

Abstract
The inflammatory chemokine CCL5, which binds the chemokine receptor CCR5 in a two-step mechanism so as to activate signaling pathways in hematopoetic cells, plays an important role in immune surveillance, inflammation, and development, as well as in several immune system pathologies. The recently published crystal structure of CCR5 bound to a high-affinity variant of CCL5 lacks the N-terminal segment of the receptor that is post-translationally sulfated and is known to be important for high-affinity binding. Here, we report the NMR solution structure of monomeric CCL5 bound to a synthetic doubly-sulfated peptide corresponding to the missing first 27 residues of CCR5. Our structures show that two sulfated tyrosine residues, sY10 and sY14, as well as the unsulfated Y15, form a network of strong interactions with a groove on a surface of CCL5 that is formed from evolutionarily conserved basic and hydrophobic amino acids. We then use our NMR structures, in combination with available crystal data, to create an atomic model of full-length wild-type CCR5:CCL5. Our findings reveal the structural determinants involved in the recognition of CCL5 by the CCR5 N-terminus. These findings, together with existing structural data, provide a complete structural framework with which to understand the specificity of receptor:chemokine interactions.
Keywords: NMR, proteins, protein complexes, sulfated tyrosine, TRNOE, intermolecular interactions, chemokines, chemokine receptors
Introduction
A functional immune system depends on targeted cell migration, which is regulated by the association of chemokines to both chemokine receptors and glycosaminoglycans (GAGs) [1]. Chemokines constitute a family of small secreted proteins (8–10 kDa) that regulate leukocyte trafficking to sites of inflammation and cell repair, thereby playing important roles in angiogenesis and metastasis, which are hallmarks of cancer and leukocyte development [2]. Negatively charged sulfated GAGs, such as heparin sulfate, chondroitin sulfate and dermatan sulfate, are abundant on cell surfaces and extracellular matrices of several tissues and serve as nucleation sites for chemokine oligomerization and the creation of chemoattractant gradients [3,4]. Chemokine receptors, found on the surface of circulating leukocytes, bind extracellular chemokines to trigger intracellular G-protein and β-arrestin signaling pathways. These pathways, in turn, prompt cytoskeletal rearrangements leading to leukocyte motility that follows the chemokine gradient [1]. Besides their role in the immune system, chemokines and chemokine receptors are also exploited by several pathogens, including HIV-1, which uses the chemokine receptor CCR5 as the major co-receptor during fusion with host cells. Clearly, understanding the molecular basis of receptor:chemokine interactions has practical implications in the development of therapies for multiple auto-immune diseases, cancer, and a number of parasite and viral infections.
The approximately 45 chemokines and 22 chemokine receptors expressed in human cells interact promiscuously, with the same chemokine often binding different receptors and vice-versa [5]. Given the highly conserved tertiary structures of both chemokines and chemokine receptors, the determinants of specificity must be subtly encoded in the form of small sequence and structural variations. Chemokines are characterized by an unstructured N-terminal region up to a first cysteine residue (at position 10 in CCL5), after which follows an approximately 10-residue flexible loop (N-loop), a short 310-helix, three strands of an antiparallel β-sheet and a single α-helix at the C-terminus [5]. The first cysteine in the N-terminal region forms a disulfide bond with a cysteine residue in the 30’s loop connecting the two first β-strands, while the second N-terminal cysteine forms a disulfide bridge with a cysteine residue on the third β-strand. These disulfide bridges characterize the chemokine family, while the number and spacing of N-terminal cysteines further sub-divide it into multiple subclasses such as: C, CC, CXC, and CX3C. Chemokine receptors belong to the class A of G-protein coupled receptors (GPCRs), sharing a conserved architecture comprised of an extracellular N-terminal segment and a membrane-embedded core of seven transmembrane helices interspersed with extra- and intracellular loops. The N-terminal segments of chemokine receptors contain several sites available for post-translational modifications, including for tyrosine sulfation [6]. Such sulfation, which is heterogenous [6], was shown to be necessary for high-affinity binding of both chemokines and viral proteins, such as HIV-1 envelope glycoprotein gp120, and to confer specificity for different ligands [7,8].
The interaction between chemokine receptors and chemokines is thought to occur in a two-step process [9]. In the first step of the interaction, the N-terminal segment of the receptor binds to the chemokine core, defining the so-called chemokine recognition site 1 (CRS1). NMR chemical shift mapping showed the receptor N-terminus binds to a shallow groove defined mainly by residues of the N-loop and third β-strand [6]. Despite considerable sequence variation, the chemokine binding surface always contains positively charged residues, likely essential in recognizing the negatively charged receptor sulfated tyrosine residues. Receptor peptides that lacked the post-translational modification were also shown to bind to this same region, albeit considerably more weakly; this highlights the role of other interactions in determining affinity and specificity to this binding region [6]. Moreover, binding of the N-terminal receptor peptide to chemokines was shown to induce spectral changes in regions of the chemokine structure that had been previously mapped as oligomerization sites [6]. Nevertheless, despite all this information and knowledge of the binding region of receptor N-termini on chemokines, there are only four structures of these complexes, all determined by NMR: Nt-CCR3 bound to monomeric CCL11 [10], Nt-CXCR1 bound to dimeric CXCL8 [11], and Nt-CXCR4 bound to dimeric (LD) [12] and monomeric (LM) [13] CXCL12. Of the four, only CCR3:CCL11 and CXCR4:LD CXCL12 were solved containing sulfated tyrosine residues.
The second step in the binding of chemokines to chemokine receptors involves the penetration of the flexible N-terminal segment of the chemokine into a deep pocket formed by the helical bundle of the receptor. These two regions, together with the second extracellular loop of the receptor constitute the second chemokine recognition site (CRS2). Unlike CRS1, whose structures are available only from NMR experiments, atomic details of CRS2 are well-known from the high-resolution crystal structures of three receptor:chemokine complexes – CXCR4:vMIP-II [14], US28:CX3CL1 [15], and CCL5:5P7-CCL5 [16] – all solved in the last two years. This means there are no high-resolution structures of full-length receptor:chemokine complexes, containing both CRSs, which could help understand chemokine recognition and binding, and how these events translate to intracellular signaling by the receptor.
As mentioned previously, CCR5 is a chemokine receptor expressed in leukocytes and is the major co-receptor for HIV. CCL5 is a CC chemokine ligand of CCR5 involved in leukocyte trafficking and a natural HIV suppressor; it is also known as RANTES (regulated on activation, normal T cell expressed and secreted). The CCR5:CCL5 system, therefore, constitutes a promising therapeutic avenue for drug development making it an active area of research for both experimental and computational structural biology [17]. The structures of CCR5 bound to the inhibitor maraviroc and to an engineered high-affinity variant of CCL5 (5P7-CCL5) are known from crystallographic studies, [18]. The structure of CCL5, in dimeric and oligomeric forms, is known from several crystallography and NMR studies [19,20]. Chemical shift perturbations measured upon titration of CCL5 with a doubly-sulfated Nt-CCR5(1–25) peptide mapped the binding interface to a contiguous surface consisting of residues C10-H23 of the N-loop, residues R44, K45, R47 and Q48 in the β2-β3 hairpin, and the last β-strand [21]. A more recent study confirmed the importance of these same CCL5 residues in binding a doubly-sulfated Nt-CCR5(1–27) peptide [22]. Importantly, although both these studies used constructs of the chemokine that prevented the formation of higher-order oligomers of CCL5, there was still a detectable monomer/dimer equilibrium that complicated data collection and interpretation. Together with weak binding and the low solubility of the complex [21–23], this likely explains why there is still no high resolution NMR structure for the Nt-CCR5:CCL5 complex.
Recently, we applied transferred-NOE to detect intermolecular interactions between an N-terminal CCR5 peptide and CCL5 and identified a CCL5 double mutant (CCL5(E66S, P9S)) that is mostly monomeric in solution [23]. In the present study, we use this construct to assign 33 pairwise intermolecular NOE interactions to the corresponding protons of doubly sulfated Nt-CCR5(1–27) and monomeric CCL5. Dihedral angles restraints on each of the components of the complex were obtained after nearly complete sequential and backbone assignment. Together with the intermolecular NOEs, these restraints allowed us to calculate the structure of the complex. Finally, we built a full-length model of the wild-type CCR5:CCL5 complex by combining our NOE data with the known crystal structure of CCR5:5P7-CCL5. We believe our structures and full-length model provide a structural framework for future drug development efforts and for understanding the determinants of specificity of receptor:chemokine interactions.
Results
Assignment and secondary structure of CCL5(P9S) and Nt-CCR5(1–27)
As supported by previous experimental data [19] and the recent crystal structure of CCR5 bound to a high-affinity CCL5 variant [16], CCR5 is thought to interact with monomeric CCL5. At micromolar concentrations in solution, wild-type CCL5 self-associates to form high-molecular weight aggregates [23,24], which is problematic for high-resolution NMR structural characterization of the receptor:chemokine interaction. While the E66S mutation of CCL5 prevents higher-order oligomerization, it still does not prevent dimerization of the chemokine at the high concentrations required for NMR. To eliminate dimer formation and enable sequential backbone and side-chain assignment without complications from the simultaneous presence of monomeric and dimeric chemokine, we used a previously identified double mutant of CCL5 (E66S, P9S), hereafter referred to as CCL5(P9S), which was shown to be mostly monomeric under NMR conditions [23].
Using 15N and 13C uniformly-labeled (U-15N and U-13C) CCL5(P9S) in the presence of 5-fold molar concentration of unlabeled doubly-sulfated Nt-CCR5(1–27), hereafter referred to as Nt-CCR5(1–27), we performed 3D NMR experiments to obtain complete backbone and side-chain assignments for CCL5(P9S), excluding the N-terminal amine. Under the measurement conditions ~90% of CCL5(P9S) is in the bound state [23] enabling us to use the chemical shift data to assess the secondary structure of CCL5(P9S) in the bound state. We used the chemical shifts of HN, Hα, Cα, Cβ, CO to predict a total of 96 backbone (φ,ψ) and 30 side-chain (χ1) torsion angle values with the program TALOS-N [25]. These predictions indicate the presence of four antiparallel β-strands at C10-I15, K25-Y29, V39-T43, and Q48-A51, as well as an α-helix between residues K56 and L65 (data not shown). Except for the first strand (C10-I15), all these secondary structure assignments are in agreement with previously reported crystal and NMR structures of other CCL5 variants [16,19,24].
NOESY and TOCSY spectra were recorded with 13C- and 15N-filters in both F1 and F2 dimensions, to eliminate contributions of CCL5(P9S) and to assign the resonances of the Nt-CCR5(1–27) peptide, under conditions detailed above. Nearly complete backbone and side-chain assignments were obtained for the receptor N-terminal peptide. Due to fast exchange, the peptide resonances represent an average between the free and bound forms, but are dominated by the former because of the large peptide excess. Therefore, Hα and HN secondary shifts were not used to assess the conformation of Nt-CCR5(1–27). The intramolecular NOEs are dominated by the TRNOE and interactions occurring in the bound state of the peptide since the NOE of the free peptide is rather small at the temperature the spectra were measured (37 °C). Seven NN(i,i+1) NOEs, one dαN(i,i+2), 3 dαN(i,i+3) NOE and one N,N(i,i+2) medium-range NOE observed for residues I9 to S17 in the double-filtered NOESY, provide evidence for the helical propensity of this segment of the peptide. These observations agree with previous studies of the free Nt-CCR5(1–27) peptide, which revealed a tendency to adopt helical conformations at lower temperatures (274 K) and when bound to HIV gp120 [26].
Structure determination of the Nt-CCR5(1–27):CCL5(P9S) complex
Based on the backbone and side-chain assignments of CCL5(P9S) in the presence of excess doubly-sulfated Nt-CCR5(1–27), we obtained a total of 1385 intramolecular NOEs from 15N-separated-NOESY and 13C-separated-NOESY spectra using CYANA [27]. Of these, 199 belonged to Nt-CCR5(1–27), while the remaining 1186 belonged to CCL5(P9S). Details of the NOEs are provided in Tables 1 and 2. A total of 33 intermolecular NOEs (Table 2) were observed using a recently developed approach that combines the measurements of a 3D isotope-edited/isotope-filtered NOESY spectrum with the TRNOE effect [23]. Of these 33 NOEs, 32 involved the aromatic protons of residues NTsY10, NTsY14, and NTY15 of the receptor N-terminus, defining this region as the core interface of the complex. No interactions were found with NTY3.
Table 1.
Structural statistics of the ensemble of Nt-CCR5(1–27)/CCL5 structures
| Number of experimental restraints | |
|---|---|
| Intra-residue unambiguous NOEs | 314 |
| Nt-CCR5 | 63 |
| CCL5 | 251 |
| Sequential unambiguous NOEs | 257 |
| Nt-CCR5 | 40 |
| CCL5 | 217 |
| Medium-range unambiguous NOEs | 103 |
| Nt-CCR5 | 8 |
| CCL5 | 95 |
| Long-range unambiguous NOEs | 136 |
| Nt-CCR5 | 1 |
| CCL5 | 135 |
| Total unambiguous NOEs | 810 |
| Total ambiguous NOEs | 575 |
| Intermolecular NOEs | 33 |
| Dihedral angles | 126 |
| Non-bonded energy valuea after explicit water refinement (kcal.mol−1) | |
| E van der Waals | −36.1 ± 5.2 |
| E electrostatics | −312.3 ± 42.7 |
| RMSD (Å) from the meanb | |
| Nt-CCR5 backbone atoms | 0.42 ± 0.11 |
| Nt-CCR5 heavy atoms | 0.67 ± 0.11 |
| CCL5 backbone atoms | 0.30 ± 0.02 |
| CCL5 heavy atoms | 0.50 ± 0.03 |
| RMSD from experimental data | |
| Distance (Å) | 0.0386 ± 0.004 |
| Dihedral (º) | 0.681 ± 0.05 |
| Restraint violations in more than 50% of the structuresc | |
| Distance (>0.3Å) | 2 |
| Intramolecular | 2 |
| Intermolecular | 0 |
| Dihedral (>5º) | 0 |
| Ramachandran Analysis | |
| Residues in most favored regions (%) | 77.2 ± 2.2 |
| Residues in additional allowed regions (%) | 19.7 ± 1.8 |
| Residues in generously allowed regions (%) | 1.2 ± 0.8 |
| Residues in disallowed regions (%) | 1.9 ± 1.1 |
The non-bonded energies were calculated with OPLS parameters using a cutoff of 8.5Å.
The RMSD calculations were performed on residues: Nt-CCR5(9–17) and CCL5(9–66).
No NOE distance restraint was violated by more than 0.34Å/1.17Å (intramolecular/intermolecular).
Table 2.
List of intermolecular NOEs used in NMR structure calculations
| Nt-CCR5(1–27) | CCL5(P9S) |
|---|---|
| δsY10 | γ2I15, δL19, γT43, εK45, γR47, γ2V49 |
| εsY10 | γ2I15, γL19, δL19, γT43, εK45, γR47, γ2V49 |
| δsY14 | γ1I15, βA16, γL19, δL19, γ2V49 |
| εsY14 | γ1I15, γ2I15, βA16, γL19, δL19, γ2V49 |
| δY15 | γ1I15, γ2I15, βA16, δL19, γ2V49 |
| εY15 | γ1I15, γL19, δL19 |
| βS17 | γ2I15 |
Initially, we unambiguously assigned 19 intermolecular NOEs between methyl protons of CCL5(P9S) and the previously mentioned tyrosine residues of the receptor peptide (see Table 2 in ref. [23]), and used these as distance restraints for structure determination using XPLOR-NIH [28]. The initial structure calculations used these and all intramolecular distance restraints, together with 126 dihedral angle restraints obtained from TALOS-N predictions and two manually inserted distance constraints for the disulfide bonds of CCL5 (C11-C34 and C12-C50) (Table 1). No dihedral angle constraints were used for Nt-CCR5(1–27). After analysis of the ten lowest-energy structures of an initial batch of 50 structures, we added 16 hydrogen bonds that appeared in more than 50% of these structures as additional restraints in subsequent calculations. The remaining 14 NOEs were assigned in an iterative manner during these calculations. The last set of 106 structures was refined in explicit solvent [29], following the protocol implemented in HADDOCK2.2 [30], and using all intra- and intermolecular NOEs, hydrogen bond, and dihedral angle restraints. The refined structures satisfy the experimental restraints, with no distance violations larger than 0.5Å. Structurally, the ensemble is well-defined, with an average backbone RMSD from the mean of 0.42 ± 0.11Å for Nt-CCR5(1–27) (calculated on residues 9 to 17) and 0.30 ± 0.02Å for CCL5(P9S) (calculated on residues 9 to 66). In addition, more than 98% of the residues in these two ranges of residues fall within the allowed or generously allowed regions of the Ramachandran plot (Table 1).
Solution structure of the Nt-CCR5(1–27):CCL5(P9S) complex
The ensemble of the ten lowest-energy structures after water refinement is shown in Fig. 1. The structure of CCL5(P9S) is mostly well-defined, except for the N (residues 1 to 8) and C (residues 67 and 68) termini, and shows all the characteristics of the canonical chemokine fold (Fig. 1a). Overall, the ensemble of structures agrees with the observed intramolecular NOEs and with the secondary structure predictions from TALOS-N. The only exception is the absence of a β-strand between residues CCL5C10 and CCL5I15, predicted by TALOS-N, which in any event is not characteristic of the CC chemokine fold. Like other CC chemokine structures determined in the presence of a receptor N-terminal peptide, there is little structural deviation between CCL5 bound to the CCR5 N-terminal segment and free CCL5. The average backbone RMSD of our ensemble to the high-resolution crystal structure of dimeric CCL5 (PDB ID: 1EQT) [31] is 1.43 ± 0.06Å for residues 6 to 66 and 0.97 ± 0.07Å for the 32 residues in structured regions. As expected, the largest deviations lie in the N-loop and loops connecting β strands that are part of either the CCL5:CCL5 dimer or Nt-CCR5(1–27):CCL5 interfaces.
Fig. 1.
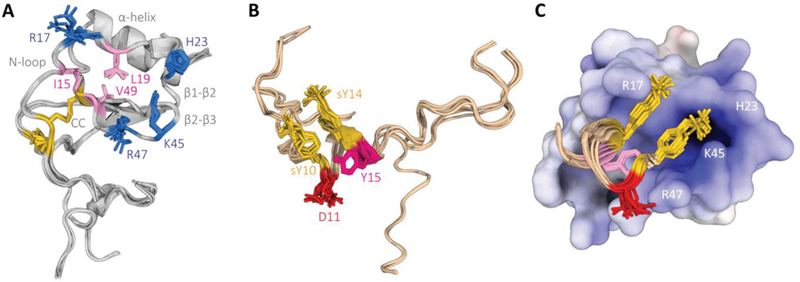
NMR structure of the Nt-CCR5(1–27):CCL5(P9S) complex. (A) Ensemble of the ten lowest-energy structures calculated for CCL5, with the characteristic features of the chemokine fold highlighted in gray, the CC disulfide bonds highlighted in gold, and important residues for interaction with Nt-CCR5(1–27) in blue (positively charged) and pink (hydrophobic). (B) Ensemble of the ten lowest-energy structures calculated for Nt-CCR5(1–27), with sulfated tyrosine residues highlighted in gold, and residues important for binding CCL5(P9S) in red (negatively charged) and pink (hydrophobic). (C) Orientation and interface of Nt-CCR5(9–17) on CCL5(P9S)(9–67), shown from the same perspective as (A).
Regarding Nt-CCR5(1–27), our measurements yielded mainly intra-residue and sequential NOEs, resulting in an ensemble of structures with significant flexibility (Fig. 1b). We could, however, assign some NOEs characteristic of a helical conformation in a central region defined by residues NTI9-NTT16, in agreement with previous NMR studies [26]. In our ensemble, residues NTN13 to NTY15 are classified by DSSP [32] as a 310 helix in 90% of the structures and as an α-helix in the remaining 10%. The neighboring residues, NTsY10, NTD11, NTI12, and NTT16, although not strictly assigned as helical by DSSP, form hydrogen-bonded turns that contribute to a longer helix-like structure, which is further stabilized by intermolecular contacts with CCL5(P9S). Indeed, residues NTP8 to NTS17 of Nt-CCR5(1–27) were shown to have a weak helical tendency at low temperatures (274K) that folded into a stable helix when bound to HIV gp120 [26]. The remaining residues of the peptide, NTM1-NTI9 and NTS17-NTQ27, are mostly disordered, consistent with the lack of medium- and long-range intramolecular NOEs, as well as of intermolecular NOEs.
As for the binding interface between the two molecules, our 3D edited/filtered experiment revealed two regions in CCL5(P9S) involved in binding Nt-CCR5(1–27): a first region comprised of residues CCL5I15-CCL5I25, containing the N-loop, and a second region comprised of residues CCL5T43 to CCL5V49, containing the BBXB motif. This is in line with previous NMR studies that measured chemical shift perturbations on CCL5 upon titration of Nt-CCR5 peptides [21,22]. Two regions form a continuous cleft lined with polar and basic residues, several of which participate in direct electrostatic interactions with the receptor N-terminus, mostly NTD11, NTsY10, and NTsY14 (Fig. 1c). The two sulfate groups point to opposite ends of the cleft, with NTsY10 interacting mainly with the imidazole of CCL5H23, which is positively charged in our experiments, and the ε-ammonium of CCL5K45; whereas NTsY14 forms a salt bridge with the guanidinium group of CCL5R17 in nearly all members of the ensemble, in agreement with previous NMR studies [21]. Notably, the (φ,ψ) angles of NTsY10 are well into the disallowed region of the Ramachandran plot for all members of the ensemble, a distortion likely due to the charged interactions anchoring it to CCL5(P9S). The negatively charged side-chain of NTD11 makes a salt bridge with CCL5R47, further anchoring the receptor peptide to the chemokine. This is supported by experiments showing that mutating NTD11 causes a 80–100% decrease in CCL5 binding [33,34]. Besides these charged interactions, the aromatic moieties of both modified tyrosine residues and of NTY15 make extensive hydrophobic contacts with apolar side-chains on the inside of the chemokine interface cleft, namely CCL5I15, CCL5L19, and CCL5V49, mutation of which have been shown to reduce binding affinity of CCL5 [21,35].
Data-driven model of the wild-type full-length CCR5:CCL5 structure
In light of the recently published high-resolution structure of CCR5 in complex with an engineered high-affinity CCL5 variant (5P7-CCL5) [16], we built a full-length model of the wild-type CCR5:CCL5 complex that includes both sites (CSR1 and CSR2) modeled from very reliable experimental data: intermolecular NOEs for CSR1 and crystal coordinates for CSR2 (see Material and Methods).
We used homology modeling and data-driven flexible refinement to generate 500 final full-length models (for details see Material and Methods for details); 495 of these share more than 60% of the pairwise residue contacts at the interface. These 495 models are grouped into a single cluster using a clustering metric that compares the models disregarding regions of the proteins that are not part of the interface (such as the flexible chain termini). The ten lowest-energy models are remarkably similar, with an average backbone RMSD to the mean, calculated for the same regions as the NMR structures, of 0.35 ± 0.14Å for CCR5 and 0.25 ± 0.07Å for CCL5 (Table 3 and Fig. 2a). The interface backbone RMSD to the mean, calculated on all atoms belonging to any residue within 10Å of the binding partner, is 0.58 ± 0.24Å, reflecting a strong structural conservation of both CRS1 and CRS2 and highlighting the convergence of the calculations and the robustness of the NOE restraints (Table 3). Overall, the interface and conformation of the N-terminal of CCR5 are in accord with the orientation of CCL5 derived from the structure of 5P7-CCL5 in complex with CCR5, in which the first fifteen CCR5 residues were not observed [16]. Much like this crystal structure, the conserved motif NTP19-NTC20 of the receptor packs against the conserved disulfide bridge of the chemokine (CCL5C11-CCL5C50). This region, named CSR1.5 [16], is further stabilized by a hydrogen bond between NTQ21 of CCR5 and CCL5Q48 of CCL5 in some of our models.
Table 3.
Statistics and energy terms of the wild-type full-length CCR5:CCL5 models
| HADDOCK Score (a.u.) | −276 ± 3 |
| Cluster Size | 495 / 500 |
| Backbone interface RMSD to average structure (Å)a | 0.58 ± 0.24 |
| van der Waals (kCal.mol-1) | −164 ± 12 |
| Electrostatics (kCal.mol-1) | −744 ± 45 |
| Restraints Violation (kCal.mol-1) | 200 ± 10 |
| Desolvation Energy (a.u.) | 16.4 ± 5.5 |
| Buried Surface Area (Å2) | 4293 ± 57 |
The RMSD calculations were performed on residues with at least one heavy atom within 10Å of the other chain. This set comprises residues 4–5, 7–29, 33–34, 36–37, 40, 78–94, 100–109, 112–113, 162–164, 166–196, 198, 247–248, 251, 254–209 for CCR5 and 1–13, 15, 17, 20–21, 23–63, 65–67 for CCL5.
Fig. 2.

Full-length wild-type model of CCR5:CCL5 based on our NMR data and the crystal structure of CCR5:5P7-CCL5[PDB Ref]. (A) Ensemble of the ten lowest-energy models calculated with HADDOCK, with CCR5 colored gold and CCL5 colored grey. (B) and (C) Chord plots of pairwise residue electrostatic and van der Waals energies, respectively, calculated for the lowest-energy model. The thickness of each chord represents the strength of the pairwise interaction relative to the total interaction energy, while the color gradient indicates the repulsive (red)/attractive (blue) character of the interaction (red being repulsive; blue attractive). For simplicity, only the interactions contributing 90% of the total energy are shown.
The interactions at the CRS1 interface resemble those observed for the NMR structures, which is expected as the same set of restraints was used to calculate both ensembles of structures. Of note is that the sulfated tyrosine residues contribute most of the binding surface by forming several electrostatic contacts with positively charged residues of CCL5 (Fig. 2b), while their aromatic moieties contact several hydrophobic side-chains inside the binding pocket (Fig. 2c). As found for CRS2, the interactions resemble those of the wild-type CCR5:CCL5 model built by Zheng and co-workers [16], with a few significant differences. In our models, the charged terminal amine of CCL5S1 interacts either with the hydroxyl group of Y2516.51 or the carbonyl of E2837.39, both previously shown to be important to receptor:chemokine binding [34]. In addition, CCL5 CCL5Y3 is involved in either π-stacking or hydrophobic interactions with a cluster of aromatic residues on several receptor transmembrane helices (Fig. 2c), most notably Y371.39, W862.60, and Y1083.32, mutants of which have been shown to diminish or abrogate binding of the chemokine [34,36]. Finally, it is important to highlight that the sequence of 5P7-CCL5 differs substantially from wild-type CCL5 (only one of the first nine residues – CCL5P2 – is conserved). While this might impact the modelling of CRS2, we are confident our model is relevant to the wild-type interactions given its agreement with mutagenesis data.
Discussion
Observation of intermolecular TRNOE enables determination of the structure of the Nt-CCR5(1–27):CCL5(P9S) complex
In this study, we calculated the structure of monomeric CCL5(P9S) bound to a doubly-sulfated peptide corresponding to the first 27 residues of the chemokine receptor CCR5 based on solution NMR data. This is the second structure of a CC chemokine bound to a sulfated peptide of a chemokine receptor, and is of particular interest due to the role of CCR5 in HIV-1 infection [33].
Two critical factors allowed us to observe intermolecular NOEs between Nt-CCR5(1–27) and CCR5(P9S). First, the P9S mutation, in addition to the E66S mutation, produced a CCL5 construct that is mostly monomeric under NMR conditions. Unlike previous constructs, which existed in a monomer/dimer equilibrium, CCL5(P9S) enabled us to assign our NMR spectra unambiguously. Second, this assignment was possible due to the combination of TRNOE with f1-edited-f2-filtered NOE measurements of CCL5(P9S) in the presence of a large excess of Nt-CCR5(1–27), together with iterative structure calculations. We could assign 33 intermolecular NOEs for the weakly bound monomeric Nt-CCR5(1–27):CCL5(P9S) complex [23], despite using high protein concentrations comparable to the KD of the complex of 70 μM.
Comparing our structures with earlier chemical shift perturbations on CCL5(P9S) upon titration with Nt-CCR5(1–27) [22] shows overall agreement with some differences. Residues CCL5I15, CCL5L19, CCL5K45, and CCL5R47 showed some of the strongest chemical shift perturbations, in agreement with our structures where they form strong interactions with residues in Nt-CCR5(1–27). Other residues with significant chemical shift perturbations are located on the N- and β2-β3 loops. Although these residues do not participate in direct interactions with the receptor in our structures, they are part of the scaffold of the groove where the sulfated tyrosine residues dock and are close in sequence to residues that interact with Nt-CCR5(1–27). On the other hand, residue CCL5V49, for which we measured several NOEs, had not been considered significantly perturbed in the previous study. These observations underline the importance of measuring intermolecular NOEs when calculating the structure of protein complexes. It is our contention that, for the majority of proteins where chemical shift perturbations are measurable, intermolecular NOEs can be detected using the transferred NOE approach [23,37]. Such pairwise interactions clarify the underlying basis for the chemical shift perturbation and whether it is due to a direct or indirect effect.
Comparison with the NMR structure of Nt-CCR3(8–23):CCL11
To date, there is only one other known structure of a CC chemokine bound to the sulfated N-terminal segment of one of its cognate receptors: Nt-CCR3(8–23):CCL11 [10]. Given that both CCR3 and CCR5 can bind CCL5 and CCL11, it is particularly interesting to compare the structure of both these complexes. It is important to highlight, however, that our structure of Nt-CCR5(1–27):CCL5 was refined in explicit solvent and using electrostatics and both attractive and repulsive van der Waals energy terms. Traditional NMR structure calculation protocols run in vacuum and often use only a repulsive van der Waals term. These differences might have an impact on the structure, particularly in the orientation and interaction of charged residues.
At first glance, there are two obvious differences between Nt-CCR5(1–27):CCL5 and Nt-CCR3(8–23):CCL11 (Figure 3A and3B): (1) the conformation of the receptor N-terminal peptide and (2) its orientation relative to the chemokine ligand. Our Nt-CCR5(1–27) peptide shows a helical turn between residues NTN13 and NTY15 and is oriented along the binding pocket of CCL5 C- to N-terminal, so that its C-terminal residues interact with the N-loop of the chemokine and its N-terminal residues bind the β2-β3 loop. On the other hand, Nt-CCR3(8–23) does not have a well-defined secondary structure and lies along the chemokine binding pocket in the opposite orientation, from N- to C- terminal. The differences in conformation between both peptides may be ascribed to the sulfation of different tyrosine residues. In Nt-CCR3(8–23), the sulfation of consecutive residues, CCR3-NTsY16 and CCR3-NTsY17, likely leads to steric and electrostatic repulsion between the side-chains and forces the backbone in this region to adopt a nonstandard turn, while in Nt-CCR5(1–27) the spacing between NTsY10 and NTsY14 allows the formation of a standard helical turn so that both tyrosine residues face the chemokine surface.
Fig. 3.

Sequence and structural conservation of receptor:chemokine CRS1 interactions. Residue-residue contacts at receptor:chemokine interfaces in (A) our Nt-CCR5(1–27):CCL5(P9S) structure and (B) CCR3:CCL11 structure [10], respectively. Amino acids are colored by character: charged in blue/red, hydrophobic in pink, and sulfated tyrosine residues in gold. Interactions in the schematics are colored according to their physicochemical character: electrostatic in blue, hydrophobic in pink. (C) Alignment of human CC chemokine sequences. Columns are colored if the amino acid character is conserved in at least 60% of the sequences (except for glycine and proline). Asterisks indicate key residues involved in chemokine receptor binding, as shown in our and other NMR structures. The gray box highlights CCR5 binders, identified in [38], ordered by binding affinity.
Nevertheless, despite the differences in orientation, conformation, and sulfation pattern, the receptor peptides bind equivalent residues on their respective chemokine partners (Fig. 3a & 3b). Interestingly, only the core hydrophobic interactions between the receptor and the chemokine are conserved pairwise: the aromatic rings of NTsY10 and NTsY14 in Nt-CCR5(1–27) both interact with the side-chains of CCL5L19 and CCL5V49 in CCL5, while the aromatic rings of CCR3-NTsY16 and CCR3-NTsY17 in Nt-CCR3(8–23) both interact with the side chains of CCL11I18 and CCL11I49 in CCL11. Other more peripheral hydrophobic interactions, such that of the aromatic ring of NTsY14 with CCL5I15 in Nt-CCR5(1–27):CCL5, are not conserved in Nt-CCR3(8–32):CCL11. In this latter structure, CCL11A14 is interacting intramolecularly with CCL11I49. On the other hand, the charged sulfate groups on the receptor peptides interact differently with the same basic residues of the chemokines. The sulfate groups of NTsY10 and NTsY14 in Nt-CCR5(1–27) form stable salt-bridges with CCL5K45 and CCL5R17, respectively, while NTD11 interacts with CCL5R47. In Nt-CCR3(8–23):CCL11, CCL11K47 is the main electrostatic contributor to the interaction, forming salt-bridges with both CCR3-NTsY16 and CCR3-NTsY17. The CCL11 equivalents of CCL5R17(CCL11R16) and CCL5K45 (CCL11L45) still interact with the sulfated tyrosine residues of Nt-CCR3(8–23) although differently: CCL11R16 makes transient cation-π or electrostatic interactions with CCR3-NTsY16, while the side-chain methyl groups of CCL11L45 interacts with the aromatic ring of CCR3-NTsY17. Finally, in Nt-CCR5(1–27):CCL5, CCL5H23 also interacts with the sulfate group of NTsY10, albeit weakly (Fig. 2B). In Nt-CCR3(8–23):CCL11, CCL11R22 is either hydrogen-bonded to the backbone carbonyl of CCR3-NTsY17 or in a cation-π interaction with its aromatic ring.
These observations offer a possible explanation for the promiscuity of both chemokine receptors and chemokine ligands. The dispersion of positive charges around the central hydrophobic core allows chemokines to bind differently sulfated forms of the same receptor, or even different receptors. On the other hand, the slight differences in the contacts made by these positively charged residues, as well as by some peripheral hydrophobic amino acids, are probable modulators of both the specificity and affinity of receptor:chemokine interactions.
Interface residues of CCL5(P9S) are structurally and evolutionarily conserved across the CC and CXC chemokine families
In the absence of further structural data, we looked at evolutionary conservation to confirm a potential role of the positively charged and hydrophobic chemokine residues in conferring specificity to receptor:chemokine interactions. To this end, we constructed a multiple sequence alignment of all annotated human CC chemokines deposited in the Uniprot database [38] (Fig. 3c) and assessed the conservation of these residues across other chemokines.
The hydrophobic residues anchoring the two sulfated tyrosines in both Nt-CCR5(1–27):CCL5 and Nt-CCR3(8–23):CCL11 are highly conserved across the CC chemokine family. CCL5L19 (CCL11I18) is either strictly conserved or replaced by an isoleucine residue, and CCL5V49 is conserved in 46% of all human sequences or replaced by other hydrophobic amino acids such as isoleucine (35%), phenylalanine (8%), leucine (8%), or alanine (4%). The basic character of residues interacting with the sulfate moieties of Nt-CCR5(1–27) and Nt-CCR3(8–23) is also conserved throughout the family. Residues equivalent to CCL5R17, CCL5H23, CCL5K45, and CCL5R47, are positively charged in 73%, 47%, 73%, and 88% of the sequences, respectively. The plasticity of interactions at the receptor:chemokine interface is further illustrated by the seemingly weak evolutionary conservation of CCL5H23. In Nt-CCR5(1–27):CCR5, CCL5H23 interacts with the sulfate group of NTsY10, while in Nt-CCR3(8–23):CCL11, the equivalent CCL11R22 interacts either with the backbone carbonyl or the aromatic ring of CCR3-NTsY17. As such, in other chemokines that have a hydrophobic or aromatic residue at this position (e.g., CCL3 and CCL4), it is reasonable to assume a strong hydrophobic interaction with the aromatic ring of the sulfate tyrosine.
Of the several chemokines interacting with CCR5 whose affinity was determined experimentally in [38], the weakest interactor is CCL7. Interestingly, CCL7 lacks a positive charge at position 45 (replaced by a leucine residue, like in CCL11), which prevents a proper stabilization of NTsY10. Then, at position 17, CCL7 has a lysine instead of the arginine observed in all other binders, which may contribute to less frequent salt-bridge formation with NTsY14 due to the shorter side-chain. As a result, the weaker affinity of CCL7 to CCR5 can be rationalized in terms of amino acid changes in these critical positions (Fig 3c). Conversely, the strongest interactors, CCL3 and CCL4, both have a phenylalanine in place of CCL5H23 and an arginine in place of CCL5K45, which might both help stabilize the bound receptor peptide.
The generality of the above types of chemokine-receptor contacts is also seen in the CXC family. Despite sequence and structural divergence, the chemokines of the Nt-CXCR1:CXCL8 [11] and both Nt-CXCR4:CXCL12 [12,13] complexes solved previously have an arginine at position 47 interacting either with a tyrosine residue of their respective receptors – LD CXCR4-NTsY21, LM CXCR4-NTY21 and CXCR1-NTY2 – or an immediately adjacent aspartate (equivalent to CCR5D11). Although in these experiments only the peptide used to determine the structure of Nt-CXCR4 bound to dimeric (LD) CXCL12 was sulfated, CXCR1-NTY2 is flanked by negatively charged amino acids, a motif used to predict sulfation sites [39]. Moreover, equivalent residues of CCL5V49 in CXC chemokines, CXCL12V49 and CXCL8L49, form hydrophobic contacts with the aromatic moiety of tyrosine residues that may be sulfated in the receptor. Together with the structures of Nt-CCR5(1–27):CCL5(P9S) and Nt-CCR3(8–23):CCL11, these observations support and help rationalize the general promiscuity between receptor and chemokine ligands, in particular amongst the CC family.
Rationalizing the absence of density for the receptor N-terminal segment in crystallography studies
As mentioned in the introduction, the crystal structures of all receptor:chemokine complexes published to date lack most of the N-terminal segment of the receptor, including the critical sulfated tyrosine residue we find responsible for high-affinity binding of the native chemokine ligands. The authors of the structures of CCR5:5P7-CCL5 [16] and US28:CX3CL1 [15] ascribe the lack of density to the flexibility or disorder of the N-terminal segment, while those of CXCR4:vMIP-II [14] suggest the moderate stability of the CRS1 interaction is the culprit. The underlying causes of this apparent flexibility or moderate affinity are, however, never discussed.
It is known that tyrosine sulfation of chemokine receptors in eukaryotes is heterogeneous [6]. As such, at any given time, the cell expresses differentially sulfated chemokine receptors with a narrow range of molecular mass and charge. There are significant differences in binding affinity for CCR5/CCL5 complexes depending on the sulfation pattern of the receptor N-terminus [8]. This is true even within mono-sulfated variants, so it is reasonable to assume that is a generalized feature or receptor:chemokine interactions. Moreover, our analysis of the conformation of chemokine-bound Nt-CCR5(1–27) and comparison to others highlighted the plasticity of receptor N-terminal peptides, particularly Nt-CCR3(8–23), and its likely dependence on the identity of the sulfated tyrosines.
We suggest, therefore, that a plausible cause for the lack of discernible electron density for the sulfate-containing N-terminal region of chemokine receptors in existing crystal structures is the heterogeneous sulfation process that leads to a diverse population with a range of conformations and affinities at CRS1. In fact, in all three crystal structures, the authors resort to different strategies to stabilize the binding of the chemokine to the receptor. In CCR5:5P7-CCL5, the chemokine construct is a highly-engineered variant of CCL5 with an anti-HIV potency twice that of PSC-RANTES, itself a variant of CCL5 with twice the affinity for CCR5 as that of the native chemokine [40]. In CXCR4:vMIP-II, the authors engineered a direct disulfide bridge between the receptor and the chemokine (CXCR4D187C-v-MIPIIW5C) [14]. Finally, in US28:CX3CL1, the viral receptor (US28) is quite promiscuous and naturally forms high-affinity interactions with several human chemokines, even when the N-terminal is mostly truncated as in this structure (lacks the first 10 residues) [15].
In addition to tyrosine sulfation, crystallization conditions and packing may also play a role in displacing the N-terminal segment from its binding site on a chemokine. As noted by the authors, the crystal packing of CCR5:5P7-CCL5 causes the chemokines of neighboring dimers to interact via their N-loop, which is an important binding region in our NMR structure. This crystal packing dislocates the Nt-CCR5 segment from it binding site on the chemokine that could result an increased flexibility. Coupled to the likely mixed population of sulfated receptors, this increased flexibility could impede structure determination of Nt-CCR5.
In contrast to the endogenous expression of the receptors for crystallography, the receptor N-terminal peptides used in the NMR structure determination here and in other studies are either chemically synthetized (Nt-CCR5, Nt-CCR3) or enzymatically sulfated in vitro and purified to give peptides with selected tyrosine sulfation patterns (Nt-CXCR4). This eliminates heterogeneous sulfation patterns in the receptor peptide surrogates. Moreover, NMR is a particularly well-suited technique for structure determination of flexible or disordered proteins. Together, these strategies enabled us to obtain important insights into the interaction of the N-terminus of CCR5 with CCL5, which complement the recently solved crystal structure.
An improved model of the wild-type CCR5:CCL5 reconciles experimental data on both CRS1 and CRS2
The recently published crystal structure of CCR5 bound to 5P7-CCL5 enabled us to build a model of the full-length wild-type CCR5:CCL5 complex (Fig. 2a). The authors of the CCR5:5P7-CCL5 structure also built a wild-type CCR5:CCL5 model, starting at NTN13 and containing sulfated tyrosine residues placed at positions 14 and 15. In these models, the upstream sulfated tyrosine NTsY14 interacts with CCL5K45 and CCL5R47, while NTsY15 forms a salt-bridge with CCL5R17, again capturing key residues on the chemokine surface and interactions that we also observe in our NMR structures and models. The conformation of the Zheng N-terminal receptor is mostly unstructured and thus different from ours, owing either to the different sulfation pattern or to the lack of upstream stabilizing residues such as NTD11.
Our NMR restraints derived from the Nt-CCR5(1–27):CCL5(P9S) interaction allowed us to model with high accuracy the receptor N-terminus up to residue NTI9 and more importantly, the interaction of the sulfated tyrosine residues with the chemokine. As such, it provides a structural framework with which to understand a large body of experimental data both on the heterogeneity of tyrosine sulfation [8] and on the role of accessory residues on the receptor N-terminal, such as NTD11 [33,34]. More importantly, it provides a high-confidence model to understand the allosteric link between CRS1 and CRS2, which has been documented before but is poorly understood due to the lack of structural data [5].
Conclusions
In this study, we report the structure of monomeric CC chemokine 5 - CCL5(P9S) - bound to a doubly-sulfated chemokine receptor fragment – Nt-CCR5(1–27) – as well as a model of the wild-type CCR5:CCL5 complex. Our data and models support the proposed two-site model, rationalize previous literature on the importance of tyrosine sulfation for CCL5 binding, and highlight the existence of a network of contacts between conserved residues that confer both high-affinity and specific binding of receptors to chemokine ligands. Moreover, our CCR5:CCL5 model is the first to reconcile high-resolution structural data for both CRS1 and CRS2, presenting a unique framework to understand the interactions of chemokine receptors with chemokine ligands.
Material and Methods
Nt-CCR5(1–27) peptide and CCL5(P9S)
The Nt-CCR5(1–27) peptide sulfated at Y10 and Y14; MDYQVSSPIY(SO3)DINY(SO3)YTSEPAQKINVKQ was synthesized by solid-phase peptide-synthesis as described previously [37]. The sulfated peptide was > 95% homogeneous on HPLC and had the expected molecular mass, as judged by ESI-MS. The yield of the peptide was 20–30%. Both HPLC and NMR analysis showed that the sulfated tyrosine moieties were stable over time under the conditions of the NMR experiments. CCL5(P9S) was expressed, labeled and purified as previously described [23].
NMR Samples conditions
CCL5(P9S)/Nt-CCR5(1–27) solutions contained 120 μM CCL5(P9S) with 5-fold molar concentration of Nt-CCR5(1–27) in 130 mM d-acetate buffer, pH 4.8. CCL5(P9S) was either U-15N, U-15N;13C or U-13C labeled.
NMR Assignment
All NMR experiments were measured on a Bruker AVIII800 spectrometer equipped with a 5 mm TCI cryoprobe. For sequential backbone resonance assignments of bound CCL5(P9S) a standard 3D triple resonance assignment approach was utilized. HNCACB, CBCA(CO)NH, HNCA and HNCO experiments were acquired on the complex of 15N-13C-CCL5(P9S) with 5-fold excess of Nt-CCR5(1–27) at pH 4.8, 37°C. Side-chain chemical shift assignments were obtained using HCCH-TOCSY, CCH-TOCSY and HCC(CO)NH-TOCSY experiments on the same complex, but with CCL5(P9S) labeled only with 13C for the HCCH-TOCSY and CCH-TOCSY experiments. The chemical shift deviations from random coil values were calculated using the random coil values of the Wishart protein database [41].
Structure calculation of Nt-CCR5:CCL5 complex
The solution structures of the Nt-CCR5(1–27):CCL5(P9S) complex were calculated using XPLOR-NIH [28]. Torsion angle restraints for CCL5(P9S) were obtained from the HN, Hα, Cα, Cβ, CO chemical shifts using TALOS-N [25]. Automatic NOE assignment was performed with CYANA [27]. The resulting NOE restraints were ported to XPLOR-NIH to calculate an initial set of 50 structures that were used to identify errors in the restraints and assignments. Iterative rounds of refinement yielded a total of 126 torsion angle restraints and 1385 inter-proton distances, 314 of which were intra-residue, 257 sequential, 103 medium-range, and 136 long-range NOEs. NOEs recorded for equivalent protons in methyl groups were used as ambiguous distance restraints in the calculations. The resulting 106 structures were then refined in explicit solvent [29] using the protocol implemented in HADDOCK2.2 [30]. The geometry of the 10 lowest-energy structures was assessed and validated using NMR-PROCHECK [42] (Table 1).
Modelling the full-length wild-type CCR5:CCL5 complex
We built initial full-length models of the wild-type CCR5:CCL5 complex using the NMR-determined ensemble of Nt-CCR5(1–27):CCL5(P9S) and the recently released crystal structure of CCR5 bound to the high potency HIV entry inhibitor 5P7-CCL5 (PDB ID: 5UIW) [16]. We used all 10 models of the NMR ensemble instead of the minimized average structure to better capture the observed conformational variability of Nt-CCR5(1–27).
The crystallized CCR5 structure contains 7 stabilizing mutations and lacks the first 15 amino acids, while the 5P7-CCL5 N-terminus differs substantially from wild-type CCL5. The NMR structures contain one mutation on CCR5 (C20A) and two mutations on CCL5 - P9S and E66S – inserted to alleviate oligomerization and dimerization. To model CCR5, we took residues 18–316 from the crystal structure, removing the fused rubredoxin and restoring the native ICL3. The N-terminal region (residues 1–17), including the two sulfated tyrosine residues, was taken from each NMR structure. For CCL5, we took residues 1–8 from the crystal structure, to preserve the conformation and orientation of the N-terminus relative to the receptor, and the remainder (residues 9–68) from the NMR structures. Despite the low sequence similarity, the crystal structure of 5P7-CCL5 agrees with pre-existing mutagenesis data for the N-terminal region of CCL5, lending credence to its use as a template to model the wild-type structure.
We used MODELLER v9.18 [43] to restore both wild-type sequences (CCR5 Uniprot AC: P51681; CCL5 Uniprot AC: P13501) and regularize the geometry of the initial models. The optimization used the loopmodel protocol and custom restraints enforcing disulfide bridges in CCR5 (C20-C269 and C101-C178) and CCL5 (C10-C34 and C11-C50). All residues but the connecting regions between the NMR and crystal structures (residues 18–23 in CCR5 and 8–10 in CCL5) were frozen to preserve their native conformations.
The resulting 10 full-length models were refined as an ensemble with HADDOCK (v2.2) [30], using all measured dihedral angle restraints, as well as intra- and intermolecular NOEs as distance restraints. The rigid-body protocol was disabled, while the semi-flexible simulated annealing and explicit solvent molecular dynamics were run with default parameters to generate 500 final models (5 per initial model). The N-terminal serine residue of CCL5 was protonated. The refined models were clustered using a fast contact-based interface similarity algorithm [44] and ranked according to the default HADDOCK scoring function, which combines van der Waals, electrostatics, and restraint energies with an empirical desolvation potential [45] and buried surface area.
The refined models were analyzed using HADDOCK built-in routines (for energetics and violation analysis). DSSP [32] was used for secondary structure calculations and Biopython (Bio.PDB) for alignment and RMSD calculations [46,47].
Sequence Alignment of CC chemokines
To assess the evolutionary conservation of residues in CC chemokines, we collected all sequences deposited in Uniprot [48] under the manually curated intecrine beta (chemokine CC) family and filtered by species (human) and database (SwissProt). We removed two entries belonging to CC-like chemokines: CCL3L1 and CCL4L1. Given the high degree of sequence conservation, the resulting 24 sequences were aligned using Clustal Omega (version 1.2.4) [49] through the EBI web portal [50].
ACKNOWLEDGMENT
We thank Dr. Tali Scherf for help in setting up some of the NMR experiments. This study was supported by the Minerva Foundation with funding from the Federal German Ministry for Education and Research, by The US-Israel Binational Science Foundation by the Comisaroff Family Trust, The estate of D. Levinson and by the Kimmelman Center (JA), by the National Institutes of Health USA (R35GM122543 to ML), and by a Niels Stensen postdoctoral fellowship (JR). J.A. is the Dr. Joseph and Ruth Owades Professor of Chemistry. F.N. was supported by the Erna and Jakob Michael Visiting Professorship while on Sabbatical at the Weizmann Institute of Science.
Abbreviations
- CCL5
CC chemokine ligand 5, also known as RANTES
- CCR5
human CC chemokine receptor 5
- DSSP
Dictionary of Secondary Structure of Proteins
- GPCR
G protein-coupled receptor
- HADDOCK
High-Ambiguity Drive DOCKing
- HIV
human immunodeficiency virus
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- Nt
CCR5 N-terminal segment of CCR5
- Nt-CCR5(1–27)
The first 27 residues of CCR5 sulfated on Y10 and Y14 with a C20A substitution.
- TRNOE
transferred nuclear Overhauser effect.
Footnotes
Residue labeling conventions: chemokine residues are designated by the chemokine name in superscript, followed by the one-letter code of the amino acid, and the residue index (Uniprot canonical sequence numbering), e.g. CCL5K45; receptor residues use the Ballesteros-Weinstein numbering scheme for transmembrane residues, e.g. Y2516.51, or are preceded by NT in superscript for N-terminal residues, e.g. NTD11. Sulfated tyrosine residues are designated by sY: e.g. NTsY14;
Bibliography
- 1.Kufareva I, Salanga CL & Handel TM (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol 93, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossi D & Zlotnik A (2000) The Biology of Chemokines and their Receptors. Annu. Rev. Immunol 18, 217–242. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y & Irvine DJ (2011) Engineering Chemoattractant Gradients Using Chemokine-Releasing Polysaccharide Microspheres. Biomaterials 32, 4903–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salanga CL & Handel TM (2011) Chemokine Oligomerization and Interactions with Receptors and Glycosaminoglycans: The Role of Structural Dynamics in Function. Exp. Cell Res 317, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufareva I (2016) Chemokines and their receptors: insights from molecular modeling and crystallography. Curr. Opin. Pharmacol 30, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ludeman JP & Stone MJ (2014) The structural role of receptor tyrosine sulfation in chemokine recognition: Tyrosine sulfation of chemokine receptors. Br. J. Pharmacol 171, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seibert C, Cadene M, Sanfiz A, Chait BT & Sakmar TP (2002) Tyrosine sulfation of CCR5 N-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc. Natl. Acad. Sci. U. S. A 99, 11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bannert N, Craig S, Farzan M, Sogah D, Santo NV, Choe H & Sodroski J (2001) Sialylated O-Glycans and Sulfated Tyrosines in the NH2-Terminal Domain of CC Chemokine Receptor 5 Contribute to High Affinity Binding of Chemokines. J. Exp. Med 194, 1661–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD & Clark-Lewis I (1997) Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J 16, 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Millard CJ, Ludeman JP, Canals M, Bridgford JL, Hinds MG, Clayton DJ, Christopoulos A, Payne RJ & Stone MJ (2014) Structural Basis of Receptor Sulfotyrosine Recognition by a CC Chemokine: The N-Terminal Region of CCR3 Bound to CCL11/Eotaxin-1. Structure 22, 1571–1581. [DOI] [PubMed] [Google Scholar]
- 11.Skelton NJ, Quan C, Reilly D & Lowman H (1999) Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Structure 7, 157–168. [DOI] [PubMed] [Google Scholar]
- 12.Veldkamp CT, Seibert C, Peterson FC, Cruz NBD la, Haugner JC, Basnet H, Sakmar TP & Volkman BF (2008) Structural Basis of CXCR4 Sulfotyrosine Recognition by the Chemokine SDF-1/CXCL12. Sci Signal 1, ra4-ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziarek JJ, Kleist AB, London N, Raveh B, Montpas N, Bonneterre J, St-Onge G, DiCosmo-Ponticello CJ, Koplinski CA, Roy I, Stephens B, Thelen S, Veldkamp CT, Coffman FD, Cohen MC, Dwinell MB, Thelen M, Peterson FC, Heveker N & Volkman BF (2017) Structural basis for chemokine recognition by a G protein–coupled receptor and implications for receptor activation. Sci Signal 10, eaah5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, Fenalti G, Wu H, Han GW, Cherezov V, Abagyan R, Stevens RC & Handel TM (2015) Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL & Garcia KC (2015) Structural basis for chemokine recognition and activation of a viral G protein–coupled receptor. Science 347, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Han GW, Abagyan R, Wu B, Stevens RC, Cherezov V, Kufareva I & Handel TM (2017) Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46, 1005–1017.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamamis P & Floudas CA (2015) Elucidating a Key Anti-HIV-1 and Cancer-Associated Axis: The Structure of CCL5 (Rantes) in Complex with CCR5. Sci. Rep 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q & Wu B (2013) Structure of the CCR5 chemokine receptor – HIV entry inhibitor Maraviroc complex. Science 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skelton NJ, Aspiras F, Ogez J & Schall TJ (1995) Proton NMR assignments and solution conformation of RANTES, a chemokine of the C-C type. Biochemistry (Mosc.) 34, 5329–5342. [DOI] [PubMed] [Google Scholar]
- 20.Chung CW, Cooke RM, Proudfoot AE & Wells TN (1995) The three-dimensional solution structure of RANTES. Biochemistry (Mosc.) 34, 9307–9314. [DOI] [PubMed] [Google Scholar]
- 21.Duma L, Häussinger D, Rogowski M, Lusso P & Grzesiek S (2007) Recognition of RANTES by Extracellular Parts of the CCR5 Receptor. J. Mol. Biol 365, 1063–1075. [DOI] [PubMed] [Google Scholar]
- 22.Schnur E, Kessler N, Zherdev Y, Noah E, Scherf T, Ding F-X, Rabinovich S, Arshava B, Kurbatska V, Leonciks A, Tsimanis A, Rosen O, Naider F & Anglister J (2013) NMR mapping of RANTES surfaces interacting with CCR5 using linked extracellular domains. FEBS J 280, 2068–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abayev M, Srivastava G, Arshava B, Naider F & Anglister J (2017) Detection of intermolecular transferred-NOE interactions in small and medium size protein complexes: RANTES complexed with a CCR5 N-terminal peptide. FEBS J 284, 586–601. [DOI] [PubMed] [Google Scholar]
- 24.Czaplewski LG, McKeating J, Craven CJ, Higgins LD, Appay V, Brown A, Dudgeon T, Howard LA, Meyers T, Owen J, Palan SR, Tan P, Wilson G, Woods NR, Heyworth CM, Lord BI, Brotherton D, Christison R, Craig S, Cribbes S, Edwards RM, Evans SJ, Gilbert R, Morgan P, Randle E, Schofield N, Varley PG, Fisher J, Waltho JP & Hunter MG (1999) Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J. Biol. Chem 274, 16077–16084. [DOI] [PubMed] [Google Scholar]
- 25.Shen Y & Bax A (2015) Protein Structural Information Derived from NMR Chemical Shift with the Neural Network Program TALOS-N. Methods Mol. Biol. Clifton NJ 1260, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnur E, Noah E, Ayzenshtat I, Sargsyan H, Inui T, Ding F-X, Arshava B, Sagi Y, Kessler N, Levy R, Scherf T, Naider F & Anglister J (2011) The Conformation and Orientation of a 27-Residue CCR5 Peptide in a Ternary Complex with HIV-1 gp120 and a CD4-Mimic Peptide. J. Mol. Biol 410, 778–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Würz JM, Kazemi S, Schmidt E, Bagaria A & Güntert P (2017) NMR-based automated protein structure determination. Arch. Biochem. Biophys. 628, 24–32. [DOI] [PubMed] [Google Scholar]
- 28.Schwieters CD, Bermejo GA & Clore GM (2017) Xplor-NIH for Molecular Structure Determination from NMR and Other Data Sources. Protein Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linge JP, Williams MA, Spronk CA, Bonvin AM & Nilges M (2003) Refinement of protein structures in explicit solvent. Proteins Struct. Funct. Bioinforma 50, 496–506. [DOI] [PubMed] [Google Scholar]
- 30.van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ & Bonvin AMJJ (2016) The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- 31.Wilken J, Hoover D, Thompson DA, Barlow PN, McSparron H, Picard L, Wlodawer A, Lubkowski J & Kent SB (1999) Total chemical synthesis and high-resolution crystal structure of the potent anti-HIV protein AOP-RANTES. Chem. Biol 6, 43–51. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W & Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 33.Navenot J-M, Wang Z, Trent JO, Murray JL, Hu Q, DeLeeuw L, Moore PS, Chang Y & Peiper SC (2001) Molecular anatomy of CCR5 engagement by physiologic and viral chemokines and HIV-1 envelope glycoproteins: differences in primary structural requirements for RANTES, MIP-1α, and vMIP-II binding11Edited by P. E. Wright. J. Mol. Biol 313, 1181–1193. [DOI] [PubMed] [Google Scholar]
- 34.Maeda K, Das D, Ogata-Aoki H, Nakata H, Miyakawa T, Tojo Y, Norman R, Takaoka Y, Ding J, Arnold GF, Arnold E & Mitsuya H (2006) Structural and Molecular Interactions of CCR5 Inhibitors with CCR5. J. Biol. Chem 281, 12688–12698. [DOI] [PubMed] [Google Scholar]
- 35.Pakianathan DR, Kuta EG, Artis DR, Skelton NJ & Hébert CA (1997) Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3, and CCR5. Biochemistry (Mosc.) 36, 9642–9648. [DOI] [PubMed] [Google Scholar]
- 36.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S & Dioszegi M (2007) Molecular Interactions of CCR5 with Major Classes of Small-Molecule Anti-HIV CCR5 Antagonists. Mol. Pharmacol 73, 789–800. [DOI] [PubMed] [Google Scholar]
- 37.Srivastava G, Moseri A, Kessler N, Akabayov SR, Arshava B, Naider F & Anglister J (2016) Detection of intermolecular transferred NOEs in large protein complexes using asymmetric deuteration: HIV-1 gp120 in complex with a CCR5 peptide. FEBS J 283, 4084–4096. [DOI] [PubMed] [Google Scholar]
- 38.Colin P, Bénureau Y, Staropoli I, Wang Y, Gonzalez N, Alcami J, Hartley O, Brelot A, Arenzana-Seisdedos F & Lagane B (2013) HIV-1 exploits CCR5 conformational heterogeneity to escape inhibition by chemokines. Proc. Natl. Acad. Sci 110, 9475–9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenquist GL & Nicholas HB (1993) Analysis of sequence requirements for protein tyrosine sulfation. Protein Sci. Publ. Protein Soc 2, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaertner H, Cerini F, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I, Nedellec R, Salkowitz J, Gorochov G & Mosier D (2008) Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci 105, 17706–17711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hafsa NE & Wishart DS (2014) CSI 2.0: a significantly improved version of the Chemical Shift Index. J. Biomol. NMR 60, 131–146. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R & Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- 43.Webb B & Sali A (2016) Comparative Protein Structure Modeling Using MODELLER: Comparative Protein Structure Modeling Using Modeller. In Current Protocols in Bioinformatics (Bateman A, Pearson WR, Stein LD, Stormo GD, & Yates JR, eds), p. 5.6.1–5.6.37. John Wiley & Sons, Inc., Hoboken, NJ, USA. [Google Scholar]
- 44.Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis P, Karaca E, Melquiond ASJ & Bonvin AMJJ (2012) Clustering biomolecular complexes by residue contacts similarity. Proteins Struct. Funct. Bioinforma, n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 45.Fernández-Recio J, Totrov M & Abagyan R (2004) Identification of Protein–Protein Interaction Sites from Docking Energy Landscapes. J. Mol. Biol 335, 843–865. [DOI] [PubMed] [Google Scholar]
- 46.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B & de Hoon MJL (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamelryck T & Manderick B (2003) PDB file parser and structure class implemented in Python. Bioinformatics 19, 2308–2310. [DOI] [PubMed] [Google Scholar]
- 48.The UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sievers F & Higgins DG (2017) Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. Publ. Protein Soc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N & Lopez R (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43, W580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]