

Abstract
Background and purpose
Some clinical studies have reported increased myocardial infarction in people living with human immunodeficiency virus (HIV) taking the antiretroviral abacavir sulphate (ABC). Given that clinical studies contain confounding variables (e.g., HIV‐associated factors), we investigated the pharmacological effects of antiretrovirals on platelet function in HIV‐negative volunteers in order to identify mechanisms of increased cardiovascular risk.
Experimental approach
Platelets were isolated from healthy volunteers and HIV‐negative subjects enrolled on a Phase I clinical trial and platelet function evaluated using aggregometry and flow cytometry. In vivo platelet thromboembolism was monitored in anaesthetized mice.
Key results
Human platelet aggregation was unaffected by all antiretrovirals tested, but ABC treatment led uniquely to increased platelet granule release. ABC also interrupted NO‐mediated inhibition of platelet aggregation and increased in vivo aggregation in mice. Another antiretroviral, tenofovir, did not affect platelet function. Furthermore, aggregation and activation of platelets isolated from 20 subjects taking clinically relevant doses of tenofovir were comparable to baseline samples.
Conclusions and implications
ABC can enhance platelet activation, independently of variables that confound clinical studies, suggesting a potential pharmacological effect that is absent with tenofovir. Mechanistically, we propose that ABC enhances platelet degranulation and interrupts NO‐mediated platelet inhibition. The interaction of ABC with NO signalling is demonstrated by ABC‐mediated enhancement of aggregation in vivo and in vitro that persisted in the presence of NO. Although an association between ABC and platelet activation has not been confirmed in patients, these findings provide evidence of a mechanistic link between platelet activation and antiretroviral therapy.
Abbreviations
- ABC
abacavir sulphate
- ART
antiretroviral therapy
- CBV‐TP
carbovir triphosphate
- Ces1c
carboxyesterase1c‐deficient
- CVD
cardiovascular disease
- D:A:D
the data collection of adverse events of anti‐HIV drugs
- FTC
emtricitabine
- HIV
human immunodeficiency virus
- MI
myocardial infarction
- NRTI
nucleotide‐reverse‐transcriptase inhibitor
- PLWH
people are living with HIV
- PrEP
pre‐exposure prophylaxis
- SNAP
S‐nitroso‐N‐acetyl‐penicillamine
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumarate
- TFV
tenofovir
- TRAP6
thrombin and thrombin receptor activating peptide 6
What is already known
Cardiovascular risk is elevated in people living with HIV and hypothesized to be exacerbated by certain antiretrovirals.
What this study adds
Mechanistic insights demonstrating differential effects of antiretrovirals upon platelet activation.
What is the clinical significance
Our data could ultimately lead to improved management of multimorbidity in people living with HIV.
1. INTRODUCTION
An estimated 36 million people are living with human immunodeficiency virus (HIV; PLWH) globally, and improvements in the diagnosis and management of the disease mean that life expectancy is now close to that of HIV‐negative individuals (Antiretroviral Therapy Cohort, 2017). Drug regimens currently recommended as first‐line antiretroviral therapy (ART) for HIV consist of a combination of two nucleotide reverse‐transcriptase inhibitors (NRTIs), typically either tenofovir disoproxil fumarate (TDF) or tenofovir alafenamide (TAF) and emtricitabine (FTC), or abacavir sulphate (ABC) and lamivudine, plus a third agent such as an integrase inhibitor, non‐NRTI or protease inhibitor (European AIDS Clinical Society [EACS], 2017; World Health Organization, 2016). ART is highly effective, and <5% of PLWH in Western countries will die from AIDS‐related illness. In contrast, 31% of deaths are attributed to cardiovascular disease (CVD), making it the leading co‐morbidity for PLWH. The relative risk of CVD for PLWH is 1.26–1.61, rising to 2.00 when ART regimens are considered. Within this population, myocardial infarction (MI) is the leading clinical presentation (D:A:D Study Group et al., 2008; Friis‐Moller, Weber, et al., 2003; Islam, Wu, Jansson, & Wilson, 2012).
The data collection of adverse events of anti‐HIV drugs (D:A:D) collaborative cohort study has analysed data from over 33,000 patients (D:A:D Study Group et al., 2008). D:A:D analyses highlight an increased relative rate of MI that is associated with exposure to ART (Friis‐Moller, Sabin, et al., 2003). Furthermore, recent use of the NRTI ABC was associated with a 90% increase in the relative rate of MI (D:A:D Study Group et al., 2008). ABC‐associated risk persisted in a follow‐up study, despite a channelling bias away from ABC in PLWH who presented with a higher cardiovascular risk profile (Sabin et al., 2016). Incident cardiovascular risk was also associated with ABC but not TDF in later cohort studies (Choi et al., 2011; D:A:D Study Group et al., 2008). Subsequent studies have confirmed the link between ABC and MI whereas, in contrast, others have not (see Alvarez, Orden, et al., 2017). A FDA‐led meta‐analysis did not find an association between ABC and MI (Ding et al., 2012), so that a consistent link between ABC and cardiovascular risk in PLWH has not been established.
As MI is platelet‐driven, several studies have investigated the effects of ART on platelet function. Clinical studies have reported enhanced ex vivo platelet aggregation in PLWH compared with matched HIV‐negative controls (Satchell et al., 2010). Furthermore, ABC was associated with enhanced platelet aggregation compared to patients on alternative therapies (Satchell et al., 2011), including TDF, which had no effect (Munoz et al., 2012). There are contrasting reports demonstrating that ABC has no effect on platelet activation in response to ADP and collagen (Alvarez, Rios‐Navarro, et al., 2017; Diallo et al., 2016). Indeed, Alvarez, Rios‐Navarro, et al. (2017) proposed that ABC‐mediated enhancement of thrombosis is via a direct interaction of the drug with the vascular endothelium. Direct comparison between these studies is complicated by differences in HIV‐associated factors, lifestyle, biological, and medical characteristics of participants and parameters assessed. Thus, a pharmacological link between ABC and enhanced platelet activation has not been shown and remains hypothetical. We have therefore assessed the comparative pharmacological effects of the alternative ART backbone therapies ABC, TDF, and the newer tenofovir (TFV)‐based compound TAF on platelet activation, independently of confounding factors associated with HIV infection by recruiting HIV‐negative volunteers for in vitro and ex vivo studies, and in vivo using inbred mouse strains.
2. METHODS
2.1. Human platelet preparation
This study was approved by the Imperial College London Research Ethics Committee and informed consent was obtained from healthy, HIV‐negative volunteers, in accordance with the Declaration of Helsinki. Volunteers, male and female, had not taken aspirin, steroids, antihistamines or used an inhaler or an investigational product within the last 6 months; they were also non‐smoker. Volunteers confirmed that they had not recently taken NSAIDs or SSRIs or suffered from a chronic condition such as diabetes. Blood was collected in acid citrate dextrose (9:1) and PRP was collected following centrifugation (175× g, 15 min). Where indicated, PRP was washed twice (1400× g, 10 min, 2 μM PGE1) and platelets were resuspended in a modified Tyrode's–HEPEs buffer (134‐mM NaCl, 2.9‐mM KCl, 20‐mM HEPES, 4.5‐mM glucose, 12‐mM NaHCO3, 0.34‐mM Na2HPO4, and 1‐mM MgCl2) and rested for 30 min prior to use.
2.2. Microplate aggregation assay
Platelet suspensions were preincubated with NRTIs or vehicle control, as indicated, for 30 min at 37°C. Platelets were added to a 96‐well plate (VWR, Leicestershire, UK) containing agonists, and the plate was read at 20‐s intervals for 16 min in a Power Wave X5 plate reader (Bio‐TEK, Swindon, UK). The machine was maintained at 37°C for the duration of the experiment with shaking for 7 s before each reading. Maximum aggregation at 16 min was calculated as a percentage change in absorbance, compared to baseline.
2.3. Light transmission aggregometry
For selected experiments, platelet aggregation was monitored in a Model 400 aggregometer (Chrono‐Log, Labmedics, Abingdon, UK) as described previously (Jones et al., 2010).
2.4. Flow cytometry
Kinetic flow cytometry was performed using methodology described previously (Jones et al., 2014), with slight modification. Briefly, PRP was diluted 1:200 into Tyrode's–HEPES buffer containing fluorescently conjugated PAC‐1 (Cat# 340507, RRID:AB_2230769), CD62P (Cat# 550888, RRID:AB_398475), and CD63 antibodies (Cat# 556019, RRID:AB_396297; BD biosciences, Belgium). Samples were analysed under continuous flow in an Accuri C6 Plus flow cytometer (BD Biosciences). Collagen‐evoked (10 μg·ml−1) activation was induced 30 s into the acquisition and responses monitored for 3 min. Colour compensation values were calculated using Ultra Comp ebeads (Thermo Fisher, Hemel Hempstead, UK) following the manufacturer's protocol. Data were analysed within Flow Jo (v10.4; Oregon, USA) and the relative increase of fluorescence calculated.
2.5. Animals
All animal care and experimental procedures were approved by the Imperial College London Animal Welfare and Ethical Review Board, and appropriate licences were obtained from the UK Home Office. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology. Three‐month‐old wild‐type male C57BL/6 mice (MGI Cat# 5654507, RRID:MGI:5654507) weighing 20–25 g were supplied by Harlan (Bicester, UK). For experiments investigating the effect of TAF, 3‐month‐old male and female carboxyesterase1‐deficient (Ces1c −/−) mice from a colony maintained by Gilead Sciences at Jackson Laboratories (RRID:MGI:4938613) were used. All mice were housed in IVC cages (2–5 per cage) containing bedding materials (wood shavings and shredded paper with a cardboard tube for environmental enrichment) within the Central Biomedical Sciences facility at Imperial College London.
2.6. In vivo platelet aggregation
In vivo platelet aggregation was assessed in real time as the thromboembolic accumulation of In111‐radiolabelled (Mallinckrodt Pharmaceuticals, Surrey, UK) mouse platelets in the pulmonary region of anaesthetized (25% urethane; 10 μl·g−1; Sigma‐Aldrich; Dorset, UK) mice via an external scintillation probe as described previously (Tymvios et al., 2008). Animals were randomized to treatment groups. To account for the higher metabolic rate of mice compared with humans, NRTI doses were administered to achieve 10× the human Cmax levels such that mice were given a single i.p. injection of either 1,755 μg·kg−1 ABC, 372 μg·kg−1 TDF, or 139 μg·kg−1 TAF, estimated to achieve plasma concentrations of 30, 3.3, and 2.2 μg·ml−1 respectively. 30 min later, platelet thromboembolism was induced by i.v. injection of collagen (50 μg·kg−1). For this protocol, blinding was not feasible as experiments were conducted by a single experimenter. Each procedure was non‐recovery, and, following procedures, mice were killed by cervical dislocation and death confirmed by exsanguination.
2.7. Ex vivo platelet aggregation
Regulatory and ethical approvals were obtained before initiating the study. Subjects signed written informed consent prior to being enrolled in the study. The study was registered at ClinicalTrials.gov (NCT03186482). Twenty‐one healthy volunteers were recruited to a Phase I, open‐label, sequential pharmacokinetic trial with the experimental objective to investigate the comparative effects of TDF and TAF/FTC on platelet function. Blood samples were collected at Chelsea and Westminster Hospital NHS Foundation Trust (London, UK) between June and December 2017. Eligible subjects were HIV‐negative, aged between 18 and 60 years without any clinically significant acute or chronic medical illness. On Day 1, participants started TAF 25 mg coformulated with FTC 200 mg once daily for 28 days. They subsequently discontinued TAF/FTC and received TDF 300 mg once daily for a further 28 days. Blood samples for platelet function studies were collected at baseline before starting TAF/FTC, after 28 days of TAF/FTC administration and after 28 days of TDF intake. Blood was processed as above, and platelet aggregation assessed by microplate assay and platelet activation by flow cytometry.
2.8. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. Data were analysed in GraphPad Prism (v7.04, RRID:SCR_002798, California, USA), presented as the mean ± SEM, and statistical significance was determined using a two‐way ANOVA or paired students' t‐test with two tails, as appropriate and indicated in the Figure legends. Curve fitting for concentration–response relationships were fitted assuming a Hill slope of 1, while dose‐inhibition curves (Figure S2) were fitted with a Hill slope of −1. Flow cytometry are presented as LOWESS curves and in vivo aggregation data are presented as representative traces. Significance is denoted as ns or * for P > 0.05 or P < 0.05 respectively
2.9. Materials
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxaline‐1‐one (ODQ), acid citrate dextrose, ADP, DMSO, PGE1, S‐nitroso‐N‐acetyl‐penicillamine (SNAP), thrombin, and thrombin receptor activating peptide 6 (TRAP6) were purchased from Sigma‐Aldrich (Dorset, UK). Selleckchem (Texas, USA) supplied ABC and TFV. Carbovir triphosphate (CBV‐TP) was purchased from US Biologicals (Massachusetts, USA). Forskolin and 9‐(tetrahydro‐2‐furanyl)‐9H‐purin‐6‐amine (SQ 22536) were obtained from Bio‐Techne (Oxford, UK). TAF/FTC (Descovy), TAF and TDF were supplied by Gilead Sciences (California, USA). Type 1 collagen from bovine tendon was from Takeda (Osaka, Japan). Unless stated, all other reagents were from Sigma‐Aldrich.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
3. RESULTS
3.1. NRTIs do not affect platelet aggregation in vitro
We adopted a medium‐throughput microplate‐based approach to enable simultaneous evaluation of the effects of NRTIs on platelet aggregation to ADP, collagen, and TRAP6. Platelets were preincubated with plasma concentrations that correspond to Cmax (i.e., 3 μg·ml−1 ABC, 0.22 μg·ml−1 TAF, and 0.33 μg·ml−1 TDF) for 30 min prior to agonist stimulation. As TDF and TAF therapy result in the presence of TFV in the systemic circulation, we also investigated the effects of TFV Cmax. Treatment with ABC, TAF, TDF, or TFV did not affect the maximum aggregation response to any concentration of agonist tested (Figure 1). Given the possibility that interactions between plasma proteins and ART may reduce the bioavailability of these drugs, we additionally evaluated the effects of NRTI on platelet activation in washed platelet suspensions. These data were comparable to those obtained in PRP with no differences observed between vehicle‐ or NRTI‐treated platelets (Figure S1).
Figure 1.
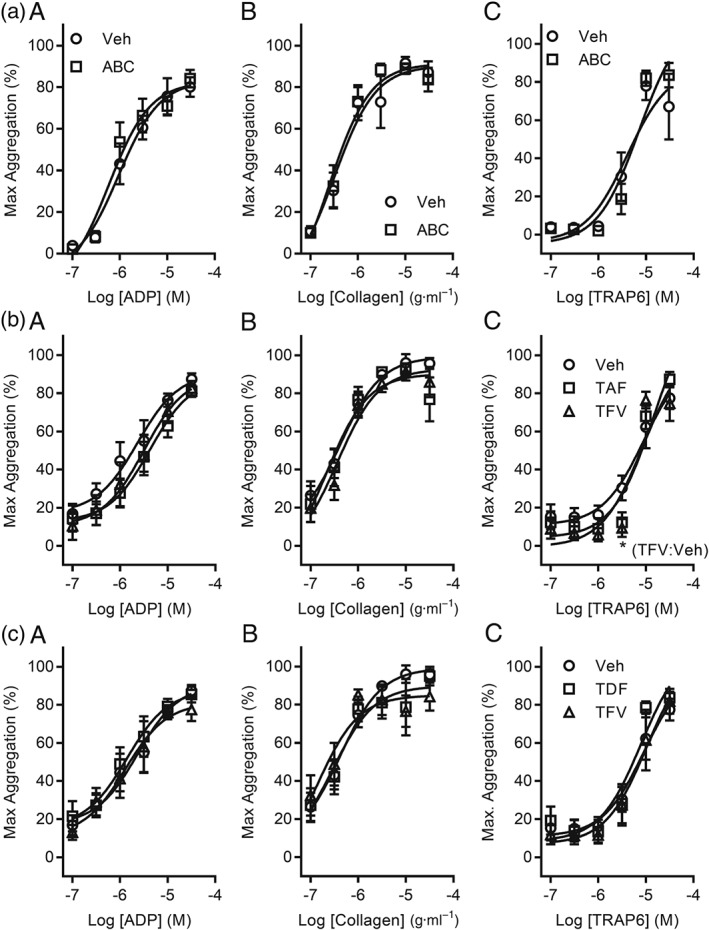
In vitro platelet aggregation responses are not affected by incubation with NRTIs. Platelet aggregation was monitored using PRP in a microplate assay. PRP was treated with plasma Cmax levels of NRTIs, or their respective anabolite, for 30 min at 37°C. Maximum aggregation is reported in responses to increasing concentrations of ADP (A), collagen (B), or TRAP6 (C). (a) Platelets were treated with 3 μg·ml−1 ABC or vehicle control (1% DMSO). (b) Aggregation was recorded following treatment with 0.22‐μg·ml−1 TAF, 15‐ng·ml−1 TFV, or vehicle control (1% DMSO). (c) Platelets were incubated with 0.33‐μg·ml−1 TDF, 0.25‐μg·ml−1 TFV, or vehicle control (2% DMSO). Data are representative of a five independent experiments for each condition. *P < 0.05, significantly different from vehicle; two‐way ANOVA
Because acute in vitro exposure to drugs may poorly model patients on chronic ART, we exposed platelets to 30–100× plasma NRTI Cmax and did not observe any significant effect of ABC, TAF, TDF, or TFV on platelet aggregation responses (Figure S2). Figure 1b shows that incubation of PRP with TFV (0.05 μM) significantly reduced the 3‐μM TRAP6‐induced aggregation response by 21.0 ± 8.7% (Figure 1bC). This inhibitory effect was not seen at other agonist concentrations, in studies using washed platelets or following exposure of platelets to a broad range of TFV concentrations (0.01–30 μM; Figure S2d). Similar data were obtained using conventional aggregometry where platelets were pretreated with Cmax levels of ABC, TAF, or TDF and stimulated by 10 μg·ml−1 collagen for 3 min (Figure S3).
3.2. Abacavir enhances human platelet granule release
Having generated no evidence of increased platelet aggregation by any of the studied NRTIs, including ABC, using an end‐point assay, we progressed to kinetic flow cytometry to evaluate real‐time expression of the platelet activation markers PAC‐1 (integrin αIIbβ3 activation), CD62P (α granule), and CD63 (dense granule; Jones et al., 2014). In the presence of the vehicle control (0.02% DMSO), application of collagen led to a rapid and sustained increase of PAC‐1, CD62P, and CD63 binding, which peaked at 1.51 ± 0.18 and 2.10 ± 0.26 respectively (Figure 2). Following incubation with 3 μg·ml−1 ABC, both α (Figure 2a) and dense (Figure 2b) granule release increased by 0.63 ± 0.18 (Figure 2a) and 0.84 ± 0.31 relative to vehicle control respectively (Figure 2b). In contrast, TAF or TDF preincubation did not alter collagen‐induced CD62P or CD63 expression (Figure 2a–b). Collagen‐induced PAC‐1 binding was not affected by any NRTI, including ABC (Figure 2c). Our observations regarding the effects of ABC on granule secretion were specific to collagen, as neither ADP nor TRAP6 had a significant effect on CD62P, CD63, or PAC‐1 binding (Figure S4).
Figure 2.
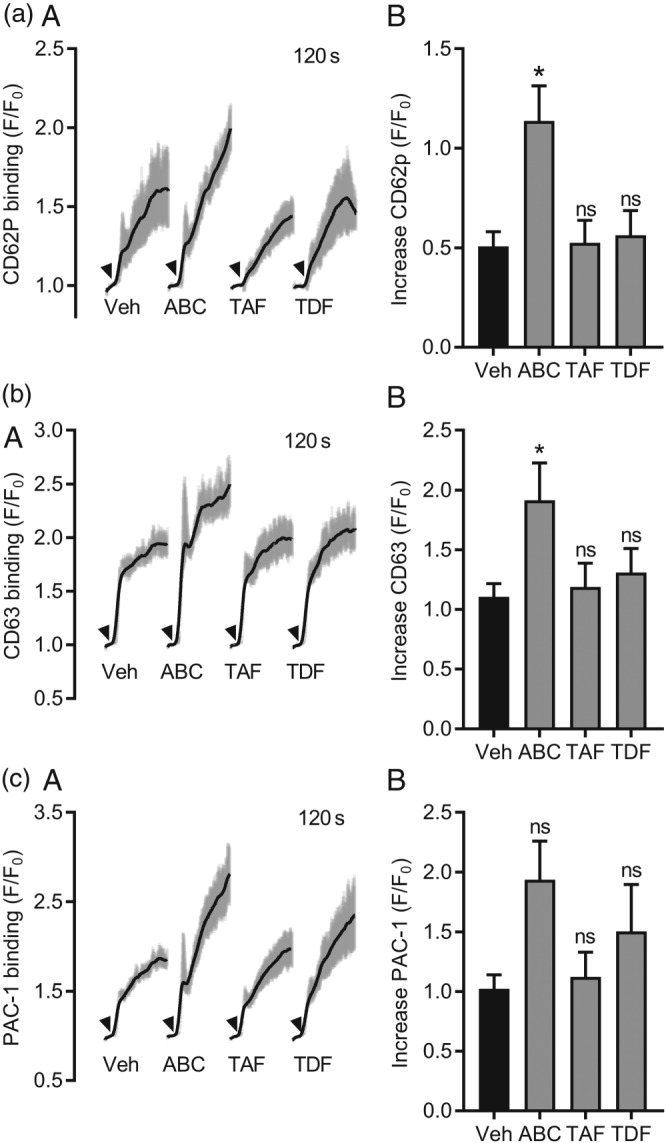
Abacavir enhances collagen‐evoked granule release. Flow cytometry was used to assess real‐time changes in surface expression of platelet granule markers. PRP was diluted 1:200 into physiological buffer containing fluorescently conjugated CD62P, CD63 and PAC‐1 antibodies. Stable baselines were achieved in the first 30 s of sample acquisition and arrowheads (A) indicate when collagen (10 μg·ml−1) was added to the samples. Addition of collagen led to a rapid and sustained increase of CD62P (a), CD63 (b), and PAC‐1 (c) binding. Responses were monitored in the presence of vehicle control (0.02% DMSO), ABC (3 μg·ml−1), TAF (0.22 μg·ml−1), or TDF (0.33 μg·ml−1). Data are representative of six independent experiments. *P < 0.05, significantly different from vehicle; two‐way ANOVA
3.3. Abacavir metabolite interrupts NO‐mediated inhibition of platelet aggregation
The active anabolite of ABC, CBV‐TP, is a guanosine analogue and antagonism of NO‐cGMP‐mediated inhibitory pathways in the platelet has been suggested as a potential mechanism by which ABC may affect platelet function (Baum, Sullam, Stoddart, & McCune, 2011). We hypothesized that CBV‐TP would interrupt NO‐mediated inhibition of aggregation whereas TFV, an adenosine analogue, would not. The NO donor SNAP significantly reduced ADP‐induced platelet aggregation by 37.3 ± 6.9% (Figure 3a). Inhibition was significantly reversed upon application of the soluble guanylate cyclase inhibitor ODQ (Figure 3a) and also by CBV‐TP. Parallel experiments were conducted in the presence of TFV, the active anabolite of both TAF and TDF. TFV‐treated platelets remained susceptible to SNAP‐mediated inhibition and were not significantly different to SNAP in the presence of vehicle control (Figure 3b). We wished to confirm that the effect of ABC was specific to the NO/cGMP axis and evaluate the effect of TFV, an adenosine analogue, upon adenylate cyclase/cAMP signalling. Preincubation with the adenylate cyclase activator, forskolin, reduced ADP‐evoked aggregation from 36.6 ± 6.7% to 6.0 ± 4.0% (Figure 3c; n = 5). Aggregation responses were preserved in the presence of the adenylate cyclase inhibitor SQ22536 (Figure 3c, n = 5) confirming the induction of adenylate cyclase by forskolin. Neither CBV‐TP nor TFV reversed forskolin‐mediated platelet inhibition as the maximum aggregation responses were 4.4 ± 2.0% and 4.0 ± 2.1% respectively (Figure 3c). These data suggest that neither CBV‐TP nor TFV interfere with adenylate cyclase‐mediated inhibition of platelet aggregation (Figure 3c; n = 5).
Figure 3.
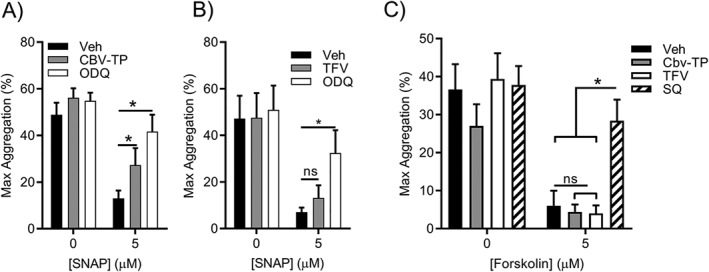
Abacavir interrupts NO‐mediated inhibition of platelet activation. Platelet aggregation was monitored by light transmission aggregometry in response to 3‐μM ADP. Platelets were preincubated for 3 min with 5 μM SNAP (a,b), 5‐μM forskolin (c), or vehicle control (0.5% DMSO). 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxaline‐1‐one (ODQ; 20 μM) was applied to assess the contribution of soluble GC to the SNAP‐mediated inhibition (a,b), while the adenylate cyclase pathway was probed by SQ22536 (SQ; 100 μM; c). The effect of CBV‐TP (a,c; 50 μM) or tenofovir (TFV: b,c; 50 μM) on NO‐ (a,b) or adenylate cyclase‐mediated (c) platelet inhibition was assessed. Data are representative of seven (a,b) and five (c) independent experiments. *P < 0.05, significantly different as indicated; two‐way ANOVA
Taken together, these data support the hypothesis that CBV‐TP, but not TFV, is able to selectively interrupt NO‐mediated inhibition of platelet aggregation. This effect was independent of adenylate cyclase activity. To investigate the physiological relevance of this finding in the context of endothelial generation of NO, we employed an in vivo mouse model of platelet aggregation in which radiolabelled platelets freely circulate and upon i.v. injection of a platelet agonist, aggregate and become trapped in the pulmonary vasculature (Tymvios et al., 2008). Dynamic platelet aggregation can be determined in real time via an external scintillation probe suspended over the pulmonary region. Platelet aggregation in this model has previously been shown to be highly sensitive to inhibition of endogenous NO production (Moore, Tymvios, & Emerson, 2010).
Collagen elicited a rapid accumulation of radiolabelled platelets in the thoracic region of vehicle (DMSO) control mice that resolved over time (Figure 4). In the presence of ABC, aggregation responses achieved a greater peak and took longer than vehicle control to resolve, which was reflected in a higher AUC (Figure 4a). Conversely, exposure of mice to TDF did not alter the peak aggregation response or AUC compared to vehicle‐treated mice (Figure 4b).
Figure 4.
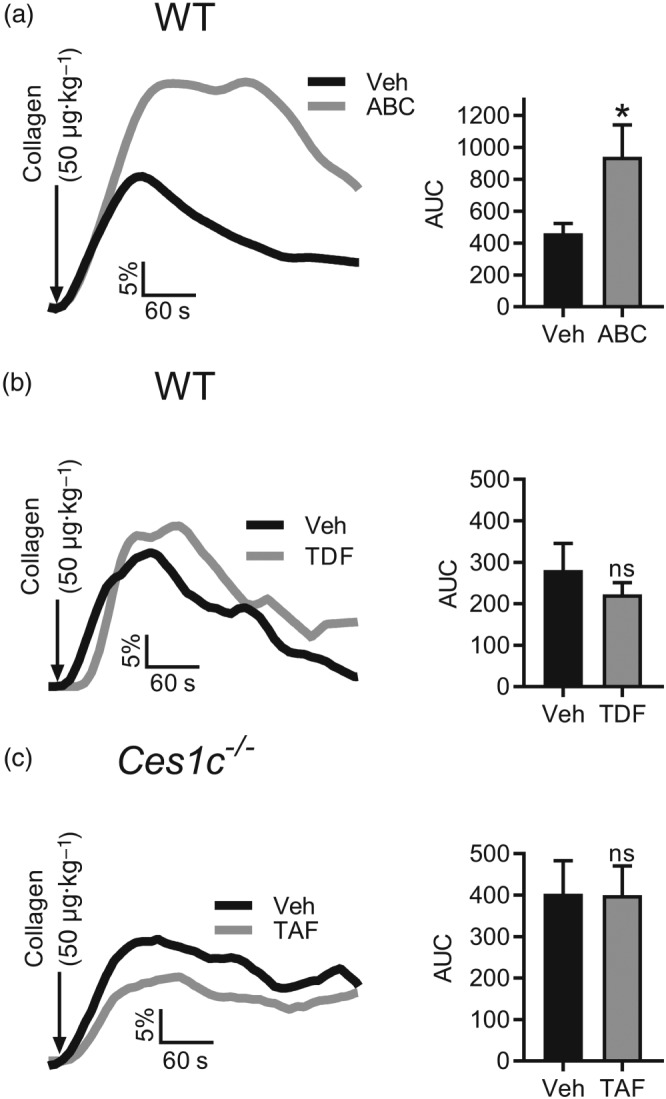
Abacavir enhances collagen‐evoked platelet aggregation in vivo. Platelet aggregation responses were monitored in vivo using terminally anaesthetized mice. Mouse platelets were isolated, radiolabelled (1.8‐MBq indium tropolone), and transfused into a recipient mouse. ABC (a; 30 μg·ml−1), TDF (b; 3.3 μg·ml−1), or TAF (c; 2.2 μg·ml−1) was administered i.p. for 30 min. A bolus of 50 μg·kg−1 collagen was injected (arrows), and responses were monitored for 10 min. ABC and TDF experiments were conducted using wild‐type (WT) C57bl/6 mice, whilst Ces1c −/− mice were utilized for TAF. The AUC was calculated for the duration of the experiment and representative traces and summary data are shown. Data are representative of seven independent experiments. *P < 0.05, significantly different from vehicle; paired, two tailed Students' t‐test
Due to elevated levels of plasma esterase activity in mice compared to humans, TAF is not stable in wild‐type mice and is rapidly degraded. The physiological effect of TAF on platelet activation was therefore assessed in Ces1c −/− mice, in which TAF has been shown to be stable. Ces1c −/− mice were treated with TAF or vehicle (DMSO) for 30 min prior to injection of collagen. Peak aggregation responses and AUC were not significantly different between groups. Thus, ABC increased collagen‐induced platelet aggregation in vivo whereas TDF and TAF had no effect.
3.4. TFV‐based therapies do not alter platelet activation at clinically relevant dosing
Clinical studies comparing the effects of ABC and TDF on platelet function are currently being conducted in PLWH but have not yet definitively linked ART with changes in platelet activation (O'Halloran et al., 2018). It is also worth noting that TDF is currently prescribed to individuals at risk of HIV infection as pre‐exposure prophylaxis (PrEP), meaning HIV‐negative people are now taking ART regularly. TAF is a novel prodrug of TFV characterized by greater permeability to peripheral blood mononuclear cells, improved stability in plasma compared to TDF and an improved kidney and bone safety profile (Post et al., 2017). There is early evidence to suggest that TAF may have comparable efficacy to TDF in PrEP formulations (Massud et al., 2016). However, in contrast to ABC and TDF, the effects of TAF/FTC on platelet function have not been assessed in a clinically relevant setting. In the context of a Phase I clinical trial, we determined the effects of daily TAF/FTC, relative to TDF, administration upon platelet aggregation and activation. Platelet analyses were conducted predose on Day 1 (baseline) and postdose after achieving steady‐state plasma NRTI levels (28 days). Platelet aggregation was determined in response to increasing concentrations of ADP, collagen, and TRAP6. Intrasubject analyses did not reveal any change in maximum aggregation responses between baseline and TAF/FTC‐ or TDF‐treated samples (Figure 5a–c). Flow cytometric analysis of platelet activation markers was conducted in a subgroup of four (20%) subjects enrolled on the trial. These exploratory studies assessed integrin αIIbβ3 activation (Figure 5d) and granule release (Figure 5e–f) in response to ADP, collagen, and TRAP6. We did not observe any apparent effect of TAF/FTC or TDF on integrin action (Figure 5d), α (Figure 5e), or dense granule (Figure 5f) release.
Figure 5.
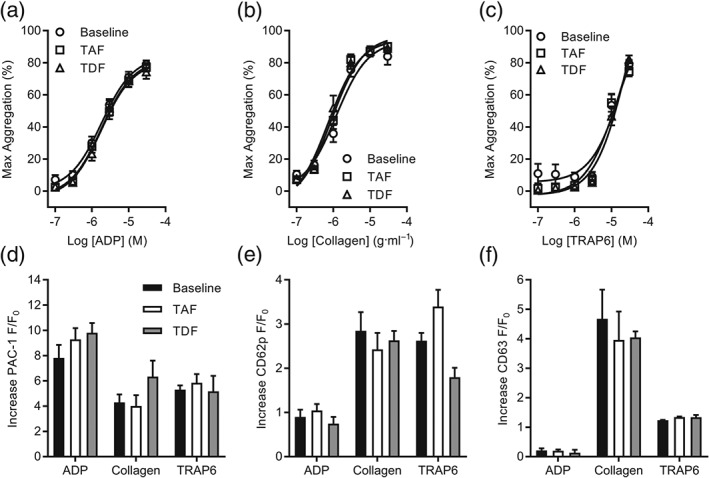
Steady‐state plasma concentrations of TAF or TDF in healthy subjects do not alter ex vivo platelet activation. (a–c) Platelet aggregation was monitored using PRP in a microplate assay. PRP was collected from healthy subjects enrolled on a Phase I clinical trial at predetermined timepoints: predose on Day 1 (baseline) and postdose following 28 days of once‐daily TAF (25 mg) or TDF (300 mg). Platelet aggregation was assessed in response to rising concentrations of ADP (a), collagen (b), or TRAP6 (c), and the maximum response is reported. (d–f) Flow cytometry was used to assess real‐time changes of platelet activation markers. PRP was collected from healthy subjects enrolled on a Phase I clinical trial at predetermined timepoints: predose on Day 1 (baseline) and postdose following 28 days of once‐daily TAF (25 mg) or TDF (300 mg). Activation of integrin αIIbβ3 (a) and surface expression of CD62P (b) and CD63 (c) were monitored following stimulation by ADP (10 μM), collagen (10 μg·ml−1), or TRAP6 (10 μM). Increases of fluorescence are reported, and intrasubject analysis was performed. Data are representative of 20 (a–c) and four (d–f) subjects enrolled on the trial. In (a‐c), no significant effects; two‐way ANOVA
4. DISCUSSION
HIV treatment has led to dramatic improvements in health so that PLWH no longer progress to AIDS. The major cause of death in PLWH is CVD (D:A:D Study Group et al., 2008; Friis‐Moller, Weber, et al., 2003; Islam et al., 2012), which is the same as the general population. Several studies, however, have linked both HIV and ART with increased risk of platelet‐driven cardiovascular events, principally MI (Friis‐Moller, Sabin, et al., 2003; Pollack & Rind, 2007; Sabin et al., 2016; Satchell et al., 2010; Satchell et al., 2011). It has also been suggested that ABC may be specifically associated with increased cardiovascular risk but there is no consensus on this topic. Cohort and observational studies demonstrating a link between ART and MI or platelet aggregation are limited by confounding factors, for example, impaired kidney function is both a contraindication for TDF use and a cardiovascular risk factor (Sax et al., 2015). In addition, a meta‐analysis did not reveal any additional effect of ABC in relation to CVD (Ding et al., 2012) so that existing data are conflicting. EACS guidelines stipulate that patients with a high cardiovascular risk profile (>20%) are now channelled away from ABC (EACS, 2017) so that matching cohorts is increasingly challenging. To test the hypothesis that ABC drives cardiovascular risk by pharmacologically enhancing platelet aggregation, we designed a study to compare the effects of ABC‐ and TFV‐based therapies, independent of major confounding factors (e.g., HIV‐associated factors and previous ART use).
We first demonstrated that incubation of platelets from HIV negative, healthy volunteers with NRTIs had no effect on subsequent platelet aggregation. We adopted a 96‐well format to allow simultaneous functional assessment of aggregation without the variability that occurs due to sample deterioration when consecutive cuvette assays are performed. The use of HIV negative donors removed the confounding influence of HIV‐associated factors such as background inflammation (Deeks, Tracy, & Douek, 2013) which could drive cardiovascular risk independently of the pharmacological effects of ART. An approach in which platelets were incubated in vitro also standardized drug exposure by avoiding pharmacokinetic variability. It is not possible to conclude definitively from these studies that NRTIs had no effects on platelet aggregation as our data are limited by the use of an end‐point, rather than a real‐time, assay. We did however additionally perform gold‐standard aggregometry experiments in cuvettes and found no effect of any NRTIs used in this study (Figure S3). In addition, although we used clinically relevant Cmax concentrations of NRTIs, the extent to which these modelled chronic exposure of drugs in patients is unclear given that PLWH have a predicted life expectancy of 79 years, Diagnosis and initiation of ART in a patient aged 19 would amount to 60 years of daily ART. To address this point to some extent, we extended our study to look at a range of drug concentrations (30–100× Cmax) and found no effects. Thus, we found no evidence for a direct effect of ART upon human platelet aggregation. Furthermore, in order to assess the effects of clinically relevant dosing, we collected samples from a Phase I clinical trial in which HIV negative volunteers took the approved doses of TAF and TDF daily over a time frame that allowed them to reach steady‐state (28 days). Intrasubject analyses showed no difference between baseline platelet aggregation or platelet activation measured by flow cytometry, confirming in a clinically relevant dosing context, our data from in vitro studies. The finding that TAF and TDF have no measurable effects on platelet activation is important, not only to PLWH but to the increasing number of individuals at risk of infection exposed to TDF (and in future probably TAF) as pre‐ or post‐exposure prophylaxis (PrEP and PEP) (Massud et al., 2016).
We employed flow cytometry to dynamically quantify the effects of NRTIs on platelet activation. We found that, in contrast to TAF and TDF, ABC exposure enhanced collagen‐evoked dense and α granule release but had no effect on granule release when platelets were stimulated by other agonists. An alternative approach to the in vitro studies reported here, is the use of switch studies in which PLWH are randomized to switch from ABC to TFV‐based therapies. Results from these studies are now emerging and have shown persistently lower collagen‐induced platelet aggregation in patients switching to TAF, compared with those remaining on ABC (Mallon et al., 2018). Other agonists either had no effect or only a transient effect. Together, these data suggest an effect of ABC on the rate of collagen‐induced granule secretion that could lead to enhanced platelet aggregation in a patient setting. This finding requires further exploration but could potentially be explained by differential sensitivities of collagen‐ compared to ADP‐ or TRAP6‐evoked granule release, cGMP‐mediated inhibition or a direct effect of ABC on collagen‐evoked platelet granule release. Given that our flow cytometry experiments were conducted in isolated platelets and therefore in the absence of NOS, which is not expressed in platelets (Tymvios et al., 2009), it is possible that ABC directly effects granule release independently of NO/cGMP (Figure 6). An overarching involvement of NO/cGMP cannot be completely excluded however, as bioactive NO has been shown to be generated in platelets via reduction of inorganic nitrite (Apostoli, Solomon, Smallwood, Winyard, & Emerson, 2014). Thus, it is possible that ABC may alter platelet activation and influence thrombus formation through multiple pathways.
Figure 6.
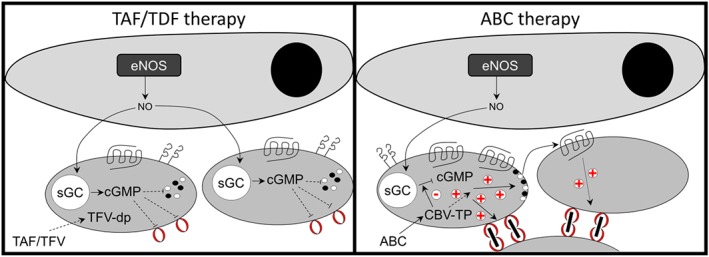
Proposed model for ABC‐dependent increase of platelet reactivity. The active metabolites of ABC and TAF/TDF are guanosine and adenosine analogues respectively. Left panel depicts the basic interaction between the endothelium and platelets in the presence of TAF/TDF. In this context, NO derived from the endothelium stimulates cGMP production in the platelet, leading to inhibition of aggregation. ABC/CBV‐TP may also directly enhance platelet granule release in a cGMP‐independent manner. Right panel depicts the proposed effect of CBV‐TP disrupting cGMP production, amplifying granule release and thrombus formation. eNOS: endothelial NOS; sGC: soluble GC
Previous studies have highlighted that CBV‐TP, the active metabolite of ABC, may, as a guanosine analogue, competitively inhibit NO‐cGMP‐mediated signalling (Baum et al., 2011). As NO is a major negative regulator of platelet activation, this provides a potential mechanism by which ABC could drive cardiovascular risk and act differently to TFV. We adopted two approaches. Firstly, we employed in vitro aggregometry and, since platelets do not express NOS enzymes (Tymvios et al., 2009), applied NO exogenously via SNAP. Secondly, we used an in vivo mouse model in which platelets circulate in the context of a functional vascular endothelium (Tymvios et al., 2008). Critically, this model has been shown to be extremely sensitive to pharmacological inhibition of endogenous NOS (Moore et al., 2010; Moore, Sanz‐Rosa, & Emerson, 2011). These complementary approaches provided further evidence that CBV‐TP, but not TFV, could block NO‐mediated inhibition of platelet aggregation and that ABC, but neither TDF nor TAF, led to enhanced platelet aggregation in vivo. Given the central role of the endothelium in generating NO and the potential effect on NO‐mediated platelet inhibition, these data suggest that ABC exposure may be problematic in the context of endothelial dysfunction, which is in line with current clinical guidelines that ABC should be used with caution in patients with elevated cardiovascular risk (EACS, 2017). The endothelium also generates prostacyclin, which inhibits platelet activation through induction of the adenylate cyclase/cAMP axis (Schwarz, Walter, & Eigenthaler, 2001). We found that NRTIs did not affect this pathway but, rather, selectively affect the NO/cGMP axis (Figure 3c). Our proposed model for ABC‐mediated cardiovascular risk is summarized in Figure 6.
In summary, the data presented here provide evidence that ABC can pharmacologically alter platelet function in vitro and in vivo, whereas the TFV‐based therapies TDF and TAF had no effect. These observations could explain epidemiological and clinical observations linking ABC with increased incidence of platelet‐driven cardiovascular events such as MI (D:A:D Study Group et al., 2008; Islam et al., 2012). Our study design utilized human HIV‐negative donors, comparative drug analysis in platelets from a single donor and inbred mice, meaning that the only variable between experimental groups was drug exposure and that effects reported here can reasonably be attributed to ABC. A caveat to our study remains the relevance of the concentrations and doses employed to patients' drug exposure. Further work should be conducted to correlate drug exposure in basic science studies with clinically relevant doses. It is important to highlight that the NRTIs used in this study are fundamentally safe drugs that can not only effectively treat HIV but also prevent infection in people who are at high risk of acquiring the virus. The conflicting findings of previous studies and the failure to consistently detect a risk signal with ABC suggest that any effect on cardiovascular health remains subtle and is more than offset by the value of ABC in the treatment of HIV and prevention of AIDS. Nonetheless, it remains important to fully understand co‐morbidities associated with HIV and the role of ART in driving them, in order to tailor ART to the changing needs and circumstances of the individual, allowing them to age and live healthily.
AUTHOR CONTRIBUTIONS
K.T., E.S., and F.R. designed experimental protocols, performed experiments, analysed data, and drafted the manuscript. M.E. conceived and acquired funding for the study, designed experimental protocols, drafted, and had final approval of the manuscript. M.B. and M.C. designed the clinical trial and drafted the manuscript. A.K., B.G., and M.N. were involved in study concept and edited the manuscript.
CONFLICT OF INTEREST
This study was funded by an Investigator‐led grant from Gilead Sciences, who manufacture two of the drugs used (TAF and TDF).
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1:
In vitro responses of washed platelet suspensions are not affected by incubation with ART.
Figure S2: In vitro platelet aggregation responses are not affected by incubation with ART
Figure S3: Plasma Cmax levels of ART do not alter collagen‐evoked platelet aggregation.
Figure S4: ADP‐ and TRAP6‐evoked expression of platelet activation markers is not altered by exposure to ART
ACKNOWLEDGEMENTS
Transgenic mice were bred and maintained by Miss Katarzyna E Smigielska. This work was funded by a Gilead Sciences investigator‐led grant, a St Stephen's AIDS Trust Research Proposal grant, and a National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs) project grant (NC/M000079/1).
Taylor KA, Smyth E, Rauzi F, et al. Pharmacological impact of antiretroviral therapy on platelet function to investigate human immunodeficiency virus‐associated cardiovascular risk. Br J Pharmacol. 2019;176:879–889. 10.1111/bph.14589
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. 10.1111/bph.13876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, A. , Orden, S. , Andujar, I. , Collado‐diaz, V. , Nunez‐Delgado, S. , Galindo, M. J. , … Esplugues, J. V. (2017). Cardiovascular toxicity of abacavir: A clinical controversy in need of a pharmacological explanation. Aids, 31, 1781–1795. 10.1097/QAD.0000000000001547 [DOI] [PubMed] [Google Scholar]
- Alvarez, A. , Rios‐Navarro, C. , Blanch‐Ruiz, M. A. , Collado‐Diaz, V. , Andujar, I. , Martinez‐Cuesta, M. A. , … Esplugues, J. V. (2017). Abacavir induces platelet‐endothelium interactions by interfering with purinergic signalling: A step from inflammation to thrombosis. Antiviral Research, 141, 179–185. 10.1016/j.antiviral.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Antiretroviral Therapy Cohort Collaboration (2017). Survival of HIV‐positive patients starting antiretroviral therapy between 1996 and 2013: A collaborative analysis of cohort studies. Lancet HIV, 4, e349–e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostoli, G. L. , Solomon, A. , Smallwood, M. J. , Winyard, P. G. , & Emerson, M. (2014). Role of inorganic nitrate and nitrite in driving nitric oxide‐cGMP‐mediated inhibition of platelet aggregation in vitro and in vivo. Journal of Thrombosis and Haemostasis, 12, 1880–1889. 10.1111/jth.12711 [DOI] [PubMed] [Google Scholar]
- Baum, P. D. , Sullam, P. M. , Stoddart, C. A. , & McCune, J. M. (2011). Abacavir increases platelet reactivity via competitive inhibition of soluble guanylyl cyclase. Aids, 25, 2243–2248. 10.1097/QAD.0b013e32834d3cc3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, A. I. , Vittinghoff, E. , Deeks, S. G. , Weekley, C. C. , Li, Y. , & Shlipak, M. G. (2011). Cardiovascular risks associated with abacavir and tenofovir exposure in HIV‐infected persons. Aids, 25, 1289–1298. 10.1097/QAD.0b013e328347fa16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D:A:D Study Group , Sabin, C. A. , Worm, S. W. , Weber, R. , Reiss, P. , El‐Sadr, W. , … Lundgren, J. D. (2008). Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV‐infected patients enrolled in the D:A:D study: A multi‐cohort collaboration. Lancet, 371, 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks, S. G. , Tracy, R. , & Douek, D. C. (2013). Systemic effects of inflammation on health during chronic HIV infection. Immunity, 39, 633–645. 10.1016/j.immuni.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo, Y. L. , Ollivier, V. , Joly, V. , Faille, D. , Catalano, G. , Jandrot‐Perrus, M. , … Ajzenberg, N. (2016). Abacavir has no prothrombotic effect on platelets in vitro. The Journal of Antimicrobial Chemotherapy, 71, 3506–3509. 10.1093/jac/dkw303 [DOI] [PubMed] [Google Scholar]
- Ding, X. , Andraca‐Carrera, E. , Cooper, C. , Miele, P. , Kornegay, C. , Soukup, M. , & Marcus, K. A. (2012). No association of abacavir use with myocardial infarction: Findings of an FDA meta‐analysis. Journal of Acquired Immune Deficiency Syndromes, 61, 441–447. 10.1097/QAI.0b013e31826f993c [DOI] [PubMed] [Google Scholar]
- European AIDS Clinical Society (2017). European AIDS Clinical Society (EACS) Guidelines Version 9.0.
- Friis‐Moller, N. , Sabin, C. A. , Weber, R. , d'Arminio Monforte, A. , El‐Sadr, W. M. , Reiss, P. , … The Data Collection on Adverse Events of Anti‐HIV Drugs (DAD) Study Group (2003). Combination antiretroviral therapy and the risk of myocardial infarction. The New England Journal of Medicine, 349, 1993–2003. 10.1056/NEJMoa030218 [DOI] [PubMed] [Google Scholar]
- Friis‐Moller, N. , Weber, R. , Reiss, P. , Thiebaut, R. , Kirk, O. , d'Arminio Monforte, A. , … DAD study group (2003). Cardiovascular disease risk factors in HIV patients‐association with antiretroviral therapy. Results from the DAD study. AIDS, 17, 1179–1193. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam, F. M. , Wu, J. , Jansson, J. , & Wilson, D. P. (2012). Relative risk of cardiovascular disease among people living with HIV: A systematic review and meta‐analysis. HIV Medicine, 13, 453–468. [DOI] [PubMed] [Google Scholar]
- Jones, C. I. , Tucker, K. L. , Sasikumar, P. , Sage, T. , Kaiser, W. J. , Moore, C. , … Gibbins, J. M. (2014). Integrin‐linked kinase regulates the rate of platelet activation and is essential for the formation of stable thrombi. Journal of Thrombosis and Haemostasis, 12, 1342–1352. 10.1111/jth.12620 [DOI] [PubMed] [Google Scholar]
- Jones, S. , Solomon, A. , Sanz‐Rosa, D. , Moore, C. , Holbrook, L. , Cartwright, E. J. , … Emerson, M. (2010). The plasma membrane calcium ATPase modulates calcium homeostasis, intracellular signaling events and function in platelets. Journal of Thrombosis and Haemostasis, 8, 2766–2774. 10.1111/j.1538-7836.2010.04076.x [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. J. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biology, 8, e1000412 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon, P. W. , Winston, A. , Post, F. , Kenny, D. , Bergin, C. , Maughan, R. T. , … Rhee, M. (2018). Platelet function upon switching to TAF vs continuing ABC: A randomised substudy. Conference on Retroviruses and Opportunistic Infections (CROI). Boston, Massachussets.
- Massud, I. , Mitchell, J. , Babusis, D. , Deyounks, F. , Ray, A. S. , Rooney, J. F. , … Garcia‐Lerma, J. G. (2016). Chemoprophylaxis with oral emtricitabine and tenofovir alafenamide combination protects macaques from rectal simian/human immunodeficiency virus infection. The Journal of Infectious Diseases, 214, 1058–1062. 10.1093/infdis/jiw312 [DOI] [PubMed] [Google Scholar]
- Moore, C. , Sanz‐Rosa, D. , & Emerson, M. (2011). Distinct role and location of the endothelial isoform of nitric oxide synthase in regulating platelet aggregation in males and females in vivo. European Journal of Pharmacology, 651, 152–158. 10.1016/j.ejphar.2010.11.011 [DOI] [PubMed] [Google Scholar]
- Moore, C. , Tymvios, C. , & Emerson, M. (2010). Functional regulation of vascular and platelet activity during thrombosis by nitric oxide and endothelial nitric oxide synthase. Thrombosis and Haemostasis, 104, 342–349. 10.1160/TH09-11-0764 [DOI] [PubMed] [Google Scholar]
- Munoz, R. P. , Gonzalez‐Correa, J. A. , Ruiz, J. , Nuno, E. , Marquez, M. , Cruz, J. P. D. L. , & Santos, J. (2012). Whole blood platelet aggregometry in HIV‐infected patients on treatment with abacavir. Open Journal of Internal Medicine, 2(2), 5. [Google Scholar]
- O'Halloran, J. A. , Dunne, E. , Tinago, W. , Denieffe, S. , Kenny, D. , & Mallon, P. W. G. (2018). Switching from abacavir to tenofovir disoproxil fumarate is associated with rises in soluble glycoprotein VI, suggesting changes in platelet‐collagen interactions. Aids, 32, 861–866. 10.1097/QAD.0000000000001783 [DOI] [PubMed] [Google Scholar]
- Pollack, T. M. , & Rind, D. M. (2007). Antiretroviral drugs and the risk of myocardial infarction. The New England Journal of Medicine, 357, 716. author reply 716‐7 [PubMed] [Google Scholar]
- Post, F. A. , Yazdanpanah, Y. , Schembri, G. , Lazzarin, A. , Reynes, J. , Maggiolo, F. , … Rhee, M. S. (2017). Efficacy and safety of emtricitabine/tenofovir alafenamide (FTC/TAF) vs. emtricitabine/tenofovir disoproxil fumarate (FTC/TDF) as a backbone for treatment of HIV‐1 infection in virologically suppressed adults: Subgroup analysis by third agent of a randomized, double‐blind, active‐controlled phase 3 trial<sup/>. HIV Clinical Trials, 18, 135–140. 10.1080/15284336.2017.1291867 [DOI] [PubMed] [Google Scholar]
- Sabin, C. A. , Reiss, P. , Ryom, L. , Phillips, A. N. , Weber, R. , Law, M. , … D:A:D Study Group (2016). Is there continued evidence for an association between abacavir usage and myocardial infarction risk in individuals with HIV? A cohort collaboration. BMC Medicine, 14, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchell, C. S. , Cotter, A. G. , O'Connor, E. F. , Peace, A. J. , Tedesco, A. F. , Clare, A. , … Mallon, P. W. (2010). Platelet function and HIV: A case‐control study. Aids, 24, 649–657. 10.1097/QAD.0b013e328336098c [DOI] [PubMed] [Google Scholar]
- Satchell, C. S. , O'Halloran, J. A. , Cotter, A. G. , Peace, A. J. , O'Connor, E. F. , Tedesco, A. F. , … Mallon, P. W. (2011). Increased platelet reactivity in HIV‐1‐infected patients receiving abacavir‐containing antiretroviral therapy. The Journal of Infectious Diseases, 204, 1202–1210. 10.1093/infdis/jir509 [DOI] [PubMed] [Google Scholar]
- Sax, P. E. , Wohl, D. , Yin, M. T. , Post, F. , DeJesus, E. , Saag, M. , … GS‐US‐292‐0104/0111 Study Team (2015). Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV‐1 infection: Two randomised, double‐blind, phase 3, non‐inferiority trials. Lancet, 385, 2606–2615. 10.1016/S0140-6736(15)60616-X [DOI] [PubMed] [Google Scholar]
- Schwarz, U. R. , Walter, U. , & Eigenthaler, M. (2001). Taming platelets with cyclic nucleotides. Biochemical Pharmacology, 62, 1153–1161. 10.1016/S0006-2952(01)00760-2 [DOI] [PubMed] [Google Scholar]
- Tymvios, C. , Jones, S. , Moore, C. , Pitchford, S. C. , Page, C. P. , & Emerson, M. (2008). Real‐time measurement of non‐lethal platelet thromboembolic responses in the anaesthetized mouse. Thrombosis and Haemostasis, 99, 435–440. 10.1160/TH07-07-0479 [DOI] [PubMed] [Google Scholar]
- Tymvios, C. , Moore, C. , Jones, S. , Solomon, A. , Sanz‐Rosa, D. , & Emerson, M. (2009). Platelet aggregation responses are critically regulated in vivo by endogenous nitric oxide but not by endothelial nitric oxide synthase. British Journal of Pharmacology, 158, 1735–1742. 10.1111/j.1476-5381.2009.00408.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2016). Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection; Recommendations for a Public Health Approach (second edition). [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1:
In vitro responses of washed platelet suspensions are not affected by incubation with ART.
Figure S2: In vitro platelet aggregation responses are not affected by incubation with ART
Figure S3: Plasma Cmax levels of ART do not alter collagen‐evoked platelet aggregation.
Figure S4: ADP‐ and TRAP6‐evoked expression of platelet activation markers is not altered by exposure to ART