

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease for which the origin and underlying cellular defects are not fully understood. Although motor neuron degeneration is the signature feature of ALS, it is not clear whether motor neurons or other cells of the motor circuit are the site of disease initiation. To better understand the contribution of multiple cell types in ALS, we made use of a Drosophila Sod1G85R knock-in model, in which all cells harbor the disease allele. End-stage dSod1G85R animals of both sexes exhibit severe motor deficits with clear degeneration of motor neurons. Interestingly, earlier in dSod1G85R larvae, motor function is also compromised, but their motor neurons exhibit only subtle morphological and electrophysiological changes that are unlikely to cause the observed decrease in locomotion. We analyzed the intact motor circuit and identified a defect in sensory feedback that likely accounts for the altered motor activity of dSod1G85R. We found cell-autonomous activation of bone morphogenetic protein signaling in proprioceptor sensory neurons which are critical for the relay of the contractile status of muscles back to the central nerve cord, completely rescues early-stage motor defects and partially rescue late-stage motor function to extend lifespan. Identification of a defect in sensory feedback as a potential initiating event in ALS motor dysfunction, coupled with the ability of modified proprioceptors to alleviate such motor deficits, underscores the critical role that nonmotor neurons play in disease progression and highlights their potential as a site to identify early-stage ALS biomarkers and for therapeutic intervention.
SIGNIFICANCE STATEMENT At diagnosis, many cellular processes are already disrupted in the amyotrophic lateral sclerosis (ALS) patient. Identifying the initiating cellular events is critical for achieving an earlier diagnosis to slow or prevent disease progression. Our findings indicate that neurons relaying sensory information underlie early stage motor deficits in a Drosophila knock-in model of ALS that best replicates gene dosage in familial ALS (fALS). Importantly, studies on intact motor circuits revealed defects in sensory feedback before evidence of motor neuron degeneration. These findings strengthen our understanding of how neural circuit dysfunctions lead to neurodegeneration and, coupled with our demonstration that the activation of bone morphogenetic protein signaling in proprioceptors alleviates both early and late motor dysfunction, underscores the importance of considering nonmotor neurons as therapeutic targets.
Keywords: ALS, BMP signaling, Drosophila, motor circuit, sensory neurons
Introduction
Amyotrophic lateral sclerosis (ALS) is currently the most common motor neuron disease, at 1.9 cases per 100,000 people, with a projected global increase over the next 25 years (Chiò et al., 2013; Arthur et al., 2016). Most ALS patients experience a rapid decline in motor function due to the loss of motor neurons that ultimately results in respiratory failure and death within 3–5 years of diagnosis (Kiernan et al., 2011). Motor neurons of ALS patients exhibit changes in excitability, have cytoplasmic inclusions, and lose synaptic connectivity at the neuromuscular junction (NMJ) (Denys and Norris, 1979; Tsujihata et al., 1984; Lowe et al., 1988; Kanai et al., 2006; Vucic et al., 2008; Iwai et al., 2016). Mutations in at least 50 genes have been linked to ALS (Taylor et al., 2016), many of which have broad expression domains encompassing many different cell types. The rapid and dramatic loss in the structural and functional integrity of motor neurons is typical of ALS; however, defects in glia, sensory neurons, cortical neurons, and skeletal muscle have all been noted in the literature (Tandan and Bradley, 1985; Theys et al., 1999; Zanette et al., 2002; Pugdahl et al., 2007; Taylor et al., 2016). Efforts to clarify the cellular origin of ALS-associated dysfunction will not only provide an understanding of the molecular and cellular basis of ALS progression, but will also inform new therapeutic strategies to treat or even prevent motor neuron degeneration and loss.
In an effort to study the consequences of ALS-associated mutations in the context of the whole organism, where all affected cells are present and neuronal connections maintained, we made use of a Drosophila Sod1G85R knock-in model (Sahin et al., 2017). This dSod1G85R model harbors a mutation in the endogenous dSod1 locus, synonymous with the human SOD1G85R mutation (Rosen et al., 1993), ensuring that its expression is physiologically relevant. SOD1 is expressed ubiquitously and SOD1G85R is compromised in its ability to eliminate the cellular build-up of superoxide while also causing a poorly understood toxic gain-of-function (Saccon et al., 2013; Sahin et al., 2017). Indicative of motor dysfunction, dSod1G85R adults fail to emerge from their pupal cases and subsequently die. Before this time, dSod1G85R third instar larvae exhibit reduced locomotion (Sahin et al., 2017). Larval crawling is a relatively simple behavior that requires a wave of contraction and relaxation of segmentally arranged muscles (Fox et al., 2006; Lahiri et al., 2011; Berni et al., 2012). A detailed knowledge of the larval motor circuit and the availability of genetic tools make Drosophila an ideal model to investigate the cellular and electrical basis of locomotor defects that arise from an ALS-causing mutation (Hughes and Thomas, 2007; Berni et al., 2012; Pulver et al., 2015).
Here, we report a detailed analysis of phenotypic defects associated with dSod1G85R at multiple time points in disease progression. Our data identify cells where defects first appear and a means to restore motor function in this ALS model. End-stage dSod1G85R animals, the motor function of which is severely impaired, show a gross disruption in NMJ morphology that is accompanied by diminished neurotransmission. By contrast, the impaired locomotion of earlier-stage animals is not accompanied by a clear degeneration of the nerve–muscle synapse. Instead, we detect a disruption in nonmotor neurons that mediate sensory feedback from the periphery to the interneuron network of the CNS. This defect acts to slow the overall locomotor pattern of dSod1G85R larvae. We also show that activation of bone morphogenetic protein (BMP) signaling in nonmotor neurons, as well as in motor neurons, can alleviate the abnormal motor function evident in both early- and end-stage dSod1G85R animals. Our findings implicate nonmotor neurons as causative in early-stage ALS-induced motor dysfunction and as potential targets of therapeutic strategies aimed at preventing or slowing the ultimate degeneration of motor neurons.
Materials and Methods
Fly stocks and culture
The w;+;dSod1WTLoxP and w;+;dSod1G85R/TM3SerGFP lines (Sahin et al., 2017), UAS-gbb9.9 (Khalsa et al., 1998), UAS-SaxA (Michael O'Connor), OK6-Gal4 (BSC#64199, RRID:BDSC_64199), OK371-Gal4 (P{GawB}VGlutOK371, BSC#26160, RRID:BDSC_26160), MD-Gal4 (P{GawB}109(2)80, BSC#8769, RRID:BDSC_8769), ChAT-Gal4 (P{ChAT-GAL4.7.4}, BSC#6798, RRID:BDSC_6798), Repo-Gal4 (BSC#7415, RRID:BDSC_7415), 2-21-Gal4 (John Thomas), UAS-mRFP (membrane bound Red Fluorescent Protein; Robert Reenan), UAS-GFP.nls (BSC#4775, RRID:BDSC_4775), and BG57-Gal4 (33) were used to construct w;+;dSod1G85R/TM6C, w;OK371-Gal4;dSod1G85R/TM6C, w;UAS-gbb9.9;dSod1G85R/TM6C, w;+; BG57-Gal4,dSod1G85R/TM6C, w;OK6-Gal4;dSod1G85R/TM6C, w;MD-Gal4;dSod1G85R/TM6C, w;+;2-21-Gal4;dSod1G85R/TM6C, w;UAS-mRFP;dSod1G85R/TM6C, w;OK371-Gal4;dSod1WTLoxP, w;+;2-21-Gal4;dSod1WTLoxP, w;UAS-mRFP;dSod1WTLoxP, and w;UAS-gbb9.9;dSod1WTLoxP. Expression patterns for Gal4 lines were found to be as described previously (Budnik et al., 1996; Salvaterra and Kitamoto, 2001; Sepp et al., 2001; Mahr and Aberle, 2006; Hughes and Thomas, 2007; Sanyal, 2009). Flies were raised at 25°C on standard cornmeal/sugar/agar food. For all experiments, two or three males were crossed to three female virgins and brooded daily to control for progeny density. Both sexes of the experimental class were used to conduct experiments.
Staging and dissection
Pharate NMJ.
Pharates (fully developed adults before eclosion) were staged based on the position of meconium along the anterior/posterior axis, with midway down the dorsal abdomen corresponding to 9–10 h before expected eclosion (Crossley, 1979). All pharates used for immunofluorescence or electrophysiology experiments were confirmed to be alive based on visible beating of the dorsal vessel. Pharate dissections essentially as described previously (Hebbar et al., 2006). Briefly, pharate was pulled from pupal case using forceps and placed in either 1× PBS (immunofluorescence experiments) or modified HL3.1 (electrophysiology). The pharate was pinned at its anterior and posterior ends dorsal side up and cut dorsally from posterior to anterior through the abdomen and thorax. Four additional pins were used to pin the pharate open, exposing the ventral muscles. Trachea, intestines, and reproductive organs were removed to expose the ventral muscles. Nerves connecting the ventral nerve cord (VNC) to body wall muscles were cut to allow clearer visualization of the body wall.
Pharate legs.
For leg extension assay, pharates were staged as for NMJ dissections and imaged using an Olympus SZX12 microscope with an OptixCam Summit D3K2–5 camera. Images were analyzed using ImageJ. Legs were dissected from pharates in PBS, mounted in 80% glycerol, and imaged using a Zeiss Axio Imager.M1 microscope. In the assignment of a leg integrity score, experimenters were blinded to the genotype and a score of nerve integrity (low = 1 to high = 5) was assigned based on the continuity of the main nerve bundle and the number of collateral projections.
Third instar larvae.
Wandering third instar larvae were identified as larvae >108 h after egg laying that had crawled out of the food and had not yet everted their anterior spiracles. Dissections were done in PBS (immunofluorescence), modified HL3.1 (NMJ electrophysiology), or in a modified saline solution (fictive crawling and patching). Wandering third instar larvae were fileted as described previously (Jan and Jan, 1976).
Immunoflourescence
Pharates and larvae.
Pharate or larval filets of the appropriate stage (see “Staging and dissection” section) were fixed for 15–20 min in 4% formaldehyde in 1× PBS (16% paraformaldehyde; Electron Microscopy Sciences). After dissection and fixation, filets were blocked for 1 h in 0.3% normal goat serum in 1× PBT (0.3% Triton X-100 in 1× PBS) at room temperature (RT) and then incubated overnight at 4°C with primary antibodies. Filets were then washed 2 × 5 min with 1× PBT and incubated for 1 h in secondary antibody solution at RT. Filets were washed 2× 5 min in 1× PBS, mounted in 80% glycerol with 0.5% N-propyl-gallate (Sigma-Aldrich), and imaged using a Zeiss LSM800 confocal microscope. The experimenter was blinded to genotype before counting boutons and those counts represent the number of boutons per NMJ. The experimenter was also blinded while performing Sholl analysis and quantifying immunofluorescence. Primary antibodies and stains used in this study were as follows: mouse anti-Dlg (4F3, DSHB, RRID:AB_528203, at 1:200), rabbit anti-HRP-Cy3 (Jackson ImmunoResearch, RRID:AB_2340262, at 1:300), goat anti-HRP-647 (Jackson ImmunoResearch, RRID:AB_2338967, at 1:300), rabbit anti-pSmad3 (Abcam ab52903, RRID:AB_882596, at 1:300), rabbit anti-pSmad1 (Peter Ten Djike, at 1:1500), rabbit anti-RFP (Rockland, RRID:AB_2209751, at 1:500), mouse anti-BRP (nc82, DSHB, RRID:AB_2314866, at 1:50), chicken anti-GFP (Life Technologies A10262, RRID:AB_2534023, at 1:000), and phalloidin-488 (Life Technologies, RRID:AB_2315147, at 1:2500). Secondary antibodies were as follows: goat anti-mouse 647 (Life Technologies, RRID:AB_2535804, at 1:300), goat anti-chicken 488 (Life Technologies, RRID:AB_2534096, at 1:300), and goat anti-rabbit 568 (Life Technologies, RRID:AB_143157, at 1:300).
Electrophysiology
Adult NMJ.
Pharates were dissected from the pupal case (see staging and dissection) in modified HL3.1 containing the following (in mm): 70 NaCl, 5 KCl, 4 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, 5 mm HEPES, and 1 mm CaCl2, pH 7.2. The four large ventral muscles (VMs) in abdominal segment 2 (A2) were recorded from using 30MΩ sharp electrodes filled with 3 M KCl. After successfully entering one of the four large ventral muscles in A2, a 200 pA current was injected for 250 ms for 50 sweeps. The average sweep of the RC curves was used to determine the resistance and capacitance of the muscle. Only muscles with an input resistance >10 MΩ and a resting membrane potential <−40 mV were used for analysis. For recordings of miniature EPSPs (mEPSPs), negative current was injected to bring the membrane potential to ∼−55 mV and mEPSPs were recorded for 3 min. Recordings were made using a Multiclamp 700A, digitized with a Digidata 1550A, filtered at 10 kHz, and analyzed using custom MATLAB (The MathWorks) scripts.
Larval NMJ.
Wandering third instar larvae were fileted in modified HL3.1 containing the following (in mm): 70 NaCl, 5 KCl, 10 MgCl2, 10 NaHCO3, 5 trehalose, 115 sucrose, 5 HEPES, and 0.5 mm CaCl2, pH 7.2. Recordings at muscle 6 in segment A3 were done using 20–25 MΩ sharp electrodes filled with 3 M KCl. After successfully entering the muscle, a 1 nA current was injected for 150 ms for 50 sweeps. The average sweep of the RC curves was used to determine the resistance and capacitance of the muscle. Only muscles with an input resistance >5 MΩ and a resting membrane potential <−60 mV were used for analysis. Evoked EPSPs were measured by suctioning a portion of the cut nerve into a suction electrode and passing a square 0.3 ms pulse of current from a stimulation unit to evoke a compound action potential. A minimum of 10 eEPSPs were averaged for each larva. mEPSPs were measured for 3 min in the absence of nerve stimulation. A second electrode was then inserted, muscle resistance was checked again, and the muscle was voltage clamped at −80 mV. EPSCs were evoked by passing a 0.3 ms pulse of current from a stimulation unit and the gain was adjusted to reduce voltage error to <5 mV. Recordings were made using an Axoclamp 2B, digitized with a Digidata 1322A, and analyzed using custom MATLAB scripts. The experimenter was blinded during data acquisition and analysis.
Muscle contraction.
Wandering third instar larvae were fileted in a modified saline solution containing the following (in mm): 128 NaCl, 2 KCl, 35 sucrose, 4 MgCl2, 5 HEPES, and 2 CaCl2, pH 7.2. The severed main nerve bundle innervating an A3 hemisegment was suctioned into an electrode as in the NMJ experiments and the nerve bundle was stimulated by applying square pulses of 0.3 ms with the same current amplitude used in the larval NMJ experiments to evoke postsynaptic potentials. The nerve bundle was then stimulated for 200 ms for three trials at 10, 25, 50, and 100 Hz. Body wall responses were observed using a Nikon FN1 microscope with an OptixCam Summit D3K2–5 camera at 105 frames/s. The experimenter was blinded to genotype and stimulus frequency. The length of the relaxed muscle was compared with the length of the muscle at maximum contraction during the stimulation. The results of the three trials for each stimulus intensity were averaged.
Fictive crawling.
Larvae of the appropriate stage were dissected in a modified saline solution containing the following (in mm): 128 NaCl, 2 KCl, 35 sucrose, 4 MgCl2, 5 HEPES, and 3 CaCl2, pH 7.2 (based on Jan and Jan, 1976), leaving the CNS intact. The majority of larvae began to fictive crawl and larvae that did not crawl or respond to a light touch were discarded. A portion of one of the nerves innervating segments A3–A7 was tightly suctioned into an electrode filled with modified saline solution and activity was recorded for 10 min. Bursts of nerve activity always correlated with peristaltic muscle activity (Cattaert and Birman, 2001; Fox et al., 2006). The paired fictive crawling recordings were done using the same modified saline solution, but with 1.5 mm CaCl2 to reduce background activity when the CNS was isolated. After recording from intact preparations for 6 min, the CNS was removed from the body wall, a portion of the severed nerve attached to the VNC was suctioned into the electrode, and recording continued for another 6 min. Recordings were done using a Multiclamp 700A amplifier, AC filtered at 1 Hz, digitized with a Digidata 1550A, and analyzed using Clampfit 10.6. The frequency of bursting events was analyzed, but the amplitude of bursting events was not analyzed because it is highly dependent upon the resistance of the electrode–nerve junction.
Motor neuron patching.
The CNS from wandering third instar larvae were dissected in modified saline solution containing the following (in mm): 128 NaCl, 2 KCl, 35 sucrose, 4 MgCl2, 5 HEPES, and 2 CaCl2, pH 7.2 and glued (Vetbond) to a Sylgard plate. After clearing the glial layer by applying 1% protease XIV (Sigma-Aldrich) locally with a broken electrode using alternating positive and negative pressure, aCC motor neurons were identified by position and morphology (Choi et al., 2004; Marley and Baines, 2011) and patched with 3–5 MΩ patch electrodes filled with internal solution containing the following (in mm): 150 KCH3SO3, 2 MgCl2, 2 EGTA, 5 KCl, and 20 HEPES, pH 7.35. After light suction to enter whole-cell configuration, series resistance was compensated and current steps (−20 pA to 160 pA in steps of 10 pA, 0.5 s duration) were applied in triplicate. Sweeps that were influenced by large EPSPs were discounted from analysis. Cells were then voltage clamped at −70 mV and cell capacitance and series resistance were compensated. Spontaneous rhythmic currents (amplitude >300 pA) were recorded for >1.5 min in gap free mode. Data were analyzed using custom MATLAB scripts.
Behavior
Larval distance.
Two to five larvae were placed on a 22-cm-diameter dish filled with 1% agarose. Ninety-second videos were taken using a Dinolite AM3111 camera and analyzed using custom MATLAB scripts.
Larval peristalsis.
Single larvae were placed on a 14 cm dish filled with 1% agarose. 60 s videos were taken using a SZX12 stereomicroscope with an OpitxCam Summit D3K2–5 camera. The experimenter was blinded for the analysis and videos were analyzed manually to determine the number of peristaltic contractions. The first five forward peristaltic motions were analyzed frame by frame to determine the crawling wave duration.
Eclosion.
dSod1G85R mutants were identified by the absence of the balancer TM6C,SbTb. Pupal cases were counted 13 d after egg laying and identified as full or empty, indicating whether the adult successfully eclosed. Successful eclosion was verified by the observation of Sb+ flies.
Experimental design and statistical analysis
Comparison of continuous data.
For comparisons of two genotypes, we used a Student's t test, accounting for the variance and pairing of the data when necessary. Two-tailed t tests where used to analyze most experiments. One-tailed tests were used when the hypothesis specified an effect direction. When a Shapiro–Wilk normality test indicated that the data were not normally distributed, we used a Wilcoxon rank-sum test. When comparing more than two groups, a Holm–Bonferroni correction for multiple hypotheses was applied. Graphs indicate samples (black dots), mean (vertical red line), and SE (horizontal red lines) in Figure 1C,D, 2A,B, 3B–E,G, 4A,C,E, 5C,D,I, 6D–F, 7D, 8C, and 9B with *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 1.
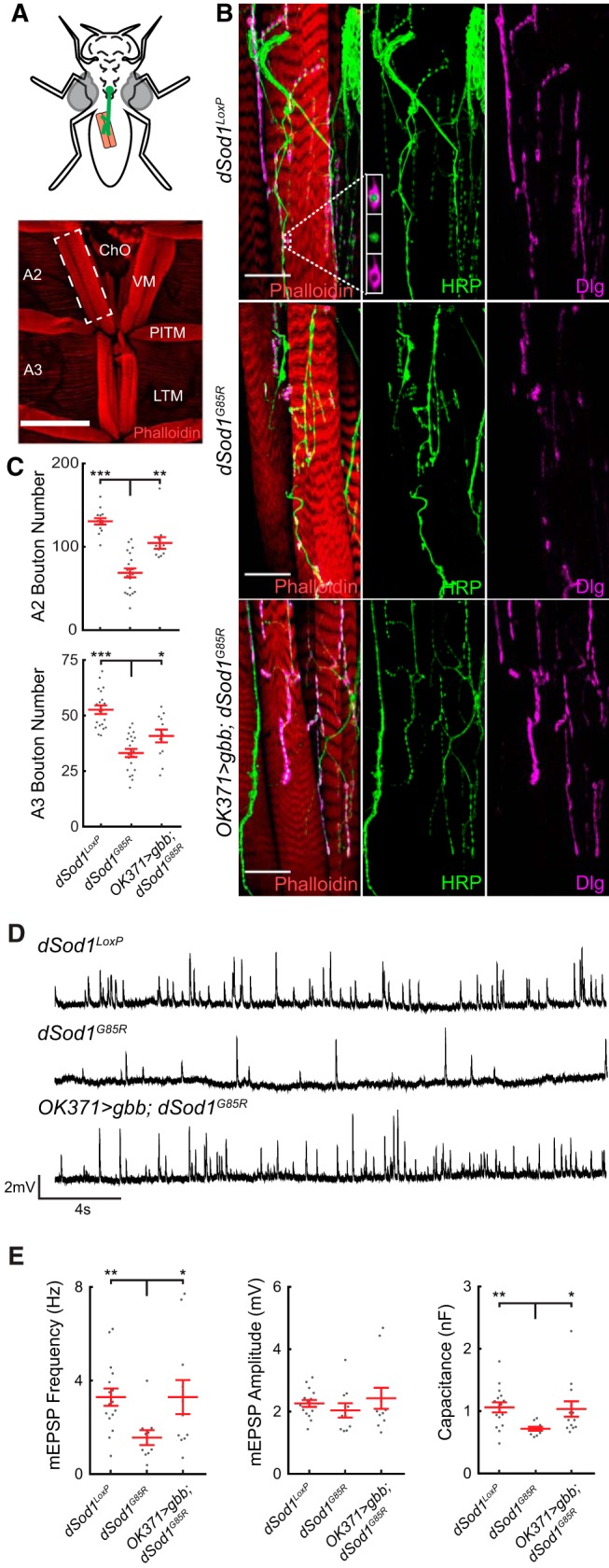
dSod1G85R mutants exhibit a reduction in bouton number, mEPSP frequency, and muscle capacitance in adult abdominal NMJs that can be rescued by the BMP ligand Gbb. A, Abdominal musculature of pharates is critical for eclosion. Ventral muscles in abdominal segments A2 and A3 are shown (red, phalloidin). B, VM NMJ in A2 hemisegment (white dashed box). High magnification of dSod1WTLoxP, dSod1G85R, and OK371-Gal4/UAS-Gbb9.9;dSod1G85R VM NMJ, presynaptic (green, anti-HRP), and postsynaptic membranes (magenta, anti-Dlg) are shown. Boutons are evident as presynaptic swellings along axonal branches. C, Bouton number is decreased in dSod1G85R mutants and increased in OK371-Gal4/UAS-gbb9.9;dSod1G85R VM NMJs (Segment A2: dSod1WTLoxP = 130.5 ± 3.8 boutons, n = 14; dSod1G85R = 68.7 ± 5.3 boutons, n = 20; OK371-Gal4/UAS-gbb9.9;dSod1G85R = 104.5 ± 7.0, n = 11; dSod1WTLoxP vs dSod1G85R: p = 2.2 × 10−10, unequal variance t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 1.8 × 10–4, one-tailed t test with Holm–Bonferroni correction. Segment A3: dSod1WTLoxP = 52.6 ± 1.9 boutons, n = 20; dSod1G85R = 33.1 ± 1.8 boutons, n = 21; OK371-Gal4/UAS-gbb9.9;dSod1G85R = 40.8 ± 2.9, n = 12; dSod1WTLoxP vs dSod1G85R: p = 1.6 × 10−8, t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 0.012, one-tailed t test with Holm–Bonferroni correction). D, Representative traces of miniature EPSPs (mEPSPs) from dSod1WTLoxP, dSod1G85R, and OK371-Gal4/UAS-Gbb9.9;dSod1G85R VM. E, mEPSP frequency and muscle capacitance are decreased in dSod1G85R, mEPSP amplitude is not. Expression of gbb in glutamatergic neurons in dSod1G85R increases mEPSP frequency and muscle capacitance (mEPSP frequency: dSod1WTLoxP = 3.29 ± 0.37 s−1, n = 16; dSod1G85R = 1.56 ± 0.32 s−1, n = 10; OK371-Gal4/UAS-gbb9.9;dSod1G85R = 3.29 ± 0.73 s−1, n = 11; dSod1G85R vs dSod1WTLoxP: p = 0.0071, t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 0.024, one-sided unequal variance t test with Holm–Bonferroni correction. mEPSP amplitude: dSod1WTLoxP = 2.26 ± 0.11 mV, n = 16; dSod1G85R = 2.04 ± 0.23 mV, n = 10; OK371-Gal4/UAS-gbb9.9;dSod1G85R = 2.43 ± 0.34 mV, n = 11; dSod1WTLoxP vs dSod1G85R: p = 0.67, t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 0.35, unequal variance t test with Holm–Bonferroni correction. muscle capacitance: dSod1WTLoxP = 1.06 ± 0.08nF, n = 16; dSod1G85R: 0.73 ± 0.03nF, n = 10; OK371-Gal4/UAS-gbb9.9;dSod1G85R: 1.03 ± 0.12nF, n = 13; dSod1WTLoxP vs dSod1G85R: p = 0.001, unequal variance t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 0.013, one-sided unequal variance t test with Holm–Bonferroni correction). Scale bars, 150 μm (A) and 25 μm (B). *p < 0.05, **p < 0.01, ***p < 0.001.
Comparison of categorical data.
For comparisons of categorical eclosion data, we used the Fisher's exact test and a Holm–Bonferroni correction for multiple hypotheses when comparing >2 groups. Graphs represent samples (black dots) and median (red line). *p < 0.05, **p < 0.01, and ***p < 0.001.
Repeated-measures data.
For comparisons of datasets with repeated measures, we assessed differences between genotypes using generalized linear mixed-effect models. Graphs in Figures 4G, 5B,E and 7B represent individual samples (lighter lines), means (darker lines), and SE (shaded area). For the analysis of action potential frequency, only the linear portion of the action potential response (0 pA to 100 pA) was used to model firing frequency. *p < 0.05, **p < 0.01, and ***p < 0.001.
Figure 4.
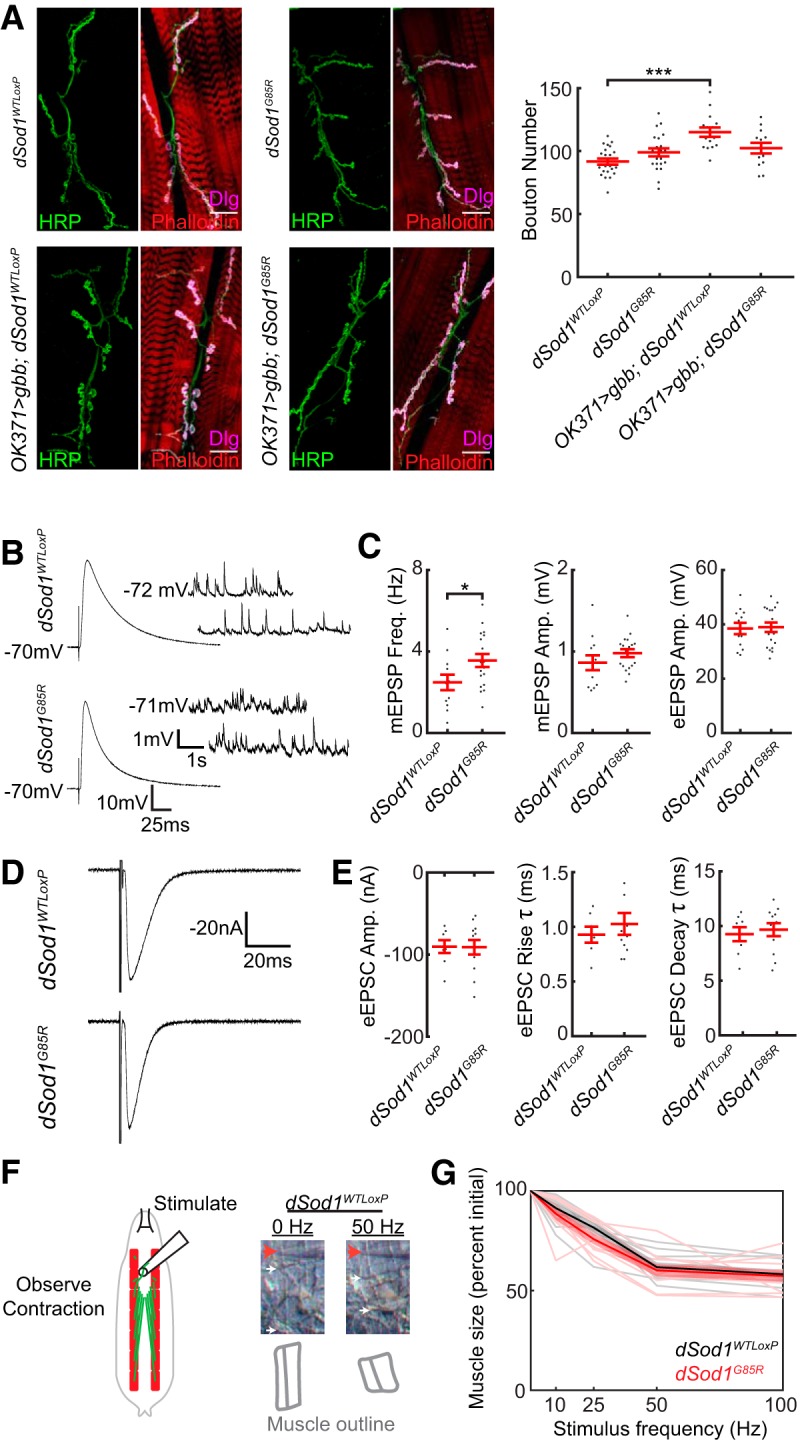
dSod1G85R larval NMJs exhibit only minor defects. A, Representative images of dSod1WTLoxP, OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP, dSod1G85R, and OK371-Gal4/UAS-gbb9.9;dSod1G85R larval NMJs at muscle 6/7 segment A2. Muscles are stained with phalloidin (red), the presynaptic membrane is marked by anti-HRP (green), and the postsynaptic membrane is marked by anti-discs-large (Dlg) (magenta). Quantification of synaptic boutons, the presynaptic swellings that contain neurotransmitter, shows no significant difference between dSod1WTLoxP and dSod1G85R (dSod1WTLoxP = 91.7 ± 2.5 boutons, n = 22; dSod1G85R = 99 ± 3.13 boutons, n = 22; p = 0.14, t test with Holm–Bonferroni correction). The NMJ is expanded in dSod1WTLoxP but not in dSod1G85R when gbb is expressed in glutamatergic neurons under the control of OK371-Gal4 (OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP = 115.0 ± 3.7 boutons, n = 15 vs dSod1WTLoxP = 91.7 ± 2.5 boutons, n = 22; p = 2.7 × 10−5, t test with Holm–Bonferroni correction; OK371-Gal4/UAS-gbb9.9;dSod1G85R: 102.3 ± 4.2 boutons, n = 13; dSod1G85R: 99 ± 3.13 boutons, n = 22; p = 0.26, t test with Holm–Bonferroni correction). B, C, Electrophysiological recordings from dSod1WTLoxP and dSod1G85R show no significant change in eEPSP or mEPSP amplitude, but mEPSP frequency is increased in dSod1G85R (mEPSP amplitude: dSod1WTLoxP = 0.86 ± 0.09 mV, n = 12; dSod1G85R = 0.98 ± 0.05 mV, n = 18; p = 0.23, t test. eEPSP amplitude: dSod1WTLoxP = 38.5 ± 2.1 mV, n = 12; dSod1G85R was 38.9 ± 1.7 mV, n = 18; p = 0.87, t test. mEPSP frequency: dSod1WTLoxP = 2.5 ± 0.38 Hz, n = 12; dSod1G85R = 3.6 ± 0.32 Hz, n = 18; p = 0.04, t test). D, E, Voltage-clamp recordings from dSod1WTLoxP and dSod1G85R show no significant changes in eEPSC amplitude, rise time, or decay time (eEPSC amplitude: dSod1WTLoxP = −90 ± 7.8 nA, n = 8; dSod1G85R = −91 ± 8.8 nA, n = 12; p = 0.96, t test. eEPSC rise time: dSod1WTLoxP = 0.93 ± 0.07 ms, n = 7; dSod1G85R = 1.02 ± 0.10 ms, n = 12; p = 0.50, t test. eEPSC decay time: dSod1WTLoxP = 9.3 ± 0.64 ms, n = 8; dSod1G85R = 9.7 ± 0.60 ms, n = 12; p = 0.65, t test). The averages of 10–15 tracings of eEPSP or eEPSC responses are shown. F, The A3 segmental nerve was stimulated for 200 ms at 10, 25, 50, and 100 Hz and the extent of muscle contraction was determined. The stimulus electrode (red arrow), segmental boundaries (white arrows), and outline of muscles 6/7 (gray) following 0 Hz and 50 Hz stimulation in dSod1WTLoxP are shown. G, Quantification of maximal muscle contraction (expressed as contracted muscle length/relaxed muscle length) in response to motor neuron stimulation (thick lines represent averages, thin lines represent individual muscles, and shaded areas represent SE) shows no difference between dSod1WTLoxP (black) and dSod1G85R (red) (dSod1WTLoxP n = 10 and dSod1G85R n = 10; generalized linear mixed-effect models: genotype effect p = 0.21, stimulus effect p = 1.5 × 10−28). Scale bars, 25 μm. *p < 0.05, ***p < 0.001.
Figure 5.

dSod1G85R larval motor neurons exhibit subtle defects. A, Action potential spikes in response to an 80 pA current step are shown. B, Quantification of the action potential response to current steps from 0 pA to +160 pA for dSod1G85R (red) and dSod1WTLoxP (black) for individual aCC neurons (thin lines) are plotted with the average number of spikes per genotype (thick line) and SE (shaded band) indicated. dSod1G85R motor neurons fire fewer action potentials than dSod1WTLoxP in response to current injection (dSod1WTLoxP n = 13 and dSod1G85R n = 10; generalized linear mixed-effect models: genotype effect p = 3.9 × 10−6, stimulus effect p = 5.4 × 10−80). C, Measurements of aCC input resistance and capacitance did not differ between dSod1G85R and dSod1WTLoxP (dSod1WTLoxP: resistance = 0.74 ± 0.08GΩ, capacitance = 27.1 ± 3.0pF, current threshold = 18.72 ± 2.69 pA, voltage threshold = −29.3 ± 1.3 mV, n = 13; dSod1G85R: resistance = 0.81 ± 0.08GΩ, capacitance = 22.2 ± 2.8pF, current threshold = 27.58 ± 5.90 pA, voltage threshold = −28.0 ± 2.1 mV, n = 10; resistance p = 0.57, capacitance p = 0.25, current threshold p = 0.15, voltage threshold p = 0.57, t tests). D, The current threshold in dSod1G85R showed a trend toward higher values, but a difference in voltage threshold between dSod1G85R and dSod1WTLoxP was not observed. E, Plot of action potential response normalized to current threshold shows no difference in action potential response between dSod1WTLoxP and dSod1G85R. F, G, Specific properties of individual action potential spikes near current threshold were analyzed and do not reveal significant differences between dSod1WTLoxP and dSod1G85R waveforms (voltage threshold, red; action potential peak, green; rise time, red to green; action potential minimum, blue; decay time, green to blue; half-width, orange). H, I, Voltage-clamp recordings of spontaneous excitatory currents were not significantly different between dSod1G85R and dSod1WTLoxP regarding peak current density or event frequency (dSod1WTLoxP = 94.2 ± 18.6 pA/pF, 11.1 events/min, n = 9; dSod1G85R = 97.8 ± 18.6 pA/pF, 11.7 ± 3.3 events/min, n = 10; amplitude p = 0.84, Wilcoxon rank-sum test; frequency p = 0.88, unequal variance t test).
Figure 7.
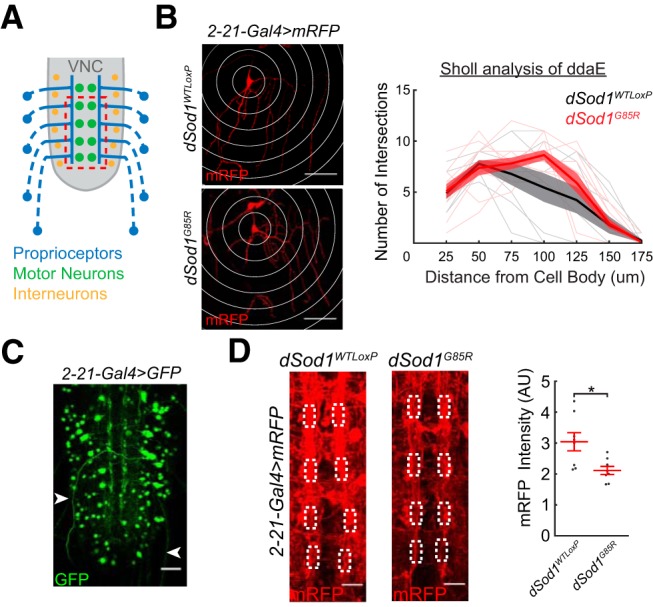
Neuronal processes are altered in dSod1G85R larval nerve cord. A, Schematic depicting axons of proprioceptors (blue) in the periphery projecting to the longitudinal tracts adjacent to the ventral nerve cord (gray) midline. B, The dendritic pattern of ddaE proprioceptors is marked by 2-21-Gal4>UAS-mRFP and quantified using Sholl analysis (white rings, 25 μm radius intervals) in both dSod1WTLoxP and dSod1G85R larvae (generalized linear mixed-effect models: genotype effect p = 0.10, radius length effect p = 2.4 × 10−13). C, In addition to peripherally located proprioceptors, 2-21-Gal4 is also expressed in cells within the larval nerve cord, as indicated by 2-21-Gal4>UAS-GFP. Axonal projections (white arrowheads) from multidendritic proprioceptor residing in the larval body wall are also visible. D, Axonal projections and other neuronal processes highlighted by 2-21-Gal4>UAS-mRFP are apparent along the VNC midline (VNC midline shown corresponds to dashed red box in A) of dSod1WTLoxP and dSod1G85R late third instar larval VNCs. White boxes define the 5 μm × 10 μm regions of interest within the neuropil where proprioceptors project (Merritt and Whitington, 1995; Grueber et al., 2007) and the area of anti-mRFP intensity quantification (dSod1WTLoxP = 3.04 ± 0.29 AU, n = 8; dSod1G85R = 2.11 ± 0.13 AU, n = 8, p = 0.017, unequal variance t test). Scale bars: 50 μm (B), 25 μm (C), and 10 μm (D). *p < 0.05.
Experimental design.
Sample size was predetermined by empirically estimating the variance and expected effect size of the parameter being measured. In all cases, n represents the number of individual animals, except in patch recording experiments, in which two different aCC motor neurons were recorded from the same VNC on three occasions.
Table 1.
Experiments were conducted as described in the “Behavior” section of the Materials and Methods. All experimental values and statistical information can be found in Table 1 and the table legend. Values of n reflect individual animals. Fisher's exact tests were used to compare genotypes and Holm–Bonferroni corrections were applied to account for multiple comparisons.
Table 1.
gbb expression partially rescues the dSod1G85R eclosion defect
| Genotype | Eclosed | Total | Percent Eclosion ± 95% CI |
|---|---|---|---|
| dSod1WTLoxP | 194 | 204 | 95.1 ± 2.96 |
| dSod1G85R | 0 | 99 | 0.0 ± 0.0 |
| UAS-gbb9.9/+;BG57-Gal4, dSod1G85R/dSod1G85R | 27 | 307 | 8.79 ± 3.17† |
| BG57,dSod1G85R/dSod1G85R | 0 | 95 | 0.0 ± 0.0 |
| UAS-gbb9.9/+;dSod1G85R | 0 | 201 | 0.0 ± 0.0 |
| dSod1WTLoxP | 397 | 400 | 99.3 ± 0.85 |
| OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP | 428 | 453 | 94.5 ± 2.10 |
| dSod1G85R | 0 | 185 | 0.0 ± 0.0 |
| OK371-Gal4/UAS-gbb9.9;dSod1G85R | 10 | 173 | 5.78 ± 3.48† |
| OK371-Gal4/+;dSod1G85R | 1 | 134 | 0.75 ± 1.46 |
| UAS-gbb9.9/+;dSod1G85R | 1 | 182 | 0.55 ± 1.07 |
| dSod1WTLoxP | 591 | 591 | 100 ± 0.0 |
| dSod1G85R | 0 | 163 | 0.0 ± 0.0 |
| OK6-Gal4/UAS-gbb9.9;dSod1G85R | 11 | 201 | 5.47 ± 3.14† |
| OK6-Gal4/+;dSod1G85R | 0 | 205 | 0.0 ± 0.0 |
| UAS-gbb9.9/+;dSod1G85R | 3 | 211 | 1.40 ± 1.60 |
dSod1G85R adults fail to emerge from the pupal case and complete eclosion (Şahin et al., 2017). Expression of gbb using two different drivers that express in motor neurons (OK371-Gal4 or OK6-Gal4) or muscles (BG57-Gal4) increases the percentage of dSod1G85R that eclose.
† indicates experimental group is statistically different (p < 0.05) from all dSod1G85R controls (dSod1WTLoxP versus dSod1G85R p = 3.1 × 10−68, UAS-gbb9.9/+;BG57-Gal4,dSod1G85R/dSod1G85R versus dSod1G85R p = 1.4 × 10−3, UAS-gbb9.9/+;BG57-Gal4,dSod1G85R/dSod1G85R versus BG57-Gal4,dSod1G85R/dSod1G85R p = 1.4 × 10−3 UAS-gbb9.9/+;BG57-Gal4,dSod1G85R/dSod1G85R versus UAS-gbb9.9/+;dSod1G85R/dSod1G85R p = 4.6 × 10−6, Fisher's exact tests with Holm–Bonferroni correction) (dSod1WTLoxP versus dSod1G85R p = 4.5 × 10−151, dSod1WTLoxP versus OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP p = 0.014, OK371-Gal4/UAS-gbb9.9;dSod1G85R versus dSod1G85R p = 2.4 × 10−3, OK371-Gal4/UAS-gbb9.9;dSod1G85R versus OK371-Gal4/+;dSod1G85R p = 0.026, OK371-Gal4/UAS-gbb9.9;dSod1G85R versus UAS-gbb9.9/+;dSod1G85R p = 0.014) (dSod1WTLoxP versus dSod1G85R p = 1.3 × 10−169, OK6-Gal4/UAS-gbb9.9;dSod1G85R versus dSod1G85R p = 2.8 × 10−3, OK6-Gal4/UAS-gbb9.9;dSod1G85R versus OK6-Gal4/+;dSod1G85R p = 1.1 × 10−3, OK6-Gal4/UAS-gbb9.9;dSod1G85R versus UAS-gbb9.9/+;dSod1G85R p = 0.029, Fisher's exact tests with Holm–Bonferroni correction).
Figure 1.
Experiments were conducted as described in the “Immunofluorescence” and “Electrophysiology” sections of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. Data were normally distributed and t tests accounting for the variance of the data were used to test differences between genotypes. A one-tailed t test was used to compare dSod1G85R and OK371-Gal4/UAS-gbb9.9;dSod1G85R because the hypothesis being tested is whether gbb rescues dSod1G85R phenotypes. Holm–Bonferroni corrections were applied to account for multiple comparisons.
Figure 2.
Experiments were conducted as described in the “Staging” and “Dissection” sections of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. Leg extension data were normally distributed and t tests accounting for the variance of the data were used to test differences between genotypes. A one-tailed t test was used to compare dSod1G85R and OK371-Gal4/UAS-gbb9.9;dSod1G85R because the hypothesis being tested is whether gbb rescues dSod1G85R leg extension. The nerve integrity score data are categorical and were therefore analyzed using Fisher's exact tests. Multiple comparisons were accounted for using Holm–Bonferroni corrections.
Figure 3.
Experiments were conducted as described in the “Behavior” and “Electrophysiology” sections of the Materials and Methods. All experimental values and statistical information can be found in Table 2. Values of n reflect individual animals. Data were normally distributed and t tests accounting for the variance of the data were used to test differences between genotypes. Holm–Bonferroni corrections were applied to account for multiple comparisons.
Table 2.
Statistical comparisons of larval locomotion in Figure 3
| Genotype 1: mean ± SEM in mm (n) | Genotype 2: mean ± SEM in mm (n) | Corrected p-value |
|---|---|---|
| dSod1WTLoxP: 63.0 ± 2.1 (63) | OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP: 49.5 ± 1.9 (45) | 1.9 × 10−5 |
| dSod1WTLoxP: 63.0 ± 2.1 (63) | dSod1G85R: 27.3 ± 2.4 (34) | 2.4 × 10−17 |
| OK371-Gal4/UAS-gbb9.9;dSod1G85R: 44.0 ± 2.5 (42) | dSod1G85R: 27.3 ± 2.4 (34) | 2.7 × 10−5 |
| OK371-Gal4/UAS-gbb9.9;dSod1G85R: 44.0 ± 2.5 (42) | OK371-Gal4/+9;dSod1G85R: 28.2 ± 2.8 (29) | 1.7 × 10−4 |
| OK371-Gal4/UAS-gbb9.9;dSod1G85R: 44.0 ± 2.5 (42) | UAS-gbb9.9/+;dSod1G85R: 33.7 ± 3.1 (36) | 0.011 |
| dSod1WTLoxP: 45.8 ± 2.3 (58) | dSod1G85R: 26.2 ± 1.8 (60) | 2.8 × 10−9 |
| OK6-Gal4/UAS-gbb9.9;dSod1G85R: 35.9 ± 2.4 (73) | dSod1G85R: 26.2 ± 1.8 (60) | 9.3 × 10−3 |
| OK6-Gal4/UAS-gbb9.9;dSod1G85R: 35.9 ± 2.4 (73) | OK6-Gal4/+9;dSod1G85R: 26.1 ± 1.7 (59) | 0.017 |
| OK6-Gal4/UAS-gbb9.9;dSod1G85R: 35.9 ± 2.4 (73) | UAS-gbb9.9/+;dSod1G85R: 28.8 ± 1.6 (59) | 4.9 × 10−3 |
The mean, SEM, and n for each genotype are listed along with the corrected p-values for comparisons of indicated genotypes.
Figure 4.
Experiments were conducted as described in the “Immunofluorescence” and “Electrophysiology” sections of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. Data were normally distributed in immunofluorescence measurements and NMJ recordings. t tests accounting for the variance of the data were used to test differences between genotypes. Holm–Bonferroni corrections were applied to bouton counts to account for multiple comparisons. A generalized linear mixed-effect model was applied to measure genotype and stimulus effects on muscle contraction.
Figure 5.
Experiments were conducted as described in the “Electrophysiology” section of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect patched cells. Most cells were taken from individual animals except on three occasions where two different aCC motor neurons were recorded from the same VNC. Data were normally distributed except for current density measurements. A Wilcoxon rank-sum test was used to analyze current density data, generalized linear mixed-effect models were used to analyze repeated measure data, and t tests were used to assess all other measurements.
Figure 6.
Experiments were conducted as described in the “Electrophysiology” section of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. Data were normally distributed and t tests accounting for variance were used to assess differences between genotypes. A one-tailed t test was used to compare dSod1G85R and OK371-Gal4/UAS-gbb9.9;dSod1G85R because the hypothesis being tested is whether gbb rescues dSod1G85R event frequency. Paired t tests were used in Figure 6F because the changes between cut and intact preparations were measured within the same animals.
Figure 6.

Peripheral feedback is disrupted in dSod1G85R larvae. A, Motor neurons (green) synapse onto muscles (orange). Sensory neurons (blue) detect stimuli, including muscle contraction, and feed back to the VNC. Interneurons (yellow) integrate sensory and central pattern generator (CPG) information within a segment and transfer signal to activate motor neurons in adjacent segment. Inputs to the interneuron network and CPG from the peripheral nervous system are removed by separating the brain/VNC from the body wall (right). B, Fictive crawling extracellular recordings from abdominal nerves in third instar larvae with an intact circuit, performed as in Figure 3. Red arrowheads mark bursts of neuronal activity. C, Extracellular recordings on abdominal nerves from isolated brain/VNC without input from peripheral feedback, after recordings were first performed with the circuit intact (B). D, Comparison of extracellular recordings from freshly dissected brains (“F”) with those that were performed on cut (“C”) nerves after first recording while the circuit was intact (i.e., in B) showed no evidence of run-down on activity over the course of the experiment (dSod1WTLoxP F = 4.8 ± 1.2 events/min, n = 6; dSod1WTLoxP C = 3.6 ± 0.4 events/min, n = 9; p = 0.41, unequal variance t test). E, Quantification of event frequency from paired intact (“I”) and cut (“C”) recordings. dSod1G85R exhibit a lower event frequency than dSod1WTLoxP when the circuit is intact (dSod1WTLoxP I = 4.5 ± 0.3 events/min, n = 9; dSod1G85R I = 3.1 ± 0.4 events/min, n = 7; p = 3.9 × 10−3, t test with Holm–Bonferroni correction). The lower event frequency of dSod1G85R is restored when gbb is expressed using OK371-Gal4 (dSod1G85R I = 3.1 ± 0.4 events/min, n = 7; OK371-Gal4/UAS-gbb9.9;dSod1G85R I = 4.4 ± 0.2 events/min, n = 6, p = 0.014, one-tailed t test with Holm–Bonferroni correction). Neural activity in the isolated CNS, reflecting output from the CPG, is higher in dSod1G85R than dSod1WTLoxP (dSod1WTLoxP C = 3.6 ± 0.4 events/min, n = 9; dSod1G85R C = 5.6 ± 0.7 events/min, n = 7; dSod1WTLoxP Cut vs dSod1G85R Cut: p = 0.041, t test with Holm–Bonferroni correction). OK371-Gal4 driven gbb does not affect this increase in CPG activity observed in dSod1G85R (dSod1G85R C = 5.6 ± 0.7 events/min, n = 7; OK371-Gal4/UAS-gbb9.9;dSod1G85R C = 5.5 ± 0.5 events/min, n = 6; p = 0.96, t test with Holm–Bonferroni correction). F, Difference between each paired recording (Cut–Intact) indicates the impact of peripheral feedback on circuit activity. Peripheral feedback does not significantly affect event frequency in dSod1WTLoxP or OK371-Gal4/UAS-gbb9.9;dSod1G85R but it slows the frequency of bursts in dSod1G85R (Cut-Intact: dSod1WTLoxP = −0.94 ± 0.59 events/min, n = 9, p = 0.15, paired t test; dSod1G85R = 2.5 ± 0.6 events, n = 7, p = 0.0054, paired t test; OK371-Gal4/UAS-gbb9.9;dSod1G85R = −1.1 ± 0.5 events, n = 6, p = 0.064, paired t test). The slowing effect of peripheral feedback in dSod1G85R appears muted when gbb is expressed. The higher rate of bursting in dSod1G85R when feedback from the periphery is absent (C, E) is not altered by OK371-Gal4-driven gbb, indicating that gbb expression does not overcome this defect. *p < 0.05, **p < 0.01.
Figure 7.
Experiments were conducted as described in the “Immunofluorescence” section of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. mRFP data were normally distributed and had unequal variance, so an unequal variance t test was used to assess difference between genotypes. Sholl analysis data were analyzed using a generalized linear mixed effect model because it is a repeated-measures dataset.
Figure 8.
Experiments were conducted as described in the “Immunofluorescence” section of the Materials and Methods. All experimental values and statistical information can be found in the figure legend. Values of n reflect individual animals. Data were normally distributed and t tests were used to assess differences between genotypes. A Holm–Bonferroni correction was applied to NMJ fluorescence measurements to account for multiple comparisons.
Figure 9.
Experiments were conducted as described in the “Behavior” section of the Materials and Methods. All experimental values and statistical information can be found in Table 3. Values of n reflect individual animals. Data were normally distributed and t tests accounting for unequal variance where appropriate were used to assess differences between genotypes. Multiple hypotheses were accounted for using Holm–Bonferroni corrections.
Table 3.
Statistical comparisons of larval locomotion in Figure 9
| Genotype 1: mean ± SEM in mm (n) | Genotype 2: mean ± SEM in mm (n) | Corrected p-value |
|---|---|---|
| dSod1WTLoxP: 48.9 ± 3.2 (24) | dSod1G85R: 19.9 ± 1.9 (22) | 1.3 × 10−8 |
| OK371-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 45.2 ± 3.2 (40) | dSod1G85R: 19.9 ± 1.9 (22) | 1.7 × 10−8 |
| OK371-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 45.2 ± 3.2 (40) | OK371-Gal4/+;dSod1G85R: 29.7 ± 1.8 (42) | 1.4 × 10−4 |
| OK371-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 45.2 ± 3.2 (40) | UAS-SaxA,dSod1G85R/dSod1G85R: 29.1 ± 3.7 (19) | 3.3 × 10−3 |
| dSod1WTLoxP: 57.3 ± 3.6 (25) | dSod1G85R: 30.8 ± 3.3 (22) | 9.7 × 10−6 |
| ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 49.7 ± 3.3 (42) | dSod1G85R: 30.8 ± 3.3 (22) | 1.6 × 10−3 |
| ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 49.7 ± 3.3 (42) | ChAT-Gal4/+;dSod1G85R: 36.4 ± 3.3 (32) | 6.7 × 10−3 |
| ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 49.7 ± 3.3 (42) | UAS-SaxA,dSod1G85R/dSod1G85R: 32.3 ± 3.9 (19) | 5.6 × 10−3 |
| dSod1WTLoxP: 50.5 ± 3.7 (19) | dSod1G85R: 30.6 ± 4.4 (35) | 2.4 × 10−3 |
| 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 48.6 ± 5.1 (30) | dSod1G85R: 30.6 ± 4.4 (35) | 4.8 × 10−3 |
| 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 48.6 ± 5.1 (30) | 2–21-Gal4,dSod1G85R/dSod1G85R: 30.8 ± 3.6 (39) | 4.2 × 10−3 |
| 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 48.6 ± 5.1 (30) | UAS-SaxA,dSod1G85R/dSod1G85R: 27.7 ± 3.9 (39) | 2.4 × 10−3 |
| dSod1WTLoxP: 64.2 ± 2.3 (51) | dSod1G85R: 41.7 ± 2.9 (44) | 9.3 × 10−8 |
| Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 36.2 ± 3.6 (33) | dSod1G85R: 41.7 ± 2.9 (44) | 0.48 |
| Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 36.2 ± 3.6 (33) | Repo-Gal4,dSod1G85R/dSod1G85R: 48.2 ± 3.8 (49) | 0.10 |
| Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 36.2 ± 3.6 (33) | UAS-SaxA,dSod1G85R/dSod1G85R: 39.8 ± 3.8 (40) | 0.51 |
| dSod1WTLoxP: 64.6 ± 2.7 (45) | dSod1G85R: 27.2 ± 2.7 (45) | 4.2 × 10−15 |
| BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 29.3 ± 2.9 (45) | dSod1G85R: 27.2 ± 2.7 (45) | 1.0 |
| BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 29.3 ± 2.9 (45) | BG57-Gal4,dSod1G85R/dSod1G85R: 26.5 ± 2.8 (38) | 1.0 |
| BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R: 29.3 ± 2.9 (45) | UAS-SaxA,dSod1G85R/dSod1G85R: 25.7 ± 2.8 (40) | 1.0 |
| dSod1WTLoxP: 48.2 ± 1.8 (60) | dSod1G85R: 27.8 ± 2.1 (45) | 1.3 × 10−10 |
| MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 33.3 ± 2.6 (34) | dSod1G85R: 27.8 ± 2.1 (45) | 0.32 |
| MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 33.3 ± 2.6 (34) | MD-Gal4/+;dSod1G85R: 29.7 ± 2.4 (35) | 0.65 |
| MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R: 33.3 ± 2.6 (34) | UAS-SaxA,dSod1G85R/dSod1G85R: 34.0 ± 3.0 (42) | 0.87 |
The mean, SEM, and n for each genotype are listed along with the corrected p-values for comparisons of indicated genotypes.
Table 2.
Experiments were conducted as described in the “Behavior” section of the Materials and Methods. All experimental values and statistical information can be found in Table 4 and the table legend. Values of n reflect individual animals. Fisher's exact tests were used to compare genotypes and Holm–Bonferroni corrections were applied to account for multiple comparisons.
Table 4.
Cell-autonomous BMP signaling in cholinergic neurons rescues dSod1G85R eclosion
| Genotype | Eclosed | Total | Percent Eclosion ± 95% CI |
|---|---|---|---|
| dSod1WTLoxP | 311 | 312 | 99.7 ± 0.61 |
| dSod1G85R | 0 | 106 | 0.0 ± 0.0 |
| OK371-Gal4/+;UAS-SaxA, dSod1G85R/dSod1G85R | 14 | 347 | 4.03 ± 2.07 |
| OK371-Gal4/+;dSod1G85R | 3 | 298 | 1.01 ± 1.13 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 4 | 134 | 2.99 ± 2.88 |
| dSod1WTLoxP | 420 | 428 | 98.1 ± 1.28 |
| dSod1G85R | 0 | 104 | 0.0 ± 0.0 |
| ChAT-Gal4/+;UAS-SaxA, dSod1G85R/dSod1G85R | 41 | 401 | 10.2 ± 2.97† |
| ChAT-Gal4/+;dSod1G85R | 2 | 213 | 0.94 ± 1.30 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 3 | 138 | 2.17 ± 2.43 |
| dSod1WTLoxP | 509 | 513 | 99.2 ± 0.76 |
| dSod1G85R | 0 | 290 | 0.0 ± 0.0 |
| 2–21-Gal4,dSod1G85R/UAS-SaxA, dSod1G85R | 96 | 270 | 35.6 ± 5.71† |
| 2.21-Gal4,dSod1G85R/dSod1G85R | 1 | 248 | 0.40 ± 0.79 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 1 | 332 | 0.30 ± 0.59 |
| dSod1WTLoxP | 1321 | 1329 | 98.3 ± 1.16 |
| dSod1G85R | 0 | 555 | 0.0 ± 0.0 |
| Repo-Gal4,dSod1G85R/UAS-SaxA, dSod1G85R | 11 | 432 | 2.55 ± 1.49 |
| Repo-Gal4,dSod1G85R/dSod1G85R | 5 | 331 | 1.51 ± 1.31 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 0 | 300 | 0.0 ± 0.0 |
| dSod1WTLoxP | 1063 | 1074 | 99.0 ± 0.60 |
| dSod1G85R | 0 | 462 | 0.0 ± 0.0 |
| BG57-Gal4,dSod1G85R/UAS-SaxA, dSod1G85R | 1 | 288 | 0.35 ± 0.68 |
| BG57-Gal4,dSod1G85R/dSod1G85R | 0 | 246 | 0.0 ± 0.0 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 4 | 445 | 0.89 ± 0.87 |
| dSod1WTLoxP | 382 | 389 | 98.2 ± 1.32 |
| dSod1G85R | 0 | 217 | 0.0 ± 0.0 |
| MD-Gal4/+;UAS-SaxA, dSod1G85R/dSod1G85R | 2 | 171 | 1.17 ± 1.61 |
| MD-Gal4/+;dSod1G85R | 1 | 186 | 0.54 ± 1.05 |
| UAS-SaxA,dSod1G85R/dSod1G85R | 1 | 209 | 0.48 ± 0.94 |
Driving UAS-SaxA using ChAT-Gal4 or 2–21-Gal4, but not OK371-Gal4, MD-Gal4, Repo-Gal4, or BG57-Gal4 increases the percent of dSod1G85R mutants that eclose.
†, experimental group is statistically different (p < 0.05) from all dSod1G85R controls (dSod1WTLoxP versus dSod1G85R p = 3.1 × 10−101; ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus dSod1G85R p = 1.5 × 10−4; ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus ChAT-Gal4/+;dSod1G85R p = 8.6 × 10−6; ChAT-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus UAS-SaxA,dSod1G85R/dSod1G85R p = 1.8 × 10−3; Fisher's exact tests with Holm–Bonferroni corrections) (dSod1WTLoxP versus dSod1G85R p = 1.3 × 10−157; MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus dSod1G85R p = 0.58; MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus MD-Gal4/+;dSod1G85R p = 1.0; MD-Gal4/+;UAS-SaxA,dSod1G85R/dSod1G85R versus UAS-SaxA,dSod1G85R/dSod1G85R p = 1.0; Fisher's exact tests with Holm–Bonferroni corrections) (dSod1WTLoxP versus dSod1G85R p = 3.3 × 10−218; 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus dSod1G85R p = 1.8 × 10−35; 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus 2–21-Gal4,dSod1G85R/dSod1G85R p = 2.3 × 10−30; 2–21-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus UAS-SaxA,dSod1G85R/dSod1G85R p = 8.6 × 10−37; Fisher's exact tests with Holm–Bonferroni corrections) (dSod1WTLoxP versus dSod1G85R p < 10−250; BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus dSod1G85R p = 1.0; BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus BG57-Gal4,dSod1G85R/dSod1G85R p = 1.0; BG57-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus UAS-SaxA,dSod1G85R/dSod1G85R p = 1.0; Fisher's exact tests with Holm–Bonferroni corrections) (dSod1WTLoxP versus dSod1G85R p < 10−250; Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus dSod1G85R p = 3.2 × 10−4; Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus Repo-Gal4,dSod1G85R/dSod1G85R p = 0.45; Repo-Gal4,dSod1G85R/UAS-SaxA,dSod1G85R versus UAS-SaxA,dSod1G85R/dSod1G85R p = 7.4 × 10−3; Fisher's exact tests with Holm–Bonferroni corrections).
All statistical comparisons were performed in MATLAB except for the multiple-category Fisher's exact test in Figure 2B, which was performed in R.
Figure 2.
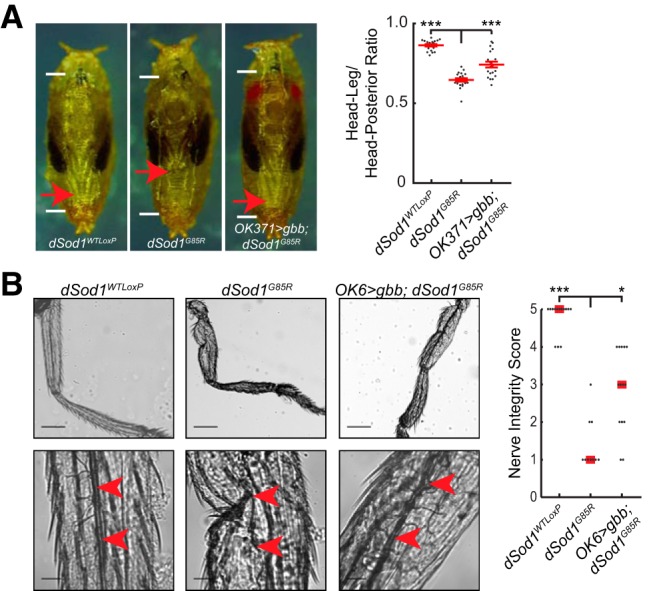
gbb expression rescues dSod1G85R leg extension and nerve integrity. A, Leg extension is compromised in dSod1G85R mutants (leg: body ratios: dSod1WTLoxP: 0.86 ± 0.01, n = 19; dSod1G85R: 0.65 ± 0.01, n = 20; OK371-Gal4/UAS-gbb9.9;dSod1G85R head:leg ratio = 0.74 ± 0.02, n = 20; dSod1WTLoxP vs dSod1G85R: p = 2.0 × 10−17, unequal variance t test with Holm–Bonferroni correction; dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R: p = 3.9 × 10−5 unequal variance one-tailed t test with Holm–Bonferroni correction). The failure of leg extension likely contributes to disruption in leg morphology (Sahin et al., 2017). The expression of gbb with OK371-Gal4 partially rescues the dSod1G85R leg extension phenotype. White lines indicate pupal length and red arrows indicate the tips of the metathoracic legs. B, dSod1G85R nerves (red arrowheads) in the metathoracic leg are severely disrupted and are rescued by expressing gbb with OK6-Gal4 (dSod1WTLoxP median score: 5, n = 14; dSod1G85R median score: 1, n = 10; OK6-Gal4/UAS-gbb9.9;dSod1G85R median score: 3, n = 14; dSod1WTLoxP vs dSod1G85R p = 5.0 × 10−5, Fisher's exact test with Holm–Bonferroni correction; dSod1G85R vs OK6-Gal4/UAS-gbb9.9;dSod1G85R p = 0.026, Fisher's exact test with Holm–Bonferroni correction). Scale bars, 200 μm in low-magnification images (B, top) and 30 μm in high-magnification images (B, bottom). *p < 0.05, ***p < 0.001.
Custom scripts
The custom MATLAB scripts used for data analysis can be found at https://github.com/ahheld/motor_circuit_dSod1ALS.
Results
End-stage dSod1G85R mutants exhibit NMJ defects consistent with motor failure
Homozygous dSod1G85R mutants die at the end of pupation, whereas wild-type dSod1WTLoxP knock-in control animals emerge (eclose) from their pupal cases and live a normal adult lifespan (Sahin et al., 2017). The majority of dSod1G85R mutants die as fully formed adults that do not complete the eclosion process to emerge from the pupal case (pharates) (Table 1). Eclosion requires a series of abdominal muscle contractions, head thrusts, and thoracic contractions to rupture the operculum and propel the adult fly from the pupal case (Kimura and Truman, 1990; McNabb et al., 1997). The failure of dSod1G85R mutants to eclose suggests a defect in muscle contractility.
We assessed the structural and functional integrity of abdominal muscle innervation given their critical role in the eclosion process. Abdominal muscle fibers of Drosophila pharates are arranged in a stereotypical manner in each A2–A6 hemisegment (Fig. 1A). We analyzed dSod1WTLoxP and dSod1G85R NMJs at the longitudinal VMs (Fig. 1A) (Crossley, 1980; Kimura and Truman, 1990; Broadie and Bate, 1991). dSod1WTLoxP exhibit a characteristic arrangement of parallel axonal branches in the VM of A2 and A3 hemisegments, visualized by presynaptic (anti-HRP) and postsynaptic (anti-Dlg) markers (Sun and Salvaterra, 1995; Budnik et al., 1996) (Fig. 1B). In contrast, the dSod1G85R A2 and A3 VM NMJs are disorganized, with kinked and discontinuous axonal projections. Furthermore, in dSod1G85R mutants, the A2 and A3 NMJs contain fewer synaptic boutons: 53% and 63%, respectively, of dSod1WTLoxP controls (Fig. 1C).
Consistent with the structural abnormalities of dSod1G85R NMJs, a substantial decrease in nerve muscle transmission in dSod1G85R pharates was evident compared with dSod1WTLoxP control animals (Fig. 1D,E). The frequency of mEPSPs recorded from the VM in A2 hemisegments of dSod1G85R was 47% of that measured in dSod1WTLoxP (Fig. 1E). mEPSP amplitudes measured in dSod1G85R and dSod1WTLoxP muscles were not distinguishable (Fig. 1E) and the dSod1G85R VM muscle capacitance, 68% of dSod1WTLoxP, suggests that muscle surface area is reduced in dSod1G85R. Although we were unable to consistently evoke EPSPs in dSod1G85R preparations due to considerable motor neuron degeneration, the reduced frequency of spontaneous synaptic vesicle fusion events and the reduced number of boutons in dSod1G85R NMJs implicate presynaptic defects.
Abdominal and thoracic muscle contractions are also necessary to force hemolymph into everting pupal leg discs and to extend newly formed legs (Mesce and Fahrbach, 2002). Consistent with a defect in abdominal and/or thoracic muscle contractility, dSod1G85R mutants fail to fully extend their legs (Fig. 2A).The main nerve tracts of dSod1G85R legs are often discontinuous, with a clear reduction in axonal arbors (Fig. 2B and Sahin et al., 2017). These defects in both leg and abdominal muscle innervation, reduced NMJ and muscle size, and the associated failures in motor-neuron-dependent processes in dSod1G85R are reminiscent of disruptions in muscle innervation, muscle wasting, and motor failure seen in other models of ALS and in human patients (Denys and Norris, 1979; Tsujihata et al., 1984; Tandan and Bradley, 1985; Azzouz et al., 1997).
BMP signaling enables dSod1G85R eclosion
Members of the TGF-β/BMP family of signaling molecules act as neurotrophic factors to promote neuronal growth and survival (Jordan et al., 1997; Krieglstein et al., 2002; Lee-Hoeflich et al., 2004; Sun et al., 2007; Hocking et al., 2009). In Drosophila, the BMP7 ortholog gbb has previously been shown to promote NMJ growth and neurotransmission (McCabe et al., 2003; Goold and Davis, 2007; James et al., 2014). We tested the ability of Gbb as a retrograde signal to suppress motor defects observed in dSod1G85R mutants. Indeed, 8.8% of dSod1G85R flies successfully eclosed when gbb was expressed in muscles (UAS-gbb9.9/+;BG57-Gal4, dSod1G85R/dSod1G85R) compared with 0% eclosion of dSod1G85R mutants (Table 1). When gbb transcription is driven using two different Gal4 lines that both express in motor neurons (OK371-Gal4 and OK6-Gal4; Mahr and Aberle, 2006; Sanyal, 2009) (OK371-Gal4/UAS-gbb9.9;dSod1G85R or OK6-Gal4 /UAS-gbb9.9;dSod1G85R), a similar rescue was observed (Table 1). In wild-type animals, muscle-driven gbb results in an increase in body size, whereas OK371-Gal4 driven gbb does not (data not shown). In subsequent experiments aimed at understanding the impact of Gbb signaling on motor neuron function, we activated signaling by driving gbb expression with OK371-Gal4, not BG57-Gal4, to avoid the effects that a change in body size may introduce and confound our interpretation of results.
Neuromuscular defects in end-stage dSod1G85R suppressed by BMP signaling
We found that disruptions in the structural integrity and function of dSod1G85R VM NMJs were both rescued by gbb expression under the control of OK371-Gal4 (OK371>gbb;dSod1G85R: OK371-Gal4/UAS-gbb9.9;dSod1G85R) (Fig. 1). Axonal projections and branching in OK371>gbb;dSod1G85R were restored to the characteristic parallel alignment of wild-type VM NMJs (Fig. 1B). An increase in bouton number was also observed at A2 and A3 VM NMJs (Fig. 1C), with a corresponding increase in mEPSP frequency and muscle capacitance from recordings of OK371>gbb;dSod1G85R compared with dSod1G85R (Fig. 1E). In addition to the rescue of dSod1G85R abdominal NMJ phenotypes, OK371>gbb;dSod1G85R animals exhibited greater leg extension and improved leg nerve integrity compared with the mutants, consistent with a gbb-mediated improvement in abdominal muscle contractions and reduced leg motor neuron degeneration (Fig. 2).
Therefore, expression of the BMP ligand Gbb in a subset of neurons that includes motor neurons (OK371>gbb;dSod1G85R) alleviates defects in axonal branching, bouton number, mEPSP frequency, and muscle membrane capacitance at the dSod1G85R VM NMJs, as well as defects in leg extension and leg nerve integrity. However, despite the nearly complete restoration of neurotransmission across OK371>gbb;dSod1G85R abdominal NMJs, relatively few individuals fully eclose (5.8%), indicating that, whereas the level of BMP signaling achieved by OK371-Gal4-driven gbb expression is sufficient to greatly improve dSod1G85R NMJ function, other abnormalities that influence successful eclosion must not be fully restored.
Reduced locomotion of dSod1G85R larvae is rescued by neuronal-driven gbb
Like pharates, wandering third instar dSod1G85R larvae exhibit compromised muscle contraction. They crawl approximately half the distance of wild-type (dSod1WTLoxP) animals during a 90 s period (Fig. 3A–C, Table 2; Sahin et al., 2017), with fewer posterior to anterior peristaltic waves of muscle contraction, each longer in duration compared with dSod1WTLoxP (Fig. 3D,E). When gbb is expressed under the control of OK371-Gal4 or OK6-Gal4, dSod1G85R larvae travel farther (Fig. 3A–C, Table 2). An increase in the frequency of peristaltic waves relative to dSod1G85R mutants and a corresponding decrease in the duration of each wave was observed when gbb was expressed in glutamatergic neurons under the control of OK371-Gal4 (Fig. 3D,E).
Figure 3.
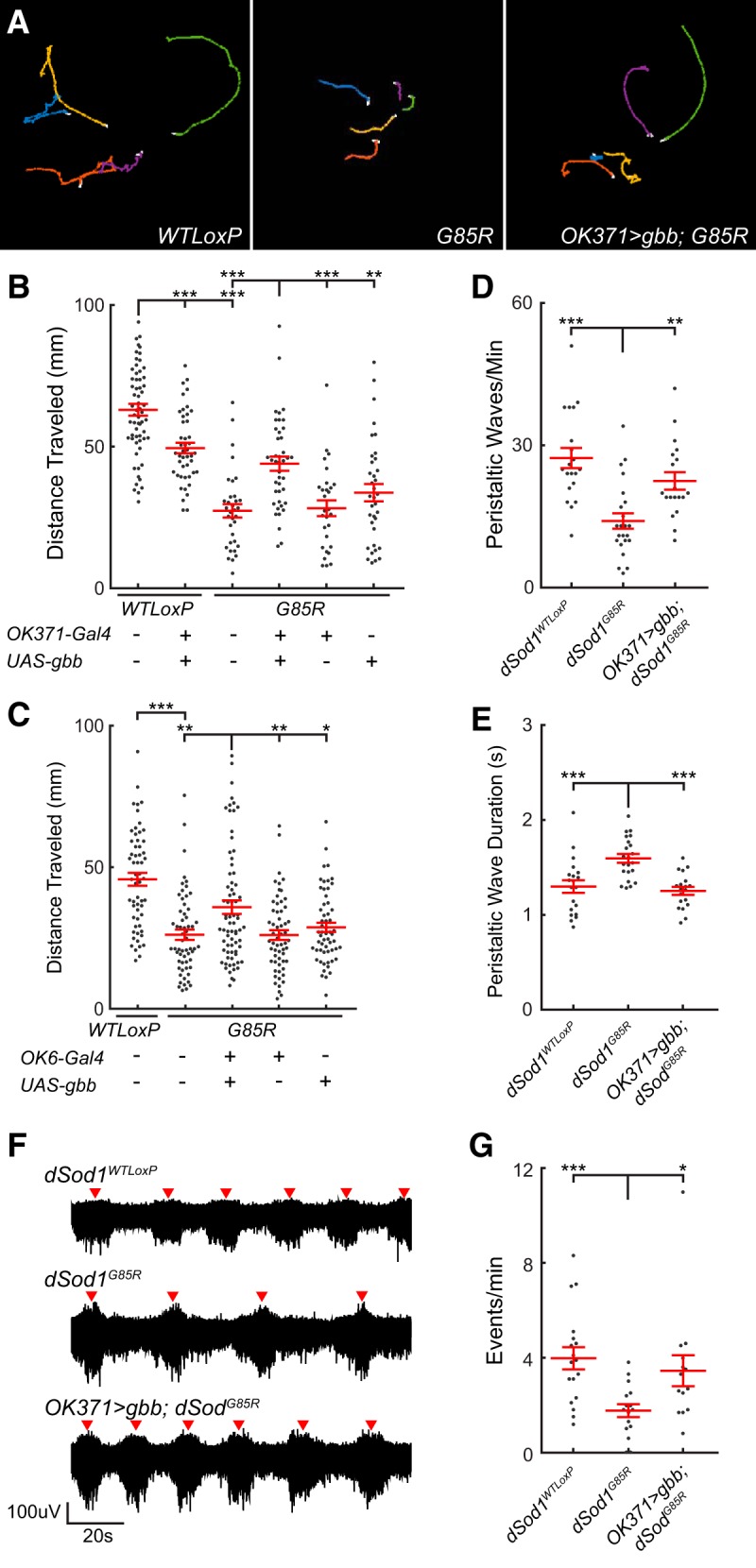
The dSod1G85R larval locomotion defect is improved by gbb expression. A, Tracks of wandering third instar larvae over 90 s. B, Plot of distance traveled over 90 s. Reduced locomotion of dSod1G85R larvae is increased by OK371-Gal4 driven gbb (OK371-Gal4/UAS-gbb9.9;dSod1G85R) despite detrimental effects of gbb overexpression in wild-type (OK371-Gal4/UAS-gbb9.9;dSod1WTLoxP). C, gbb expression using a second driver that also expresses in motor neurons, OK6-Gal4, also increases dSod1G85R larval locomotion. D, E, Quantification of number and duration of peristaltic waves of muscle contraction along the anterior/posterior axis of wandering third instar larvae. Waves are less frequent and longer in duration in dSod1G85R (Frequency: dSod1WTLoxP = 27.3 waves/min, n = 20; dSod1G85R = 14.0 waves/min, n = 23; p = 2.1 × 10−5, t test with Holm–Bonferroni correction. Duration: dSod1WTLoxP = 1.30 ± 0.07 s, n = 20; dSod1G85R: 1.59 ± 0.05 s, n = 22; p = 6.4 × 10−4, t test with Holm–Bonferroni correction). OK371-Gal4/UAS-gbb9.9;dSod1G85R larvae have increased peristaltic wave frequency and decreased wave duration compared with dSod1G85R (Frequency: dSod1G85R = 14.0 waves/min, n = 23; OK371-Gal4/UAS-gbb9.9;dSod1G85R = 22.47 ± 1.84 waves/min, n = 19; p = 6.9 × 10−4, one-sided t test with Holm–Bonferroni correction. Duration: dSod1G85R: 1.59 ± 0.05 s, n = 22; OK371-Gal4/UAS-gbb9.9;dSod1G85R: 1.25 ± 0.04 s, n = 19; p = 3.9 × 10−6, one-sided t test with Holm–Bonferroni correction). F, G, Extracellular measurements of neural activity from segmental nerve in dissected larva with intact motor circuit. Bursts of neural activity (red arrowhead) are reduced in dSod1G85R reminiscent of the decrease in crawling wave frequency in vivo, and OK371-Gal4 driven gbb increases the frequency of events. *p < 0.05, **p < 0.01, ***p < 0.001.
Consistent with a previous report showing that hyperactivation of BMP signaling can negatively affect neurons (Nahm et al., 2013), we found that expressing gbb using OK371-Gal4 led to a reduction in wild-type larval locomotion (Fig. 3B, Table 2), as well as a reduction in the number of adults that eclose (Table 1). Interestingly, despite the negative impact of overexpressed gbb in the wild-type, we found that OK371-Gal4-driven gbb improved dSod1G85R larval locomotion and enabled the eclosion of some dSod1G85R flies (Fig. 3A–E, Table 1).
Bursts of neural activity that correlate with segmental muscle contractions in larvae can be monitored in fictive crawling preparations in which the motor circuit of the dissected larva is left intact (Cattaert et al., 2001; Fox et al., 2006). Wandering third instar larvae were fileted, exposing the VNC and its connections through segmentally organized nerves to the body wall musculature. The concordance of muscle contraction and neural activity is evident when bursts of electrical activity are recorded from a single segmental nerve bundle using an extracellular suction electrode. Each burst corresponds to one round of muscle contractions for that body hemisegment as the larval preparation “crawls” ex vivo. Consistent with the reduced crawling behavior exhibited by dSod1G85R larvae, we found that the bursts of neural activity in fictive crawling preparations of dSod1G85R were less frequent than in controls (Fig. 3F,G). When gbb expression was driven by OK371-Gal4 in dSod1G85R, the bursts of neural activity were more frequent (Fig. 3F,G). These fictive crawling measurements paralleled our in vivo larval locomotion analyses, demonstrating that the dSod1G85R crawling defect and its rescue by OK371-Gal4 driven gbb can be observed and recorded ex vivo.
Minor NMJ defects detected in dSod1G85R larvae unlikely to account for reduced locomotion
Despite the abnormal locomotor behavior of dSod1G85R larvae (Fig. 3), we did not find a substantial change in the structure or function of NMJs in mutant third instar larvae (Fig. 4). This is in contrast to the severe disruption of NMJ structure and function observed in abdominal muscles of dSod1G85R pharates (Fig. 1). In dSod1G85R larvae, the number of boutons at muscle 6/7 NMJs is not different from wild-type (Fig. 4A). Similarly, the electrophysiological properties of the dSod1G85R muscle 6 NMJ did not reveal differences in the amplitude of excitatory events [mEPSPs, evoked EPSPs (eEPSPs), and eEPSCs] or the kinetics of eEPSCs compared with wild-type (Fig. 4B–E). We did observe a slight increase in the frequency of mEPSPs in dSod1G85R, suggesting a mild defect in the dSod1G85R NMJ (Fig. 4B,C). Interestingly, overexpressing gbb in dSod1G85R motor neurons did not lead to an increase in bouton number, a phenotype typically seen in wild-type NMJs when gbb expression is driven in motor neurons (James and Broihier, 2011) (Fig. 4A), suggesting that dSod1G85R NMJs are defective in some manner, consistent with their altered mEPSP frequency. Although dSod1G85R larval NMJs may be slightly defective, the fact that the excitatory event amplitudes remain normal suggests that the reduced locomotion of dSod1G85R larvae is unlikely to result from an NMJ defect.
Because larval muscle contraction requires a coordinated response from the muscle as well as repeated motor neuron firing and sufficient neurotransmitter release (Cattaert et al., 2001), we tested for the possibility that muscle contractility itself is abnormal in dSod1G85R. We measured the response of muscles 6/7 to a range of motor neuron stimulation frequencies. No alterations in contraction were observed between dSod1G85R and wild-type third instar larvae (Fig. 4F,G).Therefore, the reduced locomotion of dSod1G85R third instar larvae is not easily attributed to abnormalities at the NMJ and/or defects in muscle contraction.
dSod1G85R larval motor neurons exhibit mild hypoexcitability
Alterations in motor neuron excitability have been observed in ALS patients and in some ALS models (Kanai et al., 2006; Iwai et al., 2016; King et al., 2016). Given the relatively minor NMJ defects detected in dSod1G85R larvae, we investigated the possibility that altered motor neuron excitability could account for the observed reduction in locomotor activity. Patch recordings followed by current steps revealed a slight reduction in the excitability of aCC motor neurons in dSod1G85R compared with dSod1WTLoxP (Fig. 5A,B).Although we did not detect differences in resistance, capacitance, voltage threshold, or action potential dynamics between dSod1G85R and dSod1WTLoxP, a slightly increased but not statistically significant current threshold was observed in dSod1G85R neurons that could account for their lower excitability (Fig. 5C–G).
After the current step protocol, aCC motor neurons were voltage clamped at the chloride reversal potential (−70 mV) to isolate EPSCs. No major changes in EPSC amplitude or frequency were apparent, indicating that excitatory signals from the central pattern generator (CPG) interneurons must not be different in dSod1G85R compared with wild-type (Fig. 5H,I). In summary, our electrophysiological analysis indicates that, whereas dSod1G85R aCC motor neurons exhibit slight hypoexcitablity, they receive similar excitatory inputs from CPG interneurons in both dSod1WTLoxP and dSod1G85R with an output not distinguishable in terms of eEPSP amplitude or in maximal muscle contraction (Figs. 4, 5). Therefore, no alterations in motor neuron properties were found that could easily account for the reduced motor activity observed in dSod1G85R larvae.
Peripheral feedback dysfunction is linked to a reduction in motor output in dSod1G85R
The crawling behavior of larvae depends on output from CPG interneurons to generate the rhythmic motor patterns executed by motor neurons and muscles. The rate of the rhythmic signals produced by the CPG can then be modified by feedback from peripheral proprioceptors (Hughes and Thomas, 2007; Song et al., 2007; Berni et al., 2012). Any change in motor circuit output could arise from a disruption in the function of the motor neuron, NMJ, or muscle or from defects in other circuit components (Hughes and Thomas, 2007; Song et al., 2007). Targeted electrophysiological measurements failed to uncover defects in dSod1G85R motor neuron and NMJ function (Figs. 4, 5) that correlate with the clear reduction in neural activity evident in fictive crawling preparations (Fig. 3F,G). Therefore, we considered the possibility that some aspect of the motor pattern initiated by the CPG is abnormal in dSod1G85R or that feedback from the periphery to the CNS is defective. To distinguish between these possibilities, we compared the activity of an intact motor circuit to the activity generated by the CPG when the CNS is separated from, and no longer receives, proprioceptive or other sensory feedback.
Extracellular recordings were obtained from a segmental nerve in intact fictive crawling preparations of dSod1WTLoxP and dSod1G85R third instar larvae (Fig. 6). To eliminate peripheral feedback, segmental nerves that contain both motor and sensory axonal tracts were cut distally (deafferented) to prevent sensory input to the CNS. Extracellular recordings were then obtained proximally from axonal tracts extending out of the VNC. First, to exclude the possibility that sequential recordings could affect data quality due to a run-down effect, we compared the frequency of activity bursts from the proximal region of segmental nerves in freshly dissected CNS samples with those recorded from CNS samples that had already been recorded from as part of the intact circuit. We found no difference in their activity indicating no evidence of run-down in this experimental paradigm (Fig. 6D).
The frequency of activity bursts detected by extracellular recordings before and after severing the segmental nerve were compared between dSod1WTLoxP and dSod1G85R (Fig. 6E,F). Although a change in event frequency was not observed in dSod1WTLoxP, the event frequency of dSod1G85R doubled after deafferenting. The doubling of the burst frequency following the removal of ascending input from the periphery suggested that, in an intact dSod1G85R animal, sensory feedback slows the motor pattern produced by the CPG. When this apparently defective peripheral feedback is eliminated by severing the segmental nerve, the motor pattern in dSod1G85R speeds up.
When we compared the rhythmic motor pattern produced by the CPG between dSod1WTLoxP vs dSod1G85R as measured from the isolated CNS, we found a higher rate of bursting in dSod1G85R compared with dSod1WTLoxP (Fig. 6E, cut “C”), indicating that two components of the dSod1G85R larval motor circuit exhibit altered functions. Not only does sensory feedback slow motor circuit output, but the dSod1G85R CPG appears to fire more frequently than the control dSod1WTLoxP CPG. Although this higher CPG output would be predicted to increase the frequency of muscle contraction and rate of dSod1G85R larval locomotion, we observed that, when the circuit is intact, the defect in peripheral feedback appears to override this CPG increase and slows the overall motor circuit output, resulting in reduced locomotor activity typical of dSod1G85R larvae.
We have shown that expressing gbb in glutamatergic neurons under the control of OK371-Gal4 can increase dSod1G85R larval locomotion (Fig. 3) and, consistent with this suppression of dSod1G85R-induced motor dysfunction, we found that the event frequency recorded from segmental nerves in the intact preparations of OK371>gbb; dSod1G85R was higher than that observed in dSod1G85R (Fig. 6B,E, intact “I”). Removing peripheral feedback by deafferenting OK371>gbb; dSod1G85R preparations did not produce a significant difference in the frequency of bursts compared with the intact preparation (Fig. 6E,F), indicating that, when gbb is expressed in motor neurons and other glutamatergic neurons, sensory feedback is restored. These findings are consistent with a model in which activation of BMP signaling affects the efficacy of sensory feedback in dSod1G85R to improve motor function.
Neuronal processes are altered in the dSod1G85R larval nerve cord
Defects in peripheral feedback could arise from dysfunction in the ascending sensory neurons themselves and/or from a disruption in the processing of sensory input by the interneuron network within the dSod1G85R VNC. Proprioceptors play a critical role in relaying peripheral feedback and disrupting normal proprioceptor function slows the larval locomotor pattern by increasing peristaltic wave duration (Hughes and Thomas, 2007; Song et al., 2007). Because our data indicate that peripheral feedback slows dSod1G85R locomotion and peristaltic wave duration is increased in dSod1G85R, we considered the possibility that the proprioceptors are defective (Fig. 7). 2-21-Gal4 is expressed in proprioceptors (Hughes and Thomas, 2007), the cell bodies of which reside on the body wall and project axons into the medial region of the nerve cord, where they are thought to synapse with interneurons (Fig. 7A) (Merritt and Whitington, 1995; Grueber et al., 2007; Schneider-Mizell et al., 2016). The dendritic arbors of proprioceptors could be visualized in 2-21-Gal4>mRFP and no gross abnormalities were apparent in dSod1G85R. The dendritic branching of the ddaE proprioceptor was examined in detail by Sholl analysis in both dSod1WTLoxP and dSod1G85R larvae. The ddaE cell body was recognized in the larval body wall based on its position (Fig. 7B). The number of ddaE dendritic intersections changed with distance from the cell body, but no significant difference was observed between genotypes, suggesting that, in dSod1G85R, ddaE dendritic projections are not different from dSod1WTLoxP (Fig. 7B).
In addition to its expression in proprioceptors, 2-21-Gal4 is also expressed in a subset of interneurons within the nerve cord (Fig. 7C; GFP-positive cell bodies in 2-21-Gal4>GFP). 2-21-Gal4-positive axonal projections in the longitudinal tracts adjacent to the VNC midline are clearly visualized by mRFP (Fig. 7D). The intensity of RFP within the boxed regions of the longitudinal tracts was measured in dSod1WTLoxP and dSod1G85R VNCs (Fig. 7D). The mRFP signal is less intense in dSod1G85R, suggestive of a reduction in 2-21-Gal4-labeled neuronal processes. Such a change in 2-21-Gal4-labeled neuronal processes in the dSod1G85R larval VNC is consistent with our finding that nonmotor neuron function is compromised in dSod1G85R (Fig. 6).
BMP signaling acts in specific components of the motor circuit to rescue dSod1G85R-induced dysfunction
Gbb, the BMP7 ortholog, has been shown to act as a retrograde signal at the Drosophila larval NMJ (McCabe et al., 2003). Transduction of BMP signaling results in phosphorylation of the R-Smad Mad in Drosophila (Hoodless et al., 1996; Tanimoto et al., 2000). As expected, pMad is detected in motor neuron cell bodies in the VNC, as well as at low levels at the NMJ of the wild-type (Fuentes-Medel et al., 2012). In dSod1G85R, the level of pMad in the motor neuron cell body is not different from the wild-type, but a significant increase in pMad is apparent at the dSod1G85R NMJ (Fig. 8A,B). The consequence of an increase in pMad at the dSod1G85R NMJ is not yet clear, but this observation, in addition to the fact that an increase in gbb does not lead to an overgrowth of dSod1G85R NMJ (Fig. 4A), suggests that the NMJ is not completely normal despite maintaining relatively normal function.
Figure 8.

gbb expressed under the control of OK371-Gal4 broadly induces pMad in VNC. A, pMad (gray) levels in motor neuron cell bodies (white arrows), as determined by anti-PS3 pMad antibody (Abcam ab52903, PS3), are unchanged in dSod1G85R compared with dSod1WTLoxP (MN cell body: dSod1WTLoxP: 1.0 ± 0.12AU, n = 9; dSod1G85R: 1.2 ± 0.79AU, n = 10; p = 0.18, t test). The expression of gbb in dSod1G85R led to broadened and increased pMad staining in cells and processes along the nerve cord midline. pMad in the VNC recognized by a different antibody (anti-pSmad1, a gift from Peter tenDjike) gave a similar staining pattern (data not shown). B, At the NMJ (6/7), pMad is significantly higher in dSod1G85R compared with dSod1WTLoxP (NMJ log2(pMad/HRP) ratio: dSod1WTLoxP = −3.1 ± 0.24, n = 9; dSod1G85R = −2.1 ± 0.28, n = 11; dSod1WTLoxP vs dSod1G85R p = 0.023, unequal variance t test with Holm–Bonferroni correction). Overexpression of gbb in glutamatergic motor neurons does not lead to a further increase in synaptic pMad (OK371-Gal4>UAS-gbb) (log2(pMad/HRP) ratio: dSod1G85R = −2.1 ± 0.28, n = 11; OK371-Gal4/UAS-gbb9.9;dSod1G85R = −1.8 ± 0.30, n = 9. dSod1G85R vs OK371-Gal4/UAS-gbb9.9;dSod1G85R p = 0.50, equal variance t test with Holm–Bonferroni correction); green, presynaptic anti-HRP; red, muscle, phalloidin; gray, anti-PS1). C, The expression pattern of OK371-Gal4 is highlighted by OK371-Gal4>UAS-GFP in the medially located motor neurons (bracket) of the VNC, as well as in glutamatergic interneurons. Motor neuron innervation of muscles 6/7 is evident. Scale bars: 25 μm (A, B) and 50 μm (C). *p < 0.05.
The presence of pMad is an indicator of cells that have received a BMP signal. In response to gbb overexpression in glutamatergic neurons (OK371>gbb; dSod1G85R), we found very high levels of pMad in cells of the VNC outside of the medially located motor neurons (Fig. 8A). Although OK371-Gal4 is expressed in glutamatergic motor neurons and premotor interneurons (Fig. 8C), the broad distribution of pMad suggests that OK371>gbb may result in a nonautonomous activation of BMP signaling due to the secreted nature of Gbb (Fig. 8A). Interestingly, despite the expression of OK371-Gal4 in motor neurons, as clearly indicated by the detection of GFP in the NMJ of OK371-Gal4>UAS-GFP, overexpression of gbb did not result in a further increase in pMad over the already elevated synaptic pMad in the dSod1G85R NMJ (Fig. 8B). Therefore, it appears that Gbb produced by glutamatergic neurons can induce signaling in a nonautonomous manner within the VNC, but it is unable to induce a change in pMad levels at the motor neuron synapse. This inability of Gbb to induce synaptic pMad is in agreement with the findings of Sulkowski et al. (2016). However, the ability of Gbb to induce high levels of pMad in a nonautonomous manner within cells of the VNC raises the possibility that Gbb contributes to the rescue of dSod1G85R dysfunction by activation of BMP signaling in other cell types, such as nonmotor neurons.
To better define in which cells activation of BMP signaling is critical for the suppression of dSod1G85R-associated phenotypes, we induced the pathway in a cell-autonomous manner by expressing a constitutively active form of the Drosophila BMP type I receptor, SaxA (Xie et al., 1994; Twombly et al., 2009; Ball et al., 2010; Piccioli and Littleton, 2014) (Fig. 9A,B). Activation of BMP signaling in either glutamatergic neurons (OK371-Gal4>SaxA;dSod1G85R), which include motor neurons and a subset of premotor interneurons, or cholinergic neurons, which make up sensory and interneurons in Drosophila (ChAT-Gal4>SaxA;dSod1G85R), leads to an increase in the locomotor activity of dSod1G85R larvae (Fig. 9A,B, Table 3). Activation of BMP signaling in cholinergic neurons also resulted in an increase in successful eclosion, with 10.2% of dSod1G85R adults (ChAT-Gal4/+;UAS-SaxA dSodG85R/dSodG85R) emerging from their pupal cases (Table 4).
Figure 9.

BMP signaling rescues dSod1G85R locomotion defect when activated in nonmotor neurons. A, Traces of larvae crawling over 90 s. B, Cell-autonomous activation expression of an activated BMP receptor, SaxA, in glutamatergic neurons (OK371-Gal4), cholinergic neurons (ChAT-Gal4), or proprioceptors (2-21-Gal4) rescues the larval locomotion defect of dSod1G85R. Expression in multidendritic sensory neurons (MD-Gal4), glial cells (Repo-Gal4), or muscle (BG57-Gal4) did not. **p < 0.01, ***p < 0.001.
Expression of SaxA in multidendritic sensory neurons (MD-Gal4) did not rescue either dSod1G85R larval locomotion or eclosion (Fig. 9, Tables 3, 4). However, interestingly, driving SaxA in a subset of multidendritic sensory neurons, the proprioceptors (2-21-Gal4>SaxA; dSodG85R) enabled both an increase in dSodG85R larval locomotion and adult eclosion (Fig. 9B, Tables 3, 4). Driving expression of SaxA in either muscles (BG57-Gal4) or glial cells (Repo-Gal4) did not result in a rescue dSod1G85R larval locomotion or eclosion, indicating that the ability of BMP signaling activation to rescue dSod1G85R phenotypes is successful in only some cell types and/or with only some Gal4 drivers (Fig. 9B, Tables 3, 4).
Motor defects associated with dSod1G85R can be rescued by the cell-autonomous activation of BMP signaling in at least two different components of the motor circuit: glutamatergic and cholinergic neurons. Within the cholinergic population, we found robust suppression of dSod1G85R motor defects by the specific activation of BMP signaling in proprioceptors (2-21-Gal4), emphasizing the importance of this class of neurons in the restoration of motor circuit function. Importantly, this result, together with our findings of a defect in sensory feedback that underlies the early circuit dysfunction of dSod1G85R, underscores the importance of nonmotor neurons in the progression of ALS-associated neurodegeneration. Furthermore, our study highlights sensory neurons as a source for the identification of biomarkers indicative of the early disease state and as a site for the development of therapeutics.
Discussion
A hallmark of late-stage ALS is the profound degeneration of motor neurons; however, the site and initiating events responsible for the ultimate degeneration of motor neurons per se are not fully understood. Significant progress identifying cellular defects associated with fALS has been made, as have attempts to reduce degeneration (Boillée et al., 2006; Yamanaka et al., 2008; Casci and Pandey, 2015; Freibaum et al., 2015; Jovičić et al., 2015; Zhang et al., 2015; Nagy et al., 2016). In mice, motor neuron death can be prevented by blocking motor neuron expression of mutant FUS or TDP43 (Ditsworth et al., 2017; Scekic-Zahirovic et al., 2017). Curiously, whereas motor neuron death was prevented, locomotion was still impaired, raising the possibility that cells other than motor neurons can affect the onset and/or progression of ALS (Pramatarova et al., 2001; Boillée et al., 2006; Jaarsma et al., 2008; Yamanaka et al., 2008). Glial cells have also been shown to influence ALS progression (Di Giorgio et al., 2007; Lobsiger and Cleveland, 2007). Here, using a knock-in SOD1-ALS model, we report that nonmotor neuron dysfunction in early-stage animals correlates with disrupted locomotion in the absence of motor neuron degeneration (Fig. 10).
Figure 10.
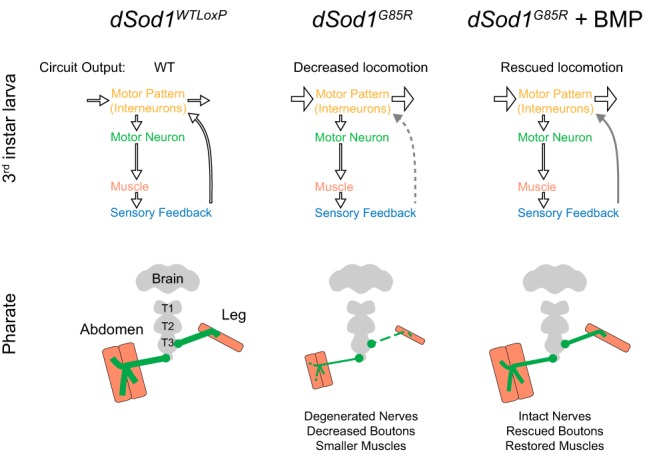
Graphical summary. Defects in dSod1G85R motor circuitry are rescued by activation of BMP signaling. Top, Schematic representation of larval motor circuit. The larval locomotor pattern (yellow) is dictated by both CPG activity (horizontal arrows) in the VNC, signals from sensory neurons (blue) in the periphery and their integration by interneurons (arrow to motor pattern) to provide feedback to the CPG. Early-stage dSod1G85R animals (third instar larvae) exhibit reduced locomotion but no degeneration of significant defects in motor neuron (green) function. Instead, an increase in CPG activity (wide horizontal arrows) in dSod1G85R was revealed along with a disruption to peripheral feedback (dotted gray line) that results in reduced locomotor activity. Activation of BMP signaling increases dSod1G85R larval locomotion. The heightened activity of dSod1G85R CPG is unchanged but feedback from the periphery is improved (solid gray arrow). Cell-autonomous activation of BMP signaling in glutamatergic neurons, cholinergic neurons, or proprioceptors increases dSod1G85R locomotion. Bottom, Adult Drosophila abdominal muscles and leg muscles are innervated by different sets of motor neurons (green). Both sets of motor neurons have disrupted morphology in end-stage dSod1G85R animals. Synaptic output at the abdominal NMJ and muscle size are decreased. These abnormalities in motor neuron morphology, motor neuron output, and muscle size likely contribute to dSod1G85R eclosion failure. The expression of gbb in dSod1G85R motor neurons improves motor neuron morphology, motor neuron output, muscle size, and allows the eclosion of some mutant animals. Driving expression of SaxA in cholinergic neurons or proprioceptors increases dSod1G85R larval locomotion and the percentage of animals that eclose.
End-stage dSod1G85R knock-in animals exhibit key features of ALS, including severely compromised locomotion, reduced motor neuron function, visible motor neuron degeneration, and decreased muscle size (Sahin et al., 2017; Fig. 10). Earlier, at the first sign of compromised locomotor activity, we found the functional properties of dSod1G85R motor neurons to be largely normal. Circuit analysis revealed a specific disruption in the function of nonmotor neurons, implicating this dysfunction as an indicator of early ALS. We also found that both early- and late-stage dSod1G85R motor defects can be attenuated by BMP signaling, a pathway known to stimulate neuronal growth in both invertebrates and vertebrates (Lee-Hoeflich et al., 2004; Sun et al., 2007; Hocking et al., 2009). Given the disruption in sensory feedback in the dSod1G85R larval motor circuit, it is noteworthy that both compromised locomotor activity of mutant larvae and eclosion failure of mutant adults is suppressed by activating BMP signaling within some interneurons, especially proprioceptors, the subset of sensory neurons that relay the status of muscle contraction back to the CNS (Fig. 10). These findings, coupled with those that have previously implicated nonautonomous factors in ALS degeneration (Ilieva et al., 2009), strongly support a hypothesis that nonmotor neurons such as proprioceptors are affected first in ALS and their dysfunction ultimately leads to motor neuron degeneration
Motor circuit dysfunction results in locomotor defects in a knock-in model of Sod1-ALS
Although compromised nonmotor neurons have not been formally shown to cause motor function loss in ALS, it has been suggested that neurodegenerative disorders can arise from circuit or network disorders (Warren et al., 2013; Fornito et al., 2015). Certainly, in late-stage ALS, disruptions in interneuron and/or sensory function were found to accompany severe motor deficits (Theys et al., 1999; Fischer et al., 2005; Stephens et al., 2006; Isaacs et al., 2007; Pugdahl et al., 2007; Sunico et al., 2011; McGown et al., 2013; Vinsant et al., 2013; Vaughan et al., 2015; Clark et al., 2017). Interestingly, in several models of ALS, an observed locomotor defect is associated with only a minor decrease in NMJ transmission (Diaper et al., 2013; Rocha et al., 2013; Zhang et al., 2015), one that would not have been expected to overcome the large safety factor observed at NMJs (Marrus and DiAntonio, 2005).
Recent studies on the motor neuron disease spinal muscular atrophy (SMA) highlighted sensory and interneurons in disease progression. Interneurons deficient in the SMN1 gene were found to increase the resistance and excitability of cocultured wild-type motor neurons in a stem-cell-based SMA model (Simon et al., 2016). In Drosophila, the smn gene is required in both proprioceptive sensory neurons and interneurons for proper motor neuron function (Imlach et al., 2012; Lotti et al., 2012). In mice, expression of SMN in proprioceptors was shown to dramatically influence motor neuron excitability (Fletcher et al., 2017). Disruption in the knock-in dSod1G85R ALS model of sensory and interneuron function resembles findings in models of SMA. Future studies to assess whether motor neuron and muscle deficits in ALS arise from a specific loss of proprioceptive function will be of interest. Certainly, our findings presented here, coupled with those in SMA models, support the narrative that circuitry disruption is an important consideration in motor neuron disease.
Activation of BMP signaling suppresses ALS phenotypes
In Drosophila, BMP signaling contributes to NMJ growth, neurotransmission, and synaptic plasticity (McCabe et al., 2003; Baines, 2004; Goold and Davis, 2007; Berke et al., 2013; Piccioli and Littleton, 2014). An increase in BMP signaling is sufficient to induce NMJ growth and produce more robust eEPSPs (James and Broihier, 2011; James et al., 2014; our observations). Increased BMP signaling due to a reduction in gene dosage of the Dad antagonist was shown to alleviate some NMJ abnormalities in a Drosophila SMA model and, more recently, in an ALS model (Chang et al., 2008; Deshpande et al., 2016). We initially explored the possibility that activating BMP signaling could ameliorate motor neuron degeneration in dSod1G85R pharates. Indeed, driving the expression of the BMP ligand Gbb in motor neurons and glutamatergic interneurons rescued adult dSod1G85R phenotypes at the NMJ. However, even in early-stage dSod1G85R animals in which NMJ defects are minimal, we found that gbb expression in glutamatergic neurons could still restore locomotion, presumably by acting somewhere other than the NMJ. Phenotypic suppression of dSod1G85R was not only achieved by cell-autonomous activation of BMP signaling glutamatergic motor neurons and interneurons, but also in cholinergic sensory and interneurons. The finding that nonmotor neuronal processes appear reduced in dSod1G85R points to these cells as a potential source of dysfunction in the ALS motor circuit. Attempts to record directly from the proprioceptors were unsuccessful, but the rescue of dSod1G85R by SaxA expression in a subset of sensory neurons, the proprioceptors, is concordant with the results of our circuit analysis highlighting a disruption in sensory feedback as the earliest defect detected in dSod1G85R and supports the hypothesis that nonmotor neurons play a critical role in ALS motor dysfunction. Curiously, expressing SaxA in all multidendritic sensory neurons did not rescue, whereas both broader expression of SaxA in the cholinergic neurons (sensory and interneurons) and specific expression in the proprioceptive sensory neurons resulted in restoration of function. This could reflect a differential response to BMP signaling (Follansbee et al., 2017) and, in the case of broad activation in all cholinergic neurons, the cumulative effect may on balance be positive to result in an increase in locomotion and eclosion. It is also possible the release of neuropeptides that influence muscle contraction and eclosion and that are known to be dependent on BMP signaling (Marques et al., 2003, Veverytsa and Allan, 2011) could have been differentially affected when SaxA was expressed by different Gal4 drivers.
Interestingly, several pieces of evidence indicate that BMP signaling itself is disrupted in dSod1G85R. Normally, the expression of gbb in motor neurons results in NMJ growth, a consequence not seen in dSod1G85R despite the clear indication of active signaling in cells of the VNC showing high levels of pMad. Furthermore, the NMJs of dSod1G85R show a significant elevation in synaptic pMad compared with controls. Dysregulation of BMP signaling may be a common feature of ALS because loss of function of the Drosophila TDP43 ortholog tbph and overexpression of hTDP43, both associated with NMJ disruptions, exhibit decreased synaptic pMad levels (Deshpande et al., 2016). Although disrupted in both SOD1 and TDP43 ALS models, the mechanics of how BMP signaling is regulated likely differs between these ALS models because manipulation of TDP43 levels results, not in an elevation of synaptic pMad, but in a reduction. Certainly, the molecular mechanisms controlling the production of synaptic pMad versus nuclear pMad in the motor neuron cell body appear distinct because nuclear pMad levels remain unchanged in both the dSod1G85R and TDP43 models. It is possible that multiple aspects of BMP signaling are defective in ALS. The ligand-independent nature of synaptic pMad production, its uncoupling from NMJ growth, and its proposed role in stabilizing postsynaptic type IIA glutamate receptors (Sulkowski et al., 2016) raises future research questions of exactly how endogenous BMP signaling is altered in ALS and how selective cellular activation of the pathway leads to restoration of motor function.
Overall, our findings reinforce the importance of developing disease models that recapitulate the endogenous spatial and temporal expression of disease alleles, especially as a complementary approach to targeted expression of orthologous genes. The ability to study a fully “diseased” motor circuit and to probe discrete elements in the context of the intact circuit is crucial for elucidating the interrelatedness of a system more analogous to the patient. Although additional studies are necessary, the work presented here provides a framework that highlights the consequences of disease-associated dysfunction in a relatively simple motor circuit. Importantly, our finding that ALS-like phenotypes can be alleviated by activation of the neurotrophic BMP pathway in nonmotor neurons lays the groundwork to identify targets of BMP signaling responsible for such rescue. In the future, developing strategies that target nonmotor neurons may provide additional benefits to therapeutic approaches that have focused on motor neurons in the treatment of ALS.
Footnotes
This work was supported by the National Institutes of Health (NIH Grant RO1GM068118 to K.A.W. and Grant T32DK060415 to A.H.), the ALS Finding a Cure Foundation (K.A.W., R.R., D.L.), The Judith and Jean Pape Adams Foundation (K.A.W.), and the Robert J. and Nancy D. Carney Institute for Brain Science Robin Chemers Neustein (Graduate Award to A.H.). We thank Arturo Andrade and Nara Muraro for electrophysiology training and advice; the Bloomington Drosophila Stock Center (NIH Grant P40OD018537), Michael O'Connor, John Thomas, and Heather Broihier for Drosophila lines and sharing reagents; the Developmental Studies Hybridoma Bank (DSHB), created by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the NIH and maintained at The University of Iowa, Department of Biology, for antibodies used in this study; the Leduc Bioimaging facility at Brown University for training; and members of the Lipscombe and Wharton laboratories for ongoing discussions.
The authors declare no competing financial interests.
References
- Arthur et al., 2016. Arthur KC, Calvo A, Price TR, Geiger JT, Chiò A, Traynor BJ (2016) Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun 7:12408. 10.1038/ncomms12408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz et al., 1997. Azzouz M, Leclerc n, Gurney M, Warter JM, Poindron P, Borg J (1997) Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve 20:45–51. 10.1002/(SICI)1097-4598(199701)20:1<45::AID-MUS6>3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- Baines, 2004. Baines RA. (2004) Synaptic strengthening mediated by bone morphogenetic protein-dependent retrograde signaling in the Drosophila CNS. J Neurosci 24:6904–6911. 10.1523/JNEUROSCI.1978-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball et al., 2010. Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, Haghighi AP (2010) Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron 66:536–549. 10.1016/j.neuron.2010.04.011 [DOI] [PubMed] [Google Scholar]
- Berke et al., 2013. Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H (2013) Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci 33:17937–17950. 10.1523/JNEUROSCI.6075-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berni et al., 2012. Berni J, Pulver SR, Griffith LC, Bate M (2012) Autonomous circuitry for substrate exploration in freely moving Drosophila larvae. Curr Biol 22:1861–1870. 10.1016/j.cub.2012.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée et al., 2006. Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392. 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Broadie and Bate, 1991. Broadie KS, Bate M (1991) The development of adult muscles in Drosophila: ablation of identified muscle precursor cells. Development 113:103–118. [DOI] [PubMed] [Google Scholar]
- Budnik et al., 1996. Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M (1996) Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron 17:627–640. 10.1016/S0896-6273(00)80196-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casci and Pandey, 2015. Casci I, Pandey UB (2015) A fruitful endeavor: modeling ALS in the fruit fly. Brain Res 1607:47–74. 10.1016/j.brainres.2014.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaert and Birman, 2001. Cattaert D, Birman S (2001) Blockade of the central generator of locomotor rhythm by noncompetitive NMDA receptor antagonists in Drosophila larvae. J Neurobiol 48:58–73. 10.1002/neu.1042 [DOI] [PubMed] [Google Scholar]
- Chang et al., 2008. Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S (2008) Modeling spinal muscular atrophy in Drosophila. PLoS One 3:e3209. 10.1371/journal.pone.0003209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò et al., 2013. Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, White LA (2013) Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41:118–130. 10.1159/000351153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi et al., 2004. Choi JC, Park D, Griffith LC (2004) Electrophysiological and morphological characterization of identified motor neurons in the Drosophila third instar larva central nervous system. J Neurophysiol 91:2353–2365. 10.1152/jn.01115.2003 [DOI] [PubMed] [Google Scholar]
- Clark et al., 2017. Clark RM, Blizzard CA, Young KM, King AE, Dickson TC (2017) Calretinin and neuropeptide y interneurons are differentially altered in the motor cortex of the SOD1 G93A mouse model of ALS. Sci Rep 7:44461. 10.1038/srep44461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, 1979. Crossley AC. (1979) The morphology and development of the Drosophila muscular system. In: Ashburner M, Wright TRF, eds. The genetics and biology of drosophila, Vol. 2b, pp 499–560. New York: Academic Press. [Google Scholar]
- Denys and Norris, 1979. Denys EH, Norris FH Jr (1979) Amyotrophic lateral sclerosis: impairment of neuromuscular transmission. Arch Neurol 36:202–205. 10.1001/archneur.1979.00500400056008 [DOI] [PubMed] [Google Scholar]
- Deshpande et al., 2016. Deshpande M, Feiger Z, Shilton AK, Luo CC, Silverman E, Rodal AA (2016) Role of BMP receptor traffic in synaptic growth defects in an ALS model. Mol Biol Cell 27:2898–2910. 10.1091/mbc.E16-07-0519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper et al., 2013. Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A, Ludlow ZN, Vanden Broeck L, Callaerts P, Dermaut B, Al-Chalabi A, Shaw CE, Robinson IM, Hirth F (2013) Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet 22:1539–1557. 10.1093/hmg/ddt005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio et al., 2007. Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10:608–614. 10.1038/nn1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditsworth et al., 2017. Ditsworth D, Maldonado M, McAlonis-Downes M, Sun S, Seelman A, Drenner K, Arnold E, Ling SC, Pizzo D, Ravits J, Cleveland DW, Da Cruz S (2017) Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol 133:907–922. 10.1007/s00401-017-1698-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer et al., 2005. Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M, Asress S, Adalbert R, Alexander GM, Glass JD (2005) The WldS gene modestly prolongs survival in the SOD1 G93A fALS mouse. Neurobiol Dis 19:293–300. 10.1016/j.nbd.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Fletcher et al., 2017. Fletcher EV, Simon CM, Pagiazitis JG, Chalif JI, Vukojicic A, Drobac E, Wang X, Mentis GZ (2017) Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat Neurosci 20:905–916. 10.1038/nn.4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follansbee et al., 2017. Follansbee TL, Gjelsvik KJ, Brann CL, McParland AL, Longhurst CA, Galko MJ, Ganter GK (2017) Drosophila nociceptive sensitization requires BMP signaling via the canonical SMAD pathway. J Neurosci 37:8524–8533. 10.1523/JNEUROSCI.3458-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito et al., 2015. Fornito A, Zalesky A, Breakspear M (2015) The connectomics of brain disorders. Nat Rev Neurosci 16:159–172. 10.1038/nrn3901 [DOI] [PubMed] [Google Scholar]
- Fox et al., 2006. Fox LE, Soll DR, Wu CF (2006) Coordination and modulation of locomotion pattern generators in Drosophila larvae: effects of altered biogenic amine levels by the tyramine beta hydroxlyase mutation. J Neurosci 26:1486–1498. 10.1523/JNEUROSCI.4749-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum et al., 2015. Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders n, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Medel et al., 2012. Fuentes-Medel Y, Ashley J, Barria R, Maloney R, Freeman M, Budnik V (2012) Integration of a retrograde signal during synapse formation by glia-secreted TGF-β ligand. Curr Biol 22:1831–1838. 10.1016/j.cub.2012.07.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold and Davis, 2007. Goold CP, Davis GW (2007) The BMP ligand gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron 56:109–123. 10.1016/j.neuron.2007.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber et al., 2007. Grueber WB, Ye B, Yang CH, Younger S, Borden K, Jan LY, Jan Y-n (2007) Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development 134:55–64. 10.1242/dev.02666 [DOI] [PubMed] [Google Scholar]
- Hebbar et al., 2006. Hebbar S, Hall RE, Demski SA, Subramanian A, Fernandes JJ (2006) The adult abdominal neuromuscular junction of Drosophila: a model for synaptic plasticity. J Neurobiol 66:1140–1155. 10.1002/neu.20279 [DOI] [PubMed] [Google Scholar]
- Hocking et al., 2009. Hocking JC, Hehr CL, Bertolesi G, Funakoshi H, Nakamura T, McFarlane S (2009) LIMK1 acts downstream of BMP signaling in developing retinal ganglion cell axons but not dendrites. Dev Biol 330:273–285. 10.1016/j.ydbio.2009.03.027 [DOI] [PubMed] [Google Scholar]
- Hoodless et al., 1996. Hoodless PA, Haerry T, Abdollah S, Stapleton M, Connor MB, Attisano L, Wrana JL (1996) MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell 85:489–500. 10.1016/S0092-8674(00)81250-7 [DOI] [PubMed] [Google Scholar]
- Hughes and Thomas, 2007. Hughes CL, Thomas JB (2007) A sensory feedback circuit coordinates muscle activity in Drosophila. Mol Cell Neurosci 35:383–396. 10.1016/j.mcn.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva et al., 2009. Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772. 10.1083/jcb.200908164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach et al., 2012. Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD (2012) SMN is required for sensory-motor circuit function in Drosophila. Cell 151:427–439. 10.1016/j.cell.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs et al., 2007. Isaacs JD, Dean AF, Shaw CE, Al-Chalabi A, Mills KR, Leigh PN (2007) Amyotrophic lateral sclerosis with sensory neuropathy: part of a multisystem disorder? J Neurol Neurosurg Psychiatry 78:750–753. 10.1136/jnnp.2006.098798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai et al., 2016. Iwai Y, Shibuya K, Misawa S, Sekiguchi Y, Watanabe K, Amino H, Kuwabara S (2016) Axonal dysfunction precedes motor neuronal death in amyotrophic lateral sclerosis. PLoS One 11:e0158596. 10.1371/journal.pone.0158596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma et al., 2008. Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC (2008) Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci 28:2075–2088. 10.1523/JNEUROSCI.5258-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James and Broihier, 2011. James RE, Broihier HT (2011) Crimpy inhibits the BMP homolog gbb in motoneurons to enable proper growth control at the Drosophila neuromuscular junction. Development 138:3273–3286. 10.1242/dev.066142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James et al., 2014. James RE, Hoover KM, Bulgari D, McLaughlin CN, Wilson CG, Wharton KA, Levitan ES, Broihier HT (2014) Crimpy enables discrimination of presynaptic and postsynaptic pools of a BMP at the Drosophila neuromuscular junction. Dev Cell 31:586–598. 10.1016/j.devcel.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan and Jan, 1976. Jan LY, Jan YN (1976) Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol 262:189–214. 10.1113/jphysiol.1976.sp011592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan et al., 1997. Jordan J, Böttner M, Schluesener HJ, Unsicker K, Krieglstein K (1997) Bone morphogenetic proteins: neurotrophic roles for midbrain dopaminergic neurons and implications of astroglial cells. Eur J Neurosci 9:1699–1709. 10.1111/j.1460-9568.1997.tb01527.x [DOI] [PubMed] [Google Scholar]
- Jovičić et al., 2015. Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai n, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18:1226–1229. 10.1038/nn.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai et al., 2006. Kanai K, Kuwabara S, Misawa S, Tamura n, Ogawara K, Nakata M, Sawai S, Hattori T, Bostock H (2006) Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 129:953–962. 10.1093/brain/awl024 [DOI] [PubMed] [Google Scholar]
- Khalsa et al., 1998. Khalsa O, Yoon JW, Torres-Schumann S, Wharton KA (1998) TGF-beta/BMP superfamily members, Gbb-60A and Dpp, cooperate to provide pattern information and establish cell identity in the Drosophila wing. Development 125:2723–2734. [DOI] [PubMed] [Google Scholar]
- Kiernan et al., 2011. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 377:942–955. 10.1016/S0140-6736(10)61156-7 [DOI] [PubMed] [Google Scholar]
- Kimura and Truman, 1990. Kimura KI, Truman JW (1990) Postmetamorphic cell death in the nervous and muscular systems of Drosophila melanogaster. J Neurosci 10:403–411. 10.1523/JNEUROSCI.10-02-00403.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King et al., 2016. King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2016) Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol 275:162–171. 10.1016/j.expneurol.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Krieglstein et al., 2002. Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K (2002) TGF-β and the regulation of neuron survival and death. J Physiol Paris 96:25–30. 10.1016/S0928-4257(01)00077-8 [DOI] [PubMed] [Google Scholar]
- Lahiri et al., 2011. Lahiri S, Shen K, Klein M, Tang A, Kane E, Gershow M, Garrity P, Samuel AD (2011) Two alternating motor programs drive navigation in Drosophila larva. PLoS One 6:e23180. 10.1371/journal.pone.0023180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich et al., 2004. Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L (2004) Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 23:4792–4801. 10.1038/sj.emboj.7600418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger and Cleveland, 2007. Lobsiger CS, Cleveland DW (2007) Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci 10:1355–1360. 10.1038/nn1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti et al., 2012. Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, Pellizzoni L (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151:440–454. 10.1016/j.cell.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe et al., 1988. Lowe J, Lennox G, Jefferson D, Morrell K, McQuire D, Gray T, Landon M, Doherty FJ, Mayer RJ (1988) A filamentous inclusion body within anterior horn neurones in motor neurone disease defined by immunocytochemical localisation of ubiquitin. Neurosci Lett 94:203–210. 10.1016/0304-3940(88)90296-0 [DOI] [PubMed] [Google Scholar]
- Mahr and Aberle, 2006. Mahr A, Aberle H (2006) The expression pattern of the Drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr Patterns 6:299–309. 10.1016/j.modgep.2005.07.006 [DOI] [PubMed] [Google Scholar]
- Marley and Baines, 2011. Marley R, Baines RA (2011) Whole-cell patch recording from Drosophila larval neurons. Cold Spring Harb Protoc 2011:pdb.prot065664. 10.1101/pdb.prot065664 [DOI] [PubMed] [Google Scholar]
- Marqués et al., 2003. Marqués G, Haerry TE, Crotty ML, Xue M, Zhang B, O'Connor MB (2003) Retrograde gbb signaling through bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development 130:5457–5470. 10.1242/dev.00772 [DOI] [PubMed] [Google Scholar]
- Marrus and DiAntonio, 2005. Marrus SB, DiAntonio A (2005) Investigating the safety factor at an invertebrate neuromuscular junction. J Neurobiol 63:62–69. 10.1002/neu.20120 [DOI] [PubMed] [Google Scholar]
- McCabe et al., 2003. McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB (2003) The BMP homolog gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39:241–254. 10.1016/S0896-6273(03)00426-4 [DOI] [PubMed] [Google Scholar]
- McGown et al., 2013. McGown A, McDearmid JR, Panagiotaki n, Tong H, Al Mashhadi S, Redhead n, Lyon AN, Beattie CE, Shaw PJ, Ramesh TM (2013) Early interneuron dysfunction in ALS: insights from a mutant sod1 zebrafish model. Ann Neurol 73:246–258. 10.1002/ana.23780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNabb et al., 1997. McNabb SL, Baker JD, Agapite J, Steller H, Riddiford LM, Truman JW (1997) Disruption of a behavioral sequence by targeted death of peptidergic neurons in Drosophila. Neuron 19:813–823. 10.1016/S0896-6273(00)80963-0 [DOI] [PubMed] [Google Scholar]
- Merritt and Whitington, 1995. Merritt DJ, Whitington PM (1995) Central projections of sensory neurons in the Drosophila embryo correlate with sensory modality, soma position, and proneural gene function. J Neurosci 15:1755–1767. 10.1523/JNEUROSCI.15-03-01755.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesce and Fahrbach, 2002. Mesce KA, Fahrbach SE (2002) Integration of endocrine signals that regulate insect ecdysis. Front Neuroendocrinol 23:179–199. 10.1006/frne.2002.0228 [DOI] [PubMed] [Google Scholar]
- Nagy et al., 2016. Nagy M, Fenton WA, Li D, Furtak K, Horwich AL (2016) Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad Sci U S A 113:5424–5428. 10.1073/pnas.1604885113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm et al., 2013. Nahm M, Lee MJ, Parkinson W, Lee M, Kim H, Kim YJ, Kim S, Cho YS, Min BM, Bae YC, Broadie K, Lee S (2013) Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron 77:680–695. 10.1016/j.neuron.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioli and Littleton, 2014. Piccioli ZD, Littleton JT (2014) Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J Neurosci 34:4371–4381. 10.1523/JNEUROSCI.4943-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova et al., 2001. Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA (2001) Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 21:3369–3374. 10.1523/JNEUROSCI.21-10-03369.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugdahl et al., 2007. Pugdahl K, Fuglsang-Frederiksen A, de Carvalho M, Johnsen B, Fawcett PR, Labarre-Vila A, Liguori R, Nix WA, Schofield IS (2007) Generalised sensory system abnormalities in amyotrophic lateral sclerosis: a European multicentre study. J Neurol Neurosurg Psychiatry 78:746–749. 10.1136/jnnp.2006.098533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulver et al., 2015. Pulver SR, Bayley TG, Taylor AL, Berni J, Bate M, Hedwig B (2015) Imaging fictive locomotor patterns in larval Drosophila. J Neurophysiol 114:2564–2577. 10.1152/jn.00731.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha et al., 2013. Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA (2013) Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One 8:e73846. 10.1371/journal.pone.0073846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen et al., 1993. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX (1993) Mutations in Cu/ZN superoxide disumutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. 10.1038/362059a0 [DOI] [PubMed] [Google Scholar]
- Saccon et al., 2013. Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P (2013) Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136:2342–2358. 10.1093/brain/awt097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin et al., 2017. Sahin A, Held A, Bredvik K, Major P, Achilli TM, Kerson AG, Wharton K, Stilwell G, Reenan R (2017) Human SOD1 ALS mutations in a Drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain and loss of function components. Genetics 205:707–723. 10.1534/genetics.116.190850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvaterra and Kitamoto, 2001. Salvaterra PM, Kitamoto T (2001) Drosophila cholinergic neurons and processes visualized with Gal4/UAS-GFP. Brain Res Gene Expr Patterns 1:73–82. 10.1016/S1567-133X(01)00011-4 [DOI] [PubMed] [Google Scholar]
- Sanyal, 2009. Sanyal S. (2009) Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr Patterns 9:371–380. 10.1016/j.gep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Scekic-Zahirovic et al., 2017. Scekic-Zahirovic J, Oussini HE, Mersmann S, Drenner K, Wagner M, Sun Y, Allmeroth K, Dieterlé S, Sinniger J, Dirrig-Grosch S, René F, Dormann D, Haass C, Ludolph AC, Lagier-Tourenne C, Storkebaum E, Dupuis L (2017) Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol 133:887–906. 10.1007/s00401-017-1687-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Mizell et al., 2016. Schneider-Mizell CM, Gerhard S, Longair M, Kazimiers T, Li F, Zwart MF, Champion A, Midgley FM, Fetter RD, Saalfeld S, Cardona A (2016) Quantitative neuroanatomy for connectomics in Drosophila. Elife 5:e12059. 10.7554/eLife.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp et al., 2001. Sepp KJ, Schulte J, Auld VJ (2001) Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev Biol 238:47–63. 10.1006/dbio.2001.0411 [DOI] [PubMed] [Google Scholar]
- Simon et al., 2016. Simon CM, Janas AM, Lotti F, Tapia JC, Pellizzoni L, Mentis GZ (2016) A stem cell model of the motor circuit uncouples motor neuron death from hyperexcitability induced by SMN deficiency. Cell Rep 16:1416–1430. 10.1016/j.celrep.2016.06.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song et al., 2007. Song W, Onishi M, Jan LY, Jan YN (2007) Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci U S A 104:5199–5204. 10.1073/pnas.0700895104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens et al., 2006. Stephens B, Guiloff RJ, Navarrete R, Newman P, Nikhar n, Lewis P (2006) Widespread loss of neuronal populations in the spinal ventral horn in sporadic motor neuron disease: a morphometric study. J Neurol Sci 244:41–58. 10.1016/j.jns.2005.12.003 [DOI] [PubMed] [Google Scholar]
- Sulkowski et al., 2016. Sulkowski MJ, Han TH, Ott C, Wang Q, Verheyen EM, Lippincott-Schwartz J, Serpe M (2016) A novel, noncanonical BMP pathway modulates synapse maturation at the Drosophila neuromuscular junction. PLoS Genet 12:e1005810. 10.1371/journal.pgen.1005810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun and Salvaterra, 1995. Sun B, Salvaterra PM (1995) Characterization of nervana, a Drosophila melanogaster neuron-specific glycoprotein antigen recognized by anti-horseradish peroxidase antibodies. J Neurochem 65:434–443. [DOI] [PubMed] [Google Scholar]
- Sun et al., 2007. Sun M, Thomas MJ, Herder R, Bofenkamp ML, Selleck SB, O'Connor MB (2007) Presynaptic contributions of chordin to hippocampal plasticity and spatial learning. J Neurosci 27:7740–7750. 10.1523/JNEUROSCI.1604-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunico et al., 2011. Sunico CR, Domínguez G, García-Verdugo JM, Osta R, Montero F, Moreno-López B (2011) Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol 21:1–15. 10.1111/j.1750-3639.2010.00417.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandan and Bradley, 1985. Tandan R, Bradley WG (1985) Amyotrophic lateral sclerosis: part 1. clinical features, pathology, and ethical issues in management. Ann Neurol 18:271–280. 10.1002/ana.410180302 [DOI] [PubMed] [Google Scholar]
- Tanimoto et al., 2000. Tanimoto H, Itoh S, ten Dijke P, Tabata T (2000) Hedgehog creates a gradient of DPP activity in Drosophila wing imaginal discs. Mol Cell 5:59–71. 10.1016/S1097-2765(00)80403-7 [DOI] [PubMed] [Google Scholar]
- Taylor et al., 2016. Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theys et al., 1999. Theys PA, Peeters E, Robberecht W (1999) Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques. J Neurol 246:438–442. 10.1007/s004150050379 [DOI] [PubMed] [Google Scholar]
- Tsujihata et al., 1984. Tsujihata M, Hazama R, Yoshimura T, Satoh A, Mori M, Nagataki S (1984) The motor end-plate fine structure and ultrastructural localization of acetylcholine receptors in amyotrophic lateral sclerosis. Muscle Nerve 7:243–249. 10.1002/mus.880070310 [DOI] [PubMed] [Google Scholar]
- Twombly et al., 2009. Twombly V, Bangi E, Le V, Malnic B, Singer MA, Wharton KA (2009) Functional analysis of saxophone, the Drosophila gene encoding the BMP type I receptor ortholog of human ALK1/ACVRL1 and ACVR1/ALK2. Genetics 183:563–579, 1SI–8SI. 10.1534/genetics.109.105585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan et al., 2015. Vaughan SK, Kemp Z, Hatzipetros T, Vieira F, Valdez G (2015) Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J Comp Neurol 523:2477–2494. 10.1002/cne.23848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverytsa and Allan, 2011. Veverytsa L, Allan DW (2011) Retrograde BMP signaling controls Drosophila behavior through regulation of a peptide hormone battery. Development 138:3147–3157. 10.1242/dev.064105 [DOI] [PubMed] [Google Scholar]
- Vinsant et al., 2013. Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C (2013) Characterization of early pathogenesis in the SOD1 G93A mouse model of ALS: Part II, results and discussion. Brain Behav 3:431–457. 10.1002/brb3.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic et al., 2008. Vucic S, Nicholson GA, Kiernan MC (2008) Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131:1540–1550. 10.1093/brain/awn071 [DOI] [PubMed] [Google Scholar]
- Warren et al., 2013. Warren JD, Rohrer JD, Schott JM, Fox NC, Hardy J, Rossor MN (2013) Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci 36:561–569. 10.1016/j.tins.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie et al., 1994. Xie T, Finelli AL, Padgett RW (1994) The Drosophila saxophone gene: a serine-threonine kinase receptor of the TGF-beta superfamily. Science 263:1756–1759. 10.1126/science.8134837 [DOI] [PubMed] [Google Scholar]
- Yamanaka et al., 2008. Yamanaka K, Boillee S, Roberts EA, Garcia ML, McAlonis-Downes M, Mikse OR, Cleveland DW, Goldstein LS (2008) Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A 105:7594–7599. 10.1073/pnas.0802556105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanette et al., 2002. Zanette G, Tamburin S, Manganotti P, Refatti n, Forgione A, Rizzuto n (2002) Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin Neurophysiol 113:1688–1697. 10.1016/S1388-2457(02)00288-2 [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2015. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]