiple aspects of intestinal cholesterol metabolism by controlling the expression of several target genes. For example, activation of LXRs in intestine promotes faecal cholesterol excretion by inducing expression of ABCG5 and ABCG8. Mutations in ABCG5 and ABCG8 result in sitosterolaemia, an autosomal recessive disorder characterized by increased plant sterol levels and accelerated atherosclerosis59,60. Similar to LXR activation in other peripheral tissues, intestinal LXR activation leads to increased RCT and HDL production through the induction of ABCA1 expression53–55. Furthermore, LXR activation in the intestine also reduces cholesterol absorption, probably owing to increased ABCG5 and ABCG8 expression and cholesterol excretion from enterocytes55,61. Consistent with the effects of LXR activation in intestine, overexpression of ABCG5 and ABCG8 reduces cholesterol absorption62. The reduced cholesterol absorption upon LXR activation might also be due in part to reduced NPC1L1 expression55,61. The molecular mechanism by which LXR activation reduces NPC1L1 expression remains to be determined. It is unlikely to be a direct transcriptional effect, as LXRs are not known to function as ligand-dependent repressors63.
LXRs in fatty acid metabolism
In addition to their importance in modulating cholesterol homeostasis, LXRs are also key regulators of de novo lipogenesis in the liver7,64,65. Administration of synthetic LXR agonists to mice increases hepatic and plasma levels of triglyceride owing to enhanced fatty acid biosynthesis and VLDL secretion64. The lipogenic activity of LXRs is due, in large measure, to the transcriptional induction of the gene encoding SREBP1c, a central transcription activator of fatty acid biosynthesis (FIG. 3). In addition, LXRs directly activate the transcription of several lipogenic genes via LXREs in their promoter regions, including those encoding fatty acid synthase (Fasn)65 and stearoyl-coenzyme A desaturase 1 (Scd1; also known as acyl-coenzyme A desaturase 1)66. Furthermore, LXRs also modulate the expression of another lipogenic regulator, carbohydrate-response element-binding protein (ChREBP). This regulation probably contributes to the induction of certain lipogenic genes in SREBP1c-null livers upon T0901317 treatment67. Interestingly, LXRs are also important for the ability of other signals, such as insulin68,69 and polyunsaturated fatty acids70–72, to regulate SREBP1c expression. Deletion of LXREs in the Srebf1c (which encodes SREBP1c) promoter disrupts its response to insulin68, and studies from the past 2 years have demonstrated that LXRα and CCAAT/enhancer-binding protein β (C/EBPβ) form a complex that binds to the Srebf1c promoter and is activated by insulin69. Polyunsaturated fatty acids suppress SREBP1c expression and lipogenesis through inhibiting SREBP1 cleavage72 and by competing with LXR ligands70.
Fig. 3 |. Roles of LXR-dependent phospholipid remodelling in liver.
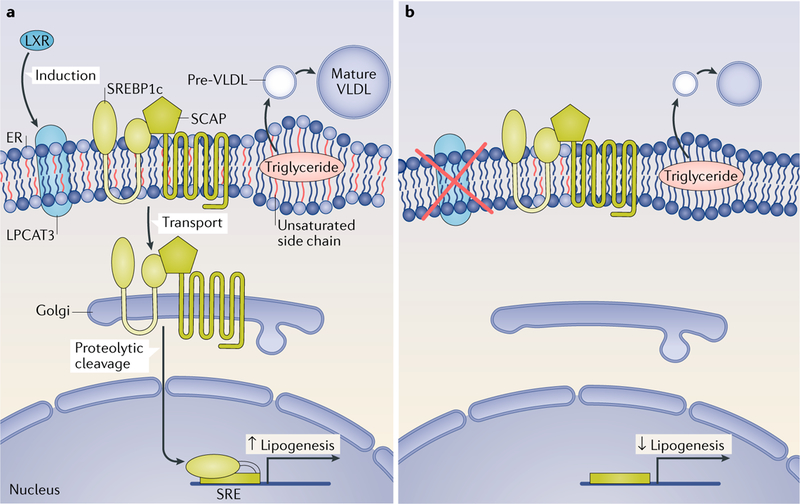
a | The effects of liver X receptor (LXR) activation. LXR activation promotes the incorporation of polyunsaturated fatty acids into phospholipids through the induction of lysophosphatidylcholine acyltransferase 3 (LPCAT3) expression. Polyunsaturated phospholipids facilitate sterol regulatory element-binding protein 1c (SREBP1c) transport from the endoplasmic reticulum (ER) to the Golgi and its proteolytic cleavage, thereby promoting lipogenesis. LXR agonists and LPCAT3 activation also promote VLDL secretion. Increased abundance of polyunsaturated phospholipids creates a dynamic membrane environment that facilitates the transfer of triglyceride to nascent apolipoprotein B (ApoB)-containing lipoprotein particles, leading to the efficient lipidation of ApoB-containing lipoproteins. b | Consequences of LPCAT3 deficiency. Loss of LPCAT3 expression reduces the composition of polyunsaturated phospholipids in the ER membrane and impairs the translocation of SREBP1c from the ER to the Golgi and its proteolytic cleavage. SCAP, sterol regulatory element-binding protein cleavage-activating protein; SRE, sterol response element.
In addition to modulating lipogenesis in the liver, LXRs also regulate fatty acid catabolism in other tissues. LXR agonists were shown to increase lipolysis and fatty acid β-oxidation in adipose tissues73,74. On a high-carbohydrate diet, LXRα-null mice exhibit higher energy expenditure and UCP1 expression in brown adipose tissue than wild-type mice75. Similarly, LXRα-null mice are resistant to high-fat and high-cholesterol diet-induced obesity owing to increased energy expenditure resulting from upregulation of UCPs in muscle and adipose tissues76. Other studies have delineated tissue-selective effects of LXR signalling pathways on lipogenesis in the setting of obesity77. As expected, LXRα-deficient and LXRβ-deficient ob/ob mice were protected from hepatic steatosis owing to impaired lipogenesis in the liver. Unexpectedly, however, loss of hepatic lipogenesis was accompanied by a reciprocal increase in lipogenesis and fat storage in adipose tissues. Moreover, ob/ob mice lacking LXRs were more insulin sensitive than ob/ob mice, despite having similar body weight, owing to enhanced adipose PPARγ and ChREBPβ activity.
LXRs in phospholipid metabolism
Phospholipids are the major constituents of biological membranes. The fatty acyl composition of component phospholipids determines the biophysical properties of membranes and influences their function78,79. In mammalian cells, saturated and monounsaturated fatty acyl chains are predominantly linked at the sn-1 position of phospholipids, whereas polyunsaturated fatty acids are most often located at the sn-2 position78,79. This asymmetrical distribution of fatty acyl chains is established largely through a remodelling process of deacylation and reacylation — a pathway referred to as the Lands’ cycle80,81. We and others showed that LXRs modulate membrane phospholipid composition through activation of lysophosphatidylcholine acyltransferase 3 (LPCAT3; also known as lysophospholipid acyltransferase 5), a phospholipid remodelling enzyme that catalyses the incorporation of polyunsaturated fatty acids at the sn-2 site of lysophospholipids82–84. The ability of LXR to alter phospholipid composition in response to changing cellular levels of sterol provides a mechanism to protect biological membranes against lipid stress. LXR activation increases LPCAT3 expression and the abundance of polyunsaturated phospholipids in cell membranes, thereby ameliorating ER stress induced by saturated free fatty acids in vitro or by hepatic lipid accumulation in vivo82.
LXR-dependent phospholipid remodelling in the liver.
LXR activation promotes VLDL secretion in mouse liver through the transcriptional induction of Srebf1c and genes downstream of SREBP1c that encode proteins involved in lipogenesis, as well as PLTP, which transfers phospholipids into nascent VLDL to enable particle expansion85,86. Since phospholipids are a major component of lipoprotein particles, phospholipid availability is known to influence the production of lipoproteins87. Studies published in the past 3 years have demonstrated that LPCAT3 and phospholipid remodelling have unexpected roles in hepatic lipoprotein production88–91, suggesting that not only the quantity but also the composition of phospholipids is important for lipoprotein secretion. Mice lacking LPCAT3 in the liver show reduced plasma levels of triglycerides, VLDL lipidation and secretion, resulting in hepatic steatosis (FIG. 3). Mechanistic studies suggest that LPCAT3 activity and polyunsaturated phospholipids probably affect the fluidity and curvature of membrane surfaces, thereby reducing triglyceride mobilization to VLDL. Liver-specific Lpcat3-knockout mice secrete less VLDL from the liver in response to synthetic LXR agonist treatment than control mice, suggesting that induction of LPCAT3 activity is required for the ability of LXRs to promote hepatic VLDL production88. Subsequent studies revealed that LPCAT3 activity is also involved in the effects of LXRs on SREBP1c-dependent lipogenesis in the liver. Incorporation of polyunsaturated fatty acids into phospholipids by LPCAT3 promotes SREBP1c processing and lipogenesis92. Interestingly, polyunsaturated phospholipid levels are selectively increased in the ER of obese mice, which seems to be owing to elevated LPCAT3 activity. Inhibition of LPCAT3 activity in obese mice reduces SREBP1c pathway activity, blunts lipogenesis and ameliorates the development of fatty liver92.
LXR-dependent phospholipid remodelling in the intestine.
In the intestine, the phospholipid acyl chain composition of enterocyte membranes is critical for dietary lipid uptake90. Loss of LPCAT3 in the intestine results in a defect in the incorporation of linoleate and arachidonate into phospholipids, which leads to marked reduction in membrane fluidity and impairs passive fatty acid transport into enterocytes and chylomicron production (FIG. 4). LPCAT3 deficiency in the setting of a triglyceride-rich diet leads to the exacerbated production of gut hormones (such as glucagon-like peptide 1 (GLP1) and peptide YY (PYY)) and the cessation of food intake despite starvation, suggesting that LXR–LPCAT3 signalling functions in a gut–brain feedback loop that couples lipid absorption to food intake.
Fig. 4 |. LPCAT3 and phospholipid remodelling in lipid absorption and intestinal homeostasis.
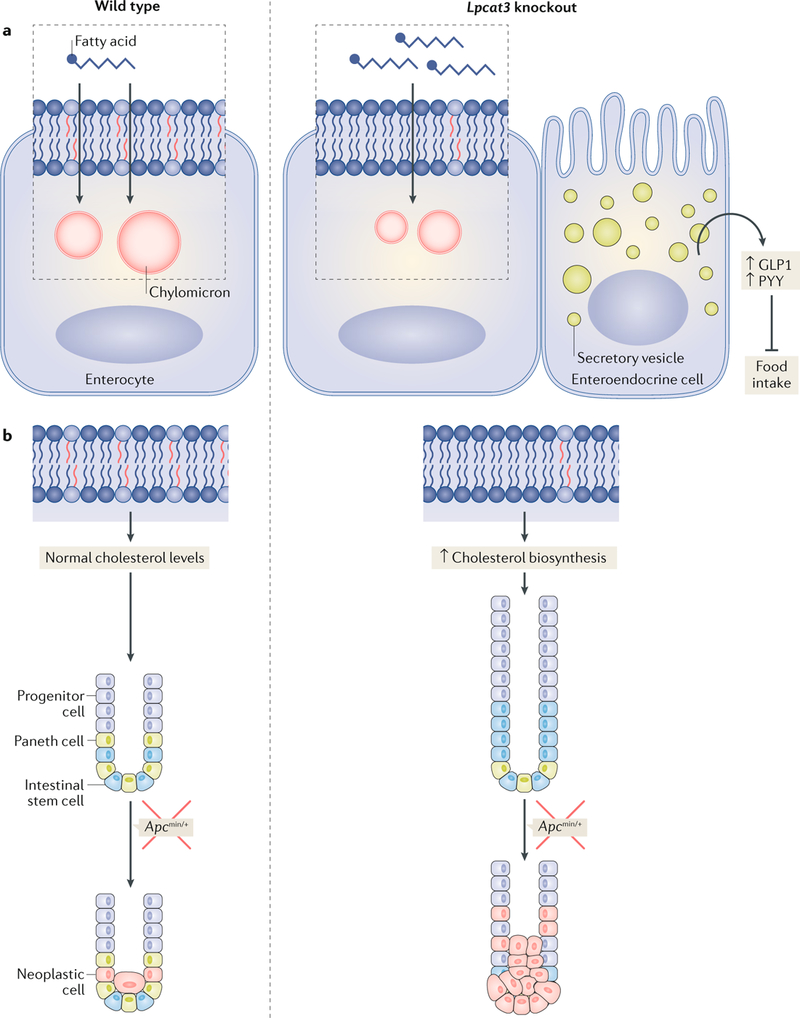
a | Lysophosphatidylcholine acyltransferase 3 (LPCAT3) activity regulates lipid absorption in the small intestine. Loss of LPCAT3 in the intestine reduces polyunsaturated phospholipid content and membrane fluidity, resulting in impaired passive fatty acid transport into enterocytes and chylomicron production. When challenged with a triglyceride-rich diet, LPCAT3-deficient mice produce more gut hormones (glucagon-like peptide 1 (GLP1) or peptide YY (PYY)), leading to the inhibition of food intake. b | Loss of LPCAT3 increases membrane saturation and stimulates cholesterol biosynthesis, thereby driving the proliferation of intestinal stem cells and progenitor cells. Consequently, LPCAT3 deficiency and cholesterol biosynthesis enhance tumour formation in Apcmin/+ mice.
Research published in 2018 also uncovered an unexpected link between membrane phospholipid composition, cholesterol biosynthesis and intestinal stem cell homeostasis93. We found that LPCAT3 deficiency in the intestine increases the saturation of fatty acyl chains in membrane phospholipids and stimulates cholesterol biosynthesis, thereby driving intestinal stem cell proliferation (FIG. 4). Interestingly, cholesterol itself appears to act as a mitogen for intestinal stem cells. Increasing cellular cholesterol content, either by promoting endogenous synthesis or providing excess exogenous cholesterol, is sufficient to drive intestinal stem cell proliferation. Loss of LPCAT3 expression or overexpression of SREBP2 in Apcmin mice markedly promoted intestinal tumour formation. Previous studies demonstrated that LXRs regulate cell apoptosis via AKT (also known as protein kinase B) survival signalling in lipid rafts94. Interestingly, polyunsaturated phospholipids inhibit cell proliferation through suppressing the binding of AKT to cell membranes95. LPCAT3 activity might mediate the effect of LXR activation on cell survival.
The mechanism whereby phospholipid remodelling is linked to cholesterol biosynthesis remains to be elucidated. It is important to note that the effects of phospholipid metabolism on SREBP1 and SREBP2 pathways appear to be tissue-specific as LPCAT3 deficiency selectively influences SREBP1c target genes in the liver, whereas it primarily affects SREBP2 target genes in the intestine. These studies highlight a mechanism whereby membrane phospholipid remodelling differentially influences SREBP maturation in response to cellular lipids.
LXRs in metabolic diseases
Atherosclerosis.
Atherosclerosis is initiated by subendothelial retention of cholesterol-rich, ApoB-containing lipoproteins in arteries96,97. Modified lipoproteins trigger a local inflammatory response in the arterial wall that activates endothelial cells and leads to the infiltration and accumulation of monocytes. These monocytes differentiate into tissue macrophages and phagocytose the modified lipoproteins through scavenger receptors and become lipid-laden foam cells98,99. Foam cells secrete pro-inflammatory mediators, including cytokines, chemokines and reactive oxygen species, which contribute to the unresolved inflammation and the progression of lesions into more advanced plaques. Considerable evidence points to LXR pathway activity as an important determinant of atherosclerosis susceptibility100–103. Loss of LXRs markedly accelerates the initiation and progression of atherosclerosis in Ldlr−/− and Apoe−/− mouse models100. Conversely, chronic administration of synthetic LXR agonists reduces the size and abundance of atherosclerotic lesions in these models101,103. Several lines of evidence suggest that the anti-atherogenic effect of LXRs is due, in large measure, to their ability to promote cholesterol efflux from macrophages within lesions through the induction of ABCA1 and ABCG1 expression. LXR agonists fail to reduce atherosclerosis progression in irradiated Ldlr−/− mice transplanted with bone marrow from LXR-null mice104, confirming that expression of LXR in macrophages is essential for the anti-atherogenic effects of LXR agonists. Accordingly, transgenic overexpression of LXRα in macrophages suppresses atherosclerosis in Ldlr−/− mice105.
Inflammation also has a substantial role in atherosclerosis progression99,106,107. In addition to exerting beneficial effects on macrophage cholesterol efflux, LXR activation also affects atherogenesis through the repression of inflammatory gene expression101,108,109. How activation of LXRs inhibits inflammatory signalling is not well understood. One proposed mechanism postulated that LXRs can be modified by sumoylation when activated by agonist and that sumoylated LXR forms a complex with transcriptional co-repressors NCoR and/or SMRT on inflammatory gene promoters110–112. Conversely, excess cholesterol crystals in macrophages initiate pro-inflammatory signalling at atherosclerotic lesions113,114. Considering that LXR activation can efficiently remove intracellular cholesterol through efflux, their ability to regulate lipid metabolism through transcriptional activation probably contributes to repressed inflammation. Indeed, a study published in the past 2 years demonstrated that LXR agonists inhibit nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signalling pathways downstream of Toll-like receptors through ABCA1-dependent changes in membrane lipid organization, suggesting that LXR transcriptional activation underlies dual biological functions in metabolism and inflammation115.
Infection and immunity.
The LXR signalling pathway modulates the function of several types of immune cells116,117. In addition, extensive documentation reveals that LXR agonists repress inflammatory gene expression triggered by Toll-like receptors in macrophages and other immune cells108,118,119. The first in vivo evidence for a role of LXR signalling in immune responses came from studies of LXR-deficient mice. Mice lacking LXRs were susceptible to infection by the intracellular pathogen Listeria monocytogenes owing to accelerated macrophage apoptosis and defective bacterial clearance120. Subsequent work outlined a role for LXRs in phagocytosis and the maintenance of immune tolerance. The phagocytosis of apoptotic cells activates LXRs and initiates a positive feedback loop that promotes apoptotic cell clearance through tyrosine-protein kinase MER, which is critical for phagocytosis121. Furthermore, LXR-deficient mice exhibit defective phagocytosis of apoptotic cells in vivo and develop autoantibodies and autoimmune glomerulonephritis121.
LXRs couple cellular cholesterol homeostasis to immune cell function. Activation of LXR inhibits mitogen-driven T cell proliferation by altering cellular sterol content through ABCG1-mediated cholesterol trafficking, whereas loss of LXRβ expression promotes lymphocyte proliferation, resulting in enhanced homeostatic and antigen-driven responses122. Furthermore, cholesterol accumulation in CD11c+ immune cells and dendritic cells triggers the development of autoimmunity123,124. Lxrβ−/− and Apoe−/− double-mutant or Lxrβ−/− mice fed a high-cholesterol diet develop hypercholesterolaemia and autoantibodies. Mechanistically, cholesterol overload in lymphoid organs enhances antigen presentation and T cell priming and stimulates the production of the B cell growth factors BAFF (B cell activating factor; also known as TNFSF13B) and APRIL (a proliferation-inducing ligand; also known as TNFSF13)123. Similarly, loss of ABCA1 and ABCG1 in dendritic cells results in cholesterol enrichment and inflammasome activation, increased cell surface levels of granulocyte–macrophage colony-stimulating factor (GM-CSF) and enhanced inflammatory cytokine production, leading to proliferation of T cells and B cells124.
Alzheimer disease.
The brain is the most cholesterol-rich organ and contains ~25% of the cholesterol in the body125. Cholesterol is synthesized locally in the brain, and its metabolism is largely separated from peripheral cholesterol metabolism because of the blood– brain barrier. In the brain, cholesterol homeostasis is maintained through a balance between biosynthesis and clearance. Dysregulation of cholesterol homeostasis in the brain is associated with several neurodegenerative diseases, including Alzheimer disease, Parkinson disease and Huntington disease126. LXRs regulate cholesterol homeostasis in the brain. For instance, activation of LXRs by synthetic or endogenous ligands, including 24(S)-hydroxycholesterol, 22(R)-hydroxycholesterol, 24(S),25-epoxycholesterol and 27-hydroxycholesterol, induces the expression of ApoE (a primary apolipoprotein in the brain) and of its lipidating transporters ABCA1 or ABCG1 to promote cholesterol efflux127.
Given that the E4 isoform of ApoE, an LXR target, is a well-documented risk factor for late-onset Alzheimer disease, the potential involvement of LXRs in Alzheimer disease pathogenesis has been extensively investigated. Loss of LXRs and its target ABCA1 in a mouse model of Alzheimer disease led to a marked increase in amyloid-β deposition and an Alzheimer-disease-like pathology128,129. Similarly, deficiency in another LXR target, IDOL, increased expression of LDLR in the brain, decreased levels of ApoE and soluble and insoluble amyloid-β, reduced amyloid plaque burden and ameliorated neuroinflammation130. In agreement with these observations, LXR agonists increase clearance of amyloid-β and improve memory in amyloid precursor protein (APP)-transgenic mice131. These studies demonstrate that LXRs and their target genes have important roles in the pathogenesis of Alzheimer disease and that pharmacological activation of LXRs might be a potential therapeutic approach for Alzheimer disease. The effect of LXRs on Alzheimer disease pathology is largely attributed to the induction of ABCA1 expression, which promotes cholesterol clearance and ApoE lipidation. ApoE, especially lipidated ApoE, interacts with amyloid-β and facilitates its proteolytic clearance132. In addition, the repression of pro-inflammatory genes by LXR activation might also contribute to the slowing of Alzheimer disease progression132
Therapeutic targeting LXRs
Given the beneficial effects of LXR activation in animal models of metabolic diseases100,101, much effort has been expended to develop small-molecule agonists of LXRs. The first two widely studied agonists, GW3965 and T0901317, are potent pan-LXR agonists that induce the expression of target genes of both LXRα and LXRβ in vitro and in vivo. Although they are extensively used in research, they proved unsuitable for clinical trial because of undesirable adverse effects, including hepatic steatosis and hypertriglyceridaemia. To minimize the adverse effects of pan-LXR activation, several strategies have been explored. The lipogenic activity of LXRs is mainly mediated by the activation of SREBP1c by LXRα in the liver. The increased plasma levels of triglycerides induced by T0901317 treatment were largely eliminated in liver-specific Lxrα-knockout mice, whereas its anti-atherogenic effects were preserved20, suggesting that LXRβ agonists might have fewer adverse effects than LXRα agonists.
The goal of identifying LXRβ-selective agonists has proved challenging owing to high homology in the ligand binding domains of LXRα and LXRβ133,134. Quinet and colleagues identified a synthetic oxysterol, N, N-dimethyl-3β-hydroxycholenamide (DMHCA), as a gene-selective LXR modulator that activates ABCA1 expression in liver, intestine and macrophages with limited effect on SREBP1c expression and serum levels of triglyceride135. Several partial agonists with considerable selectivity for LXRβ over LXRα were subsequently developed136–138. One such compound, LXR-623, lowered LDL cholesterol levels in primates and reduced the severity of atherosclerosis in mice without activating lipogenesis in the liver139. LXR-623 was the first LXR agonist to enter clinical trials (TABLE 1). Phase I studies suggested that LXR-623 activated the expression of ABCA1 and ABCG1 in healthy participants136. Unfortunately, the trial was terminated owing to unexpected adverse neurological effects136. Several other partial agonists, including CS-8080and BMS-779788, were tested for safety and pharmacodynamics in healthy volunteers. The trial for CS-8080 was terminated owing to undisclosed safety concerns140. The phase I clinical trial for BMS-779788 was completed in July 2009, but the results were not published141. Another LXRβ-selective agonist, BMS-852927, was evaluated in patients with hypercholesterolaemia rather than healthy volunteers. Preclinical studies showed that BMS-852927 had favourable profiles in cynomolgus monkeys and mice with a wide therapeutic index49. Although BMS-852927 induced similar beneficial effects on the RCT pathway in patients with hypercholesterolaemia, it also caused adverse effects, including increased plasma and hepatic levels of triglyceride and increased plasma levels of LDL cholesterol, ApoB, ApoE and CETP as well as decreased circulating levels of neutrophils49. Despite these setbacks, efforts to identify LXRβ-selective agonists with fewer adverse effects are continuing142–144.
Table 1 |.
LXR agonists tested in clinical trials for metabolic diseases
| Compound | Activity | Clinical trial | Outcomes | Refs |
|---|---|---|---|---|
| LXR-623 | Partial LXRα–LXRβ agonist | NCT00366522 (phase I in healthy adults) | Completed, no results reported | 136,147 |
| NCT00385489 (phase I in healthy Japanese adults) | Completed, no results reported | 148 | ||
| NCT00379860 (phase I in healthy adults) | Terminated owing to central nervous system-related adverse effects | 149 | ||
| CS-8080 | Unknown | NCT00613431 (phase I in healthy volunteers) | Completed, no results reported | 150 |
| NCT00796575 (phase I in healthy volunteers) | Terminated owing to undisclosed safety concerns | 140 | ||
| BMS-779788 | Unknown | NCT00836602 (phase I in healthy subjects) | Completed, no results reported | 141 |
| BMS-852927 | Unknown | NCT01651273 (phase I in patients with hypercholesterolaemia) | Terminated owing to increased plasma and hepatic levels of triglyceride, increased plasma levels of LDL cholesterol, ApoB, ApoE and CETP and decreased circulating levels of neutrophils | 49,151 |
ApoB, apolipoprotein B; ApoE, apolipoprotein E; CETP, cholesteryl ester transfer protein; LXR, liver X receptor.
Another approach to theoretically avoid the lipogenic effects of LXR would be to develop tissue-selective agonists. As previously discussed, peripheral tissues and cell types, especially intestinal cells and macrophages, contribute to RCT and systemic cholesterol homeostasis. LXR activation in the intestine reduces cholesterol absorption, stimulates RCT and protects from atherosclerosis in the absence of hepatic steatosis, suggesting that intestine-specific activation of LXRs might be sufficient to achieve beneficial effects55. Owing to its poor absorption, YT-23, an oxidized derivative of the yeast sterol ergosterol, was shown to reduce cholesterol absorption by inducing ABCG5 and ABCG8 expression in enterocytes when orally delivered without causing hepatic steatosis or hypertriglyceridaemia145. Similarly, another study demonstrated that GW6340, an intestinal-specific LXR agonist, induced the expression of LXR target genes and promoted macrophage RCT in mice146. The development of tissue-selective LXR agonists that bypass their hepatic effects might provide a new avenue for LXR therapy in metabolic diseases.
A third strategy for modulating the LXR pathway without activating SREBP1c expression is to target one or more LXR-regulated gene products directly. ABC transporters, including ABCA1, ABCG1, ABCG5 and ABCG8, are major mediators of cholesterol trafficking in different tissues upon LXR activation. Activation of one or multiple ABC transporters might recapitulate some of the beneficial effects of LXR activation on cholesterol homeostasis, such as increasing RCT. Another candidate drug target for modulating the LXR pathway is IDOL. As described in this Review, induction of IDOL by LXR agonists reduces LDLR expression and LDL uptake in primates48. Finally, it might be possible to mimic some of the effects of the LXR pathway activity on membrane phospholipid composition through the development of small molecules that regulate LPCAT3 activity. Considering the important roles that LPCAT3 has in lipid metabolism (particularly in lipid absorption, VLDL secretion and high-fat-diet consumption) inhibitors of LPCAT3 activity might provide a new therapeutic approach for obesity and associated metabolic conditions.
Conclusions
Our understanding of the LXR pathway in lipid homeostasis and metabolic diseases has advanced greatly over the past two decades. LXRs are now recognized to be fundamental to controlling the cellular and systemic homeostasis of major classes of lipids, including sterols, fatty acids and phospholipids. Given the diverse biological pathways affected by LXR-dependent transcription, the potential for harvesting the power of the LXR pathway for therapeutic ends remains great. Further basic and translational research will be needed to realize this potential and to develop novel strategies for manipulating LXRs and their targets in the setting of metabolic disease.
Key points.
Liver X receptors (LXRs) are ligand-activated nuclear receptors that modulate lipid homeostasis.
Cellular and systemic cholesterol homeostasis is maintained by LXRs through the regulation of cholesterol absorption, cellular uptake, excretion, reverse transport and biosynthesis in multiple tissues and cell types.
LXR activation increases lipogenesis through the control of sterol regulatory element-binding protein 1c and its target genes in liver.
LXRs modulate membrane phospholipid composition through inducing the expression of lysophosphatidylcholine acyltransferase 3, an enzyme that has important roles in lipid metabolism in liver and intestine.
The lipogenic activity of hepatic LXRα is a major limitation for the development of LXR agonists as therapeutics for atherosclerosis or Alzheimer disease.
Further basic and translational research is needed to develop novel strategies for manipulating LXRs and their targets in the setting of metabolic disease.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Calkin AC & Tontonoz P Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol 13, 213–224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimano H & Sato R SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol 13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Evans RM & Mangelsdorf DJ Nuclear receptors, RXR, and the big bang. Cell 157, 255–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong C & Tontonoz P Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov 13, 433–444 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Apfel R et al. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol. Cell. Biol 14, 7025–7035 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willy PJ et al. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9, 1033–1045 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Repa JJ et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14, 2819–2830 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seol W, Choi HS & Moore DD Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol. Endocrinol 9, 72–85 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Chen JD & Evans RM A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377, 454–457 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Horlein AJ et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377, 397–404 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Glass CK & Rosenfeld MG The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14, 121–141 (2000). [PubMed] [Google Scholar]
- 12.Tamura K et al. LXRalpha functions as a cAMP-responsive transcriptional regulator of gene expression. Proc. Natl Acad. Sci. USA 97, 8513–8518 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morello F et al. Liver X receptors alpha and beta regulate renin expression in vivo. J. Clin. Invest 115, 1913–1922 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janowski BA, Willy PJ, Devi TR, Falck JR & Mangelsdorf DJ An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383, 728–731 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Fu X et al. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J. Biol. Chem 276, 38378–38387 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Lehmann JM et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem 272, 3137–3140 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Yang C et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J. Biol. Chem 281, 27816–27826 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Peet DJ et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 93, 693–704 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Yu L et al. Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J. Biol. Chem 278, 15565–15570 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y et al. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Invest 122, 1688–1699 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkateswaran A et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl Acad. Sci. USA 97, 12097–12102 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy MA et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 1, 121–131 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Rust S et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet 22, 352–355 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Bodzioch M et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet 22, 347–351 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Qian H et al. Structure of the human lipid exporter ABCA1. Cell 169, 1228–1239 e1210 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Tarling EJ & Edwards PA ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl Acad. Sci. USA 108, 19719–19724 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong C et al. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: identification of ARL7 as a direct target. J. Lipid Res 52, 531–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel T et al. ADP-ribosylation factor (ARF)-like 7 (ARL7) is induced by cholesterol loading and participates in apolipoprotein AI-dependent cholesterol export. FEBS Lett 566, 241–246 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Mak PA et al. Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors alpha and beta. J. Biol. Chem 277, 31900–31908 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Laffitte BA et al. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl Acad. Sci. USA 98, 507–512 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hummasti S et al. Liver X receptors are regulators of adipocyte gene expression but not differentiation: identification of apoD as a direct target. J. Lipid Res 45, 616–625 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Laffitte BA et al. The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol. Cell. Biol 23, 2182–2191 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Y & Tall AR Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element. J. Clin. Invest 105, 513–520 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Repa JJ, Gauthier K & Mangelsdorf DJ Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J. Biol. Chem 276, 43018–43024 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Wong J, Quinn CM & Brown AJ SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem. J 400, 485–491 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sallam T et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature 534, 124–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L et al. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. eLife 6, e28766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell DW et al. Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell 37, 577–585 (1984). [DOI] [PubMed] [Google Scholar]
- 39.Brown MS & Goldstein JL A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47 (1986). [DOI] [PubMed] [Google Scholar]
- 40.Ishibashi S, Goldstein JL, Brown MS, Herz J & Burns DK Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Invest 93, 1885–1893 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishibashi S et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest 92, 883–893 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hua X et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl Acad. Sci. USA 90, 11603–11607 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zelcer N, Hong C, Boyadjian R & Tontonoz P LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong C et al. The E3 ubiquitin ligase IDOL induces the degradation of the low density lipoprotein receptor family members VLDLR and ApoER2. J. Biol. Chem 285, 19720–19726 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scotti E et al. IDOL stimulates clathrin-independent endocytosis and multivesicular body-mediated lysosomal degradation of the low-density lipoprotein receptor. Mol. Cell. Biol 33, 1503–1514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L et al. The IDOL-UBE2D complex mediates sterol-dependent degradation of the LDL receptor. Genes Dev 25, 1262–1274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calkin AC et al. FERM-dependent E3 ligase recognition is a conserved mechanism for targeted degradation of lipoprotein receptors. Proc. Natl Acad. Sci. USA 108, 20107–20112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong C et al. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab 20, 910–918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirchgessner TG et al. Beneficial and adverse effects of an LXR Agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab 24, 223–233 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Weissglas-Volkov D et al. The N342S MYLIP polymorphism is associated with high total cholesterol and increased LDL receptor degradation in humans. J. Clin. Invest 121, 3062–3071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chasman DI et al. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet 5, 257–264 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Altmann SW et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303, 1201–1204 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Brunham LR et al. Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ. Res 99, 672–674 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Brunham LR et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Invest 116, 1052–1062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo Sasso G et al. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab 12, 187–193 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Graf GA et al. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem 278, 48275–48282 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Graf GA et al. Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J. Clin. Invest 110, 659–669 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jakulj L et al. Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab 24, 783–794 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Berge KE et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290, 1771–1775 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Lu K et al. Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am. J. Hum. Genet 69, 278–290 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duval C et al. Niemann-Pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem. Biophys. Res. Commun 340, 1259–1263 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Yu L et al. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J. Clin. Invest 110, 671–680 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tontonoz P & Mangelsdorf DJ Liver X receptor signaling pathways in cardiovascular disease. Mol. Endocrinol 17, 985–993 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Schultz JR et al. Role of LXRs in control of lipogenesis. Genes Dev 14, 2831–2838 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Joseph SB et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem 277, 11019–11025 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Chu K, Miyazaki M, Man WC & Ntambi JM Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol 26, 6786–6798 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cha JY & Repa JJ The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem 282, 743–751 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Chen G, Liang G, Ou J, Goldstein JL & Brown MS Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl Acad. Sci. USA 101, 11245–11250 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tian J, Goldstein JL & Brown MS Insulin induction of SREBP-1c in rodent liver requires LXRalpha-C/EBPbeta complex. Proc. Natl Acad. Sci. USA 113, 8182–8187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshikawa T et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem 277, 1705–1711 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Hannah VC, Ou J, Luong A, Goldstein JL & Brown MS Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem 276, 4365–4372 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Yahagi N et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem 274, 35840–35844 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Ross SE et al. Microarray analyses during adipogenesis: understanding the effects of Wnt signaling on adipogenesis and the roles of liver X receptor alpha in adipocyte metabolism. Mol. Cell. Biol 22, 5989–5999 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stenson BM et al. Activation of liver X receptor regulates substrate oxidation in white adipocytes. Endocrinology 150, 4104–4113 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Korach-Andre M, Archer A, Barros RP, Parini P & Gustafsson JA Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity. Proc. Natl Acad. Sci. USA 108, 403–408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalaany NY et al. LXRs regulate the balance between fat storage and oxidation. Cell Metab 1, 231–244 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Beaven SW et al. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab 18, 106–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spector AA & Yorek MA Membrane lipid composition and cellular function. J. Lipid Res 26, 1015–1035 (1985). [PubMed] [Google Scholar]
- 79.Holzer RG et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lands WE Metabolism of glycerolipids. 2. The enzymatic acylation of lysolecithin. J. Biol. Chem 235, 2233–2237 (1960). [PubMed] [Google Scholar]
- 81.Lands WE & Merkl I Metabolism of glycerolipids. III. Reactivity of various acyl esters of coenzyme A with alpha’-acylglycerophosphorylcholine, and positional specificities in lecithin synthesis. J. Biol. Chem 238, 898–904 (1963). [PubMed] [Google Scholar]
- 82.Rong X et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab 18, 685–697 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Demeure O et al. Regulation of LPCAT3 by LXR. Gene 470, 7–11 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Ishibashi M et al. Liver x receptor regulates arachidonic acid distribution and eicosanoid release in human macrophages: a key role for lysophosphatidylcholine acyltransferase 3. Arterioscler Thromb. Vasc. Biol 33, 1171–1179 (2013). [DOI] [PubMed] [Google Scholar]
- 85.Grefhorst A & Parks EJ Reduced insulin-mediated inhibition of VLDL secretion upon pharmacological activation of the liver X receptor in mice. J. Lipid Res 50, 1374–1383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okazaki H, Goldstein JL, Brown MS & Liang G LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J. Biol. Chem 285, 6801–6810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vance DE Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol 19, 229–234 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Rong X et al. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. eLife 4, e06557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hashidate-Yoshida T et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. eLife 4, e06328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang B et al. Intestinal phospholipid remodeling is required for dietary-lipid uptake and survival on a high-fat diet. Cell Metab 23, 492–504 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Z et al. Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology 149, 1519–1529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rong X et al. ER phospholipid composition modulates lipogenesis during feeding and in obesity. J. Clin. Invest 127, 3640–3651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang B et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220 e204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pommier AJ et al. Liver X Receptor activation downregulates AKT survival signaling in lipid rafts and induces apoptosis of prostate cancer cells. Oncogene 29, 2712–2723 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Koeberle A et al. Arachidonoyl-phosphatidylcholine oscillates during the cell cycle and counteracts proliferation by suppressing Akt membrane binding. Proc. Natl Acad. Sci. USA 110, 2546–2551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lusis AJ Atherosclerosis. Nature 407, 233–241 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tabas I, Garcia-Cardena G & Owens GK Recent insights into the cellular biology of atherosclerosis. J. Cell Biol 209, 13–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moore KJ, Sheedy FJ & Fisher EA Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol 13, 709–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Libby P, Ridker PM & Hansson GK Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 (2011). [DOI] [PubMed] [Google Scholar]
- 100.Tangirala RK et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc. Natl Acad. Sci. USA 99, 11896–11901 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joseph SB et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl Acad. Sci. USA 99, 7604–7609 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calkin AC & Tontonoz P Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb. Vasc. Biol 30, 1513–1518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Verschuren L, de Vries-van der Weij J, Zadelaar S, Kleemann R & Kooistra T LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in apoE*3Leiden mice: time course and mechanisms. J. Lipid Res 50, 301–311 (2009). [DOI] [PubMed] [Google Scholar]
- 104.Levin N et al. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thromb. Vasc. Biol 25, 135–142 (2005). [DOI] [PubMed] [Google Scholar]
- 105.Teupser D et al. Effect of macrophage overexpression of murine liver X receptor-alpha (LXR-alpha) on atherosclerosis in LDL-receptor deficient mice. Arterioscler. Thromb. Vasc. Biol 28, 2009–2015 (2008). [DOI] [PubMed] [Google Scholar]
- 106.Hansson GK Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med 352, 1685–1695 (2005). [DOI] [PubMed] [Google Scholar]
- 107.Tabas I Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol 10, 36–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Castrillo A et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 12, 805–816 (2003). [DOI] [PubMed] [Google Scholar]
- 109.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ & Tontonoz P Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem 278, 10443–10449 (2003). [DOI] [PubMed] [Google Scholar]
- 110.Ghisletti S et al. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23, 681–693 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ghisletti S et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol. Cell 25, 57–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee JH et al. Differential SUMOylation of LXRalpha and LXRbeta mediates transrepression of STAT1 inflammatory signaling in IFN-gamma-stimulated brain astrocytes. Mol. Cell 35, 806–817 (2009). [DOI] [PubMed] [Google Scholar]
- 113.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rajamaki K et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE 5, e11765 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ito A et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 4, e08009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kidani Y & Bensinger SJ Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol. Rev 249, 72–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Spann NJ & Glass CK Sterols and oxysterols in immune cell function. Nat. Immunol 14, 893–900 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ & Tontonoz P Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med 9, 213–219 (2003). [DOI] [PubMed] [Google Scholar]
- 119.Glass CK & Saijo K Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol 10, 365–376 (2010). [DOI] [PubMed] [Google Scholar]
- 120.Joseph SB et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119, 299–309 (2004). [DOI] [PubMed] [Google Scholar]
- 121.N. A. G. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bensinger SJ et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ito A et al. Cholesterol accumulation in CD11c(+) immune cells is a causal and targetable factor in autoimmune disease. Immunity 45, 1311–1326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Westerterp M et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab 25, 1294–1304 e1296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dietschy JM Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol. Chem 390, 287–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Courtney R & Landreth GE LXR regulation of brain cholesterol: from development to disease. Trends Endocrinol. Metab 27, 404–414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang L et al. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc. Natl Acad. Sci. USA 99, 13878–13883 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zelcer N et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl Acad. Sci. USA 104, 10601–10606 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Burns MP et al. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J. Neurochem 98, 792–800 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Choi J et al. The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Abeta amyloidosis. Sci. Transl Med 7, 314ra184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Riddell DR et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell Neurosci 34, 621–628 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Lane-Donovan C & Herz J ApoE, ApoE receptors, and the synapse in Alzheimer’s Disease. Trends Endocrinol. Metab 28, 273–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Svensson S et al. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J 22, 4625–4633 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Janowski BA et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl Acad. Sci. USA 96, 266–271 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Quinet EM et al. Gene-selective modulation by a synthetic oxysterol ligand of the liver X receptor. J. Lipid Res 45, 1929–1942 (2004). [DOI] [PubMed] [Google Scholar]
- 136.Katz A et al. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol 49, 643–649 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Ratni H et al. Discovery of tetrahydro-cyclopenta[b] indole as selective LXRs modulator. Bioorg. Med. Chem. Lett 19, 1654–1657 (2009). [DOI] [PubMed] [Google Scholar]
- 138.Hu B et al. Identification of phenylsulfone-substituted quinoxaline (WYE-672) as a tissue selective liver X-receptor (LXR) agonist. J. Med. Chem 53, 3296–3304 (2010). [DOI] [PubMed] [Google Scholar]
- 139.Quinet EM et al. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J. Lipid Res 50, 2358–2370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00796575 (2015). [DOI] [PubMed]
- 141.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00836602 (2011). [DOI] [PubMed]
- 142.Nomura S, Endo-Umeda K, Makishima M, Hashimoto Y & Ishikawa M Development of tetrachlorophthalimides as Liver X Receptor beta (LXRbeta)-selective agonists. ChemMedChem 11, 2347–2360 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Zheng Y et al. Discovery of a novel, orally efficacious liver X receptor (LXR) beta agonist. J. Med. Chem 59, 3264–3271 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Stachel SJ et al. Identification and in vivo evaluation of liver X receptor beta-selective agonists for the potential treatment of Alzheimer’s disease. J. Med. Chem 59, 3489–3498 (2016). [DOI] [PubMed] [Google Scholar]
- 145.Kaneko E et al. Induction of intestinal ATP-binding cassette transporters by a phytosterol-derived liver X receptor agonist. J. Biol. Chem 278, 36091–36098 (2003). [DOI] [PubMed] [Google Scholar]
- 146.Yasuda T et al. Tissue-specific liver X receptor activation promotes macrophage reverse cholesterol transport in vivo. Arterioscler Thromb. Vasc. Biol 30, 781–786 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00366522 (2009). [DOI] [PubMed]
- 148.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00385489 (2009). [DOI] [PubMed]
- 149.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00379860 (2007). [DOI] [PubMed]
- 150.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT00613431 (2008). [DOI] [PubMed]
- 151.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01651273 (2013). [DOI] [PubMed]