

Abstract
Mutations in the mitochondrial GTPase mitofusin 2 (MFN2) cause Charcot-Marie-Tooth disease type 2 (CMT2A), a form of peripheral neuropathy that compromises axonal function. Mitofusins promote mitochondrial fusion and regulate mitochondrial dynamics. They are also reported to be involved in forming contacts between mitochondria and the endoplasmic reticulum. The fruit fly, Drosophila melanogaster, is a powerful tool to model human neurodegenerative diseases, including CMT2A. Here, we have downregulated the expression of the Drosophila mitofusin (dMfn RNAi) in adult flies and showed that this activates mitochondrial retrograde signalling and is associated with an upregulation of genes involved in folic acid (FA) metabolism. Additionally, we demonstrated that pharmacological and genetic interventions designed to increase the FA metabolism pathway suppresses the phenotype of the dMfn RNAi flies. We conclude that strategies to increase FA metabolism may ameliorate diseases, such as peripheral neuropathies, that are associated with loss of mitochondrial function. A video abstract for this article is available at https://youtu.be/fs1G-QRo6xI.
Introduction
Chartcot-Marie-Tooth (CMT) disease is the most common inherited neuromuscular disorder1, with no cure or treatment available. This peripheral neuropathy results from damage to neurons that transmit information from the brain and spinal cord to other parts of the body. Individuals with CMT suffer from muscle weakness and atrophy as well as mild sensory loss. There are two main types of CMT: type 1, where the damage occurs to myelin sheaths that surround the axons of neurons (CMT1) and type 2, where the damage is to the neuron itself (CMT2). Mutations in mitofusin 2 (MFN2) have been found to be one of the most common causes of CMT2, called CMT2a2.
Mitofusins are GTP-hydrolysing enzymes that promote mitochondrial fusion. They are important to ensure the dynamic balance between fusion and fission that determines mitochondria morphology . This dynamic balance also regulates mitochondrial health, since fission is crucial for recycling defective mitochondria through a process called mitophagy (reviewed in3).
Mitofusins can also act as organelle bridges. Mfn2 was reported to link mitochondria to the endoplasmic reticulum (ER)4 and the downregulation of Drosophila mitofusin (dMfn, also known as Marf) reduced contacts between the ER and mitochondria5.
The fruit fly, Drosophila melanogaster, is a powerful tool to study human neurodegenerative diseases (reviewed in6). Moreover, pharmacological approaches can be used to test therapeutic candidates in flies. Drugs can be incorporated in the food and readily delivered. Since flies lack a stringent blood-brain barrier, these drugs can easily access the nervous system (reviewed in7).
Previously, it was reported that the ubiquitous downregulation of dMfn in Drosophila is lethal at the larval stage8 and causes cellular toxicity by activating ER stress9. Here, we employed a strategy that enabled the generation of adult flies with ubiquitous downregulation of dMfn and explored the consequences of its loss. We show the downregulation of dMfn in adult flies compromises mitochondrial function and slows down the axonal transport of mitochondria in wing sensory neurons.
We used an exploratory analysis of the toxic consequences of downregulating dMfn in adult flies. We uncovered an alteration of folic acid (FA) metabolism transcripts in flies with downregulated dMfn. We show that both dietary and genetic interventions to enhance FA metabolism partially supressed the defects observed in adult flies lacking dMfn. We conclude that strategies to enhance FA metabolism may prevent or delay the axonal defects in MFN2-linked CMT2a and other peripheral neuropathies associated with mitochondrial dysfunction.
Results
Reducing the expression of dMfn compromises motor function in adult flies
The ubiquitous downregulation of dMfn by RNAi using the tubulin-Gal4 driver is lethal at the larval stage8. By using an alternative ubiquitous driver, daughterless-Gal4 (daGal4), we generated adult dMfn RNAi flies where the dMfn transcript was downregulated by 50% (Fig. 1a). This also reduced dMfn protein levels (Fig. 1b) and enabled approximately 50% of the adult flies to hatch (Fig. 1c). We then investigated the basal locomotor activity in dMfn RNAi flies and found that the loss of dMfn resulted in locomotor defects (Fig. 1d). Additionally, both the ubiquitous or pan-neuronal downregulation of dMfn resulted in climbing defects (Fig. 1e).
Fig. 1. In vivo suppression of dMfn causes motor impairment in adult flies.
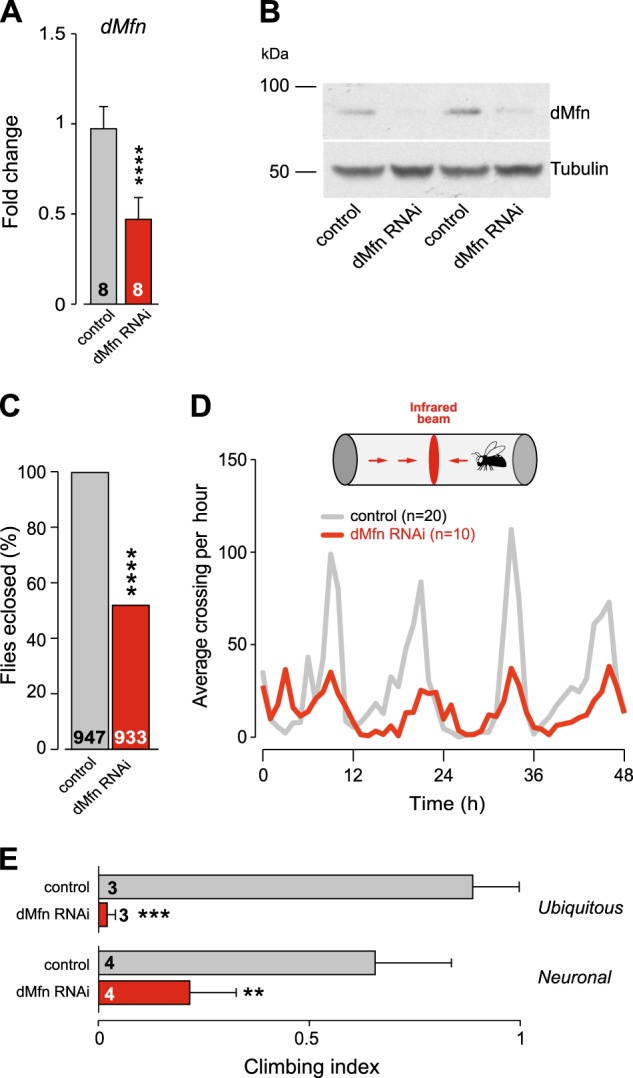
a and b RNAi-mediated suppression of dMfn. a Expression levels of the dMfn transcript were measured by real-time qPCR (mean ± SD; asterisks, two-tailed unpaired t-test compared to control). b Analysis of dMfn protein levels. Whole-fly lysates were analysed using the indicated antibodies. c Eclosion defects following RNAi-mediated suppression of dMfn (asterisks, chi-square two-tailed, 95% confidence intervals). d Suppression of dMfn decreases locomotor activity. The number of flies tested is indicated for each genotype. e Motor impairment upon RNAi-mediated suppression of dMfn. Flies were tested using a standard climbing assay (mean ± SD; asterisks, one-way ANOVA with Dunnett’s multiple comparison test). Genotypes in (a–e (ubiquitous)): Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+. (e (neuronal)): Control: w; elavGal4/+; +, dMfn RNAi: w; elavGal4/dMfn RNAi; +
Downregulation of dMfn expression results in a loss of mitochondrial cristae and reduced mitochondria axonal transport
We next performed an analysis of mitochondria in dMfn RNAi flies. A morphological analysis of the larval ventral nerve cord revealed fragmentation of mitochondria in the mechanosensory axons (Fig. 2a). Ultrastructural analysis of adult fly brains showed fragmented mitochondria cristae (Fig. 2b). To determine if the ultrastructural defects observed upon depletion of dMfn were associated with a decrease in mitochondrial density (MD), we measured the levels of the mitochondrial matrix enzyme citrate synthase10, an indirect measurement of MD, in adult flies. Citrate synthase levels in dMfn RNAi flies were not significantly altered compared to controls (Fig. 2c), indicating that the loss of mitochondrial cristae following depletion of dMfn is not accompanied by a generalised loss of mitochondrial mass. Mitochondria are responsible for the generation of the majority of cellular ATP, a source of energy for axonal mitochondrial transport (reviewed in11). We also observed a significantly lower level of ATP in dMfn RNAi flies compared to the control (Fig. 2d). Moreover, MFN2 mutations present in CMT2 patients cause defects in mitochondrial transport in cultured neurons12. We therefore assessed the effect of lowering dMfn expression on the axonal transport of mitochondria to (retrograde) and away (anterograde) from the cell nucleus in wing sensory neurons (Vagnoni et al., 2016). We observed that lowering the expression of dMfn results in decreased speeds for both anterograde and retrograde transport of mitochondria (Fig. 3).
Fig. 2. Suppression of dMfn causes a loss of mitochondrial cristae density.

a RNAi-mediated suppression of dMfn results in mitochondrial fragmentation. Confocal analysis of mitoGFP in the larval mechanosensory axons. The quantification of mitochondrial length is shown as a combined violin and box plot (p value, two-tailed unpaired t-test compared to control). b The knockdown of dMfn causes defects in mitochondrial cristae. Ultrastructural analysis of the brains in adult dMfn RNAi flies (m, mitochondria; nu, cell nuclei). Asterisks, chi-square two-tailed, 95% confidence intervals. c The knockdown of dMfn does not affect overall mitochondrial mass. Mitochondrial mass was assessed by measuring the activity of the mitochondrial matrix enzyme citrate synthase in adults (ns, p > 0.05, two-tailed unpaired t-test compared to control). d The knockdown of dMfn causes a loss of ATP in adult flies (mean ± SD, asterisks, two-tailed unpaired t-test compared to control). Genotypes in a Control: w; elavGal4/+; UASmitoGFP/+, dMfn RNAi: w; elavGal4/dMfn RNAi; UASmitoGFP/+. b–d Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+
Fig. 3. The knockdown of dMfn decreases axonal mitochondrial motility.

a Representative image of a control wing nerve with mitoGFP expression and a scheme indicating the direction of anterograde and retrograde mitochondrial transport. b Decreased mitochondrial speed in dMfn RNAi wing sensory axons. Both anterograde and retrograde mitochondrial speed were measured for the indicated genotypes. Combined violin and box plot. (n is the number of moving mitochondria analysed, asterisks, two-tailed unpaired t-test compared to control). Genotypes: Control: w; dprGal4/+; UASmitoGFP/+, dMfn RNAi: w; dprGal4/dMfn RNAi; UASmitoGFP/+
We next focused on the functional status of mitochondria in adult flies with decreased expression of dMfn. High-resolution respirometry analysis revealed a significant decrease in respiration rates following suppression of dMfn (Fig. 4a). Furthermore, we detected a loss of mitochondrial membrane potential (Δψm) (Fig. 4b) and an increase in the levels of reactive oxygen species (ROS) (Fig. 4c, d) in the brains of adult flies with decreased expression of dMfn. These results, together with the loss of mitochondrial cristae integrity (Fig. 2b), suggest that the suppression of dMfn does not affect the overall quantity of mitochondria but does compromise their function.
Fig. 4. The knockdown of dMfn reduces mitochondria respiration and leads to an increase in reactive oxygen species.

a Decreased respiration in dMfn RNAi flies. Activity was measured by high-resolution respirometry in 3-day-old flies. Data are shown as the means ± SD (n ≥ 4 in each genotype); asterisks, two-tailed unpaired t-test. b Loss of Δψm following RNAi-mediated suppression of dMfn. The data are shown as the mean ± SD; asterisks, two-tailed unpaired t-test. c and d Increased mitochondrial ROS production in dMfn RNAi flies. c Representative images of adult fly brains stained with MitoSOX using false colour rendering. d Quantification of mitoSOX signal following the RNAi-mediated suppression of dMfn (mean ± SD; asterisks, two-tailed unpaired t-test). Genotypes: a-d Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+
Decreased expression of dMfn is associated with the activation of ATF4-dependent targets associated with folic acid metabolism
Debattisti and colleagues showed that loss of dMfn activates ER stress9. We have recently demonstrated that ER stress linked to mitochondrial dysfunction causes the activation of dATF4, a transcription factor involved in cellular adaptation to stress13. To determine if a decrease in dMfn could affect dATF4 expression, we measured the levels of activation of this transcription factor in dMfn RNAi tissues using a reporter for dATF4 activation14. This analysis showed that the decreased expression of dMfn led to a marked activation of dATF4 (Fig. 5a, b). The response to ER stress involves a generalised decrease in protein translation mediated by PERK and a concomitant activation of ATF4-dependent transcriptional programmes designed to induce a cellular adaptation to stress (reviewed in15). To identify such transcriptional programmes in dMfn RNAi flies, we took an unbiased approach by using microarray analysis. The analysis of transcriptional changes in dMfn RNAi flies (Supplementary Table 1) showed a significant (FDR step-up ≤ 0.05) upregulation of 275 transcripts with fold-change ≥ 1.6 (Supplementary Table 2). Next, a pathway analysis of the upregulated transcripts showed that the decrease in dMfn expression causes an activation of genes involved in FA metabolism (Fig. 5c, d and Supplementary Table 3), including mitochondrial serine hydroxymethyl transferase (Shmt2) and mitochondrial NAD-dependent methylenetetrahydrofolate dehydrogenase (Nmdmc), two transcriptional targets of dATF4 that code for mitochondrial proteins13. The increased levels of Shmt2 and Nmdmc transcripts following dMfn RNAi were confirmed in adult flies using real-time qPCR (Fig. 5e).
Fig. 5. The knockdown of dMfn is associated with an enhanced expression of one-carbon metabolism transcripts.

a and b The RNAi-mediated suppression of dMfn activates dATF4. RNAi dMfn was targeted at the posterior compartment of the larval wing disc using hhGal4 recombined with a GFP reporter. a Representative confocal images showing the enhanced in vivo expression of the ATF4.5’UTR > dsRed reporter (lower right panel) following the RNAi-mediated suppression of dMfn (posterior to the left and anterior to the right). b Quantitative analysis of dsRed fluorescence in the wing discs of the indicated genotypes. Combined violin and box plot (asterisks, two-tailed unpaired t-test). c and d Enhanced expression of one-carbon pool by folate metabolism transcripts following the RNAi-mediated suppression of dMfn. c Top pathways, comprising functionally related genes identified using Partek Pathway Analysis software are shown. P-values were calculated using Fisher’s exact test. See also Supplementary Table 3. d Individual transcripts belonging to the one-carbon pool by folate metabolism enriched following the RNAi mediated suppression of dMfn. Red and blue correspond to, respectively, upregulated and downregulated transcripts above a statistical threshold (FDR ≤ 5%). Asterisks indicate transcripts previously shown to be controlled by dATF4. e dMfn RNAi flies show an upregulation of mitochondrial Shmt2 and Nmdmc (mean ± SD; asterisks, two-tailed unpaired t-test). Genotypes: a–b Control: w; ATF4.5′ UTR > dsRed/+; hhGal4,UASGFP/+, dMfnRNAi: w; ATF4.5’UTR > dsRed/dMfnRNAi; hhGal4,UASGFP/+. c–e Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+
A diet supplemented with folic acid suppresses mitochondrial dysfunction
Folic acid has been historically used to treat anaemia during pregnancy. It functions as a carbon donor in metabolic reactions, including those involved in the synthesis of nucleotides from purine precursors (reviewed in16), and it has been shown to counteract mitochondrial dysfunction in the Drosophila central nervous system17.
Our data show that in the context of decreased levels of dMfn, flies attempt to compensate for mitochondrial stress by inducing the expression of FA metabolism genes. We therefore tested if enhancing this metabolic pathway by increasing the bioavailability of FA could rescue cellular defects associated with the loss of dMfn.
Maintaining dMfn RNAi flies on an FA-supplemented diet during embryonic development did not alter mitochondrial fragmentation in larval mechanosensory axons (Fig. 6a) but was able to suppress loss of Δψm in the brains of adult flies (Fig. 6b). This FA-supplemented diet also suppressed the activation of dAtf4 (Fig. 6c, d) and rescued the eclosion defects of dMfn RNAi flies (Fig. 6e). However, dMfn RNAi flies raised on an FA-supplemented diet showed no significant difference in their lifespan (Fig. 6f) or locomotor activity (Fig. 6g) as compared to flies on a normal diet.
Fig. 6. Dietary supplementation with FA suppresses defects associated with RNAi-mediated suppression of dMfn.

Dietary supplementation with FA does not affect mitochondrial fragmentation in dMfn RNAi flies (a) but partially reverses the loss of Δψm (b) (mean ± SD; asterisks, one-way ANOVA with Bonferroni’s multiple comparison test). c, d Dietary supplementation with FA suppresses the increased expression of dATF4 in dMfn RNAi flies. Representative confocal images and quantitative analysis of dsRed fluorescence in the indicated wing discs are shown (asterisk, two-tailed unpaired t-test). e Decreased eclosion defects in dMfn RNAi flies, following dietary supplementation with FA (asterisks, chi-square two-tailed, 95% confidence intervals). f An FA-supplemented diet does not modify lifespan upon RNAi-mediated suppression of dMfn. Fly viability was scored over a period of 10 days. g An FA supplemented diet does not rescue the locomotor defects of dMfn RNAi flies. Flies were tested using a standard climbing assay (mean ± SD; asterisks, one-way ANOVA with Dunnett’s multiple comparison test). NF normal food; FA folic acid supplemented food. Genotypes: (a) Control: w; elavGal4/+; UASmitoGFP/+, dMfn RNAi: w; elavGal4/dMfn RNAi; UASmitoGFP/+, c–d Control: w; ATF4 5′ UTR DsRed/+; hhGal4,UASGFP/+, dMfn RNAi: w; ATF4.5′ UTR > dsRed/dMfn RNAi; hhGal4,UASGFP/+, e–f w; dMfn RNAi/+; daGal4/+ and (b and g) Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+. Datasets in (a), labelled control (NF) and dMfn RNAi (NF) are also used in Fig. 2a. Dataset in (b) labelled dMfn RNAi (NF) is also used in Fig. 7a, labelled dMfn RNAi. Dataset in (d) labelled control (NF) and dMfn RNAi (NF) are also used in Fig. 5b
Expression of Nmdmc rescues the phenotypes of dMfn RNAi flies
Genes involved in FA metabolism, such as Nmdmc, comprise a branch of mitochondrial retrograde signalling under the control of the dAtf4 transcription factor. We therefore tested the effect of enhancing the expression of Nmdmc in dMfn RNAi flies. Increasing the expression of Nmdmc rescued mitochondrial function in neurons (Fig. 7a), suppressed the activation of dAtf4 (Fig. 7b) and decreased the eclosion defects of dMfn RNAi adult flies (Fig. 7c). We also observed that the expression of Nmdmc increased the lifespan (Fig. 7d) and decreased the locomotor deficit (Fig. 7e) of dMfn RNAi flies. Taken together, these results indicate that the expression of this gene, involved in FA metabolism, has a protective role upon loss of dMfn.
Fig. 7. Expression of Nmdmc rescues defects associated with downregulation of dMfn.

Expression of Nmdmc rescues (a) the loss of Δψm in dMfn RNAi flies, mean ± SD; asterisks, one-way ANOVA with Bonferroni’s multiple comparison test and (b) the increased expression of dATF4 in dMfn RNAi flies (asterisks, one-way ANOVA with Dunnett’s multiple comparison test). c Expression of Nmdmc prevents the eclosion defects in dMfn RNAi flies (asterisks, chi-square two-tailed, 95% confidence intervals). d Expression of Nmdmc increases the lifespan of dMfn RNAi flies. The p-value is shown for the log-rank Mantel-Cox test. e Expression of Nmdmc increases locomotor activity of dMfn RNAi flies. Flies were tested using a standard climbing assay (mean ± SD; asterisks, one-way ANOVA with Dunnett’s multiple comparison test). Genotypes; (a, c-e) Control: w; +; daGal4/+, dMfn RNAi: w; dMfn RNAi/+; daGal4/+, dMfn RNAi, UAS Nmdmc: w; dMfn RNAi/UAS Nmdmc; daGal4/+, (b) Control: w; ATF4 5’UTR DsRed/+; hhGal4,UASGFP/+, dMfn RNAi: w; ATF4.5’UTR > dsRed/dMfn RNAi; hhGal4,UASGFP/+, dMfn RNAi, UAS Nmdmc: w; ATF4.5’UTR > dsRed/dMfn RNAi, UAS Nmdmc; hhGal4,UASGFP/+. Dataset in A, labelled dMfn RNAi, is also used in Fig. 6b, labelled dMfn RNAi (NF)
Discussion
Mitochondrial dynamics are coordinated by division and fusion. Fusion is coordinated by mitofusins, nuclear encoded mitochondrial transmembrane GTPases. Mitofusins act by promoting the fusion of the mitochondrial outer membrane (reviewed in18). Flies contain two genes coding for mitofusins, with dMfn being the only mitofusin expressed in the somatic tissues of adults. Here, we show that the reduction of dMfn in adult tissues leads to a fragmentation of mitochondrial cristae, compromising mitochondrial function and transport. Even though it was previously reported that the deletion of Mfn2 in cultured mouse cells increases mitochondrial mass19, we did not detect any alterations of mitochondrial mass after the in vivo depletion of dMfn. Our data also suggest that the observed fragmentation of mitochondrial cristae upon loss of dMfn is not associated with loss of mitochondria since the overall mitochondrial mass is not altered.
The loss of Δψm and activation of dAtf4 in flies depleted for dMfn could be reversed with dietary supplementation with FA, suggesting that defective mitochondria can act as upstream activators of ER stress pathways. This backs our previous work showing that mitochondrial defects activate ER stress in pink1 or parkin mutant flies5.
The climbing defects and decreased lifespan observed in dMfn RNAi flies could be improved by the expression of Nmdmc, an enzyme involved in FA metabolism, but not by dietary supplementation with FA. Since the intracellular levels of FA intermediates were not measured in our study, we reason that flies lacking dMfn might have a decreased food uptake or, alternatively, defects in the intestinal absorption of FA. This is in line with a previous study reporting that mitochondrial fragmentation triggered by the adult expression of Drp1, a protein that promotes mitochondrial fission, compromises the integrity of the fly intestinal barrier20. This, in turn, might explain why our pharmacological intervention was less efficient than a genetic enhancement approach.
A therapeutic approach to treat Charcot-Marie-Tooth disease type 2 by manipulating mitofusin conformations has been proposed recently21. Our data show that enhancement of FA metabolism might also be a viable approach to treat Charcot-Marie-Tooth disease type 2 and other diseases linked to mitochondrial dysfunction.
Methods
Genetics and Drosophila strains
Fly stocks and crosses were maintained on standard cornmeal agar media at 25 °C. The strains used were daGAL4, w1118, elavGAL4, UASmitoGFP (Bloomington Stock Centre), RNAi line dMfn (ID: 105261,Vienna Drosophila RNAi Center), UAS Nmdmc as previously described13, hhGal4, UASGFP (kind gift from H. Steller, Rockefeller University, New York, USA), dprGal4 (kind gift from S. Bullocks, MRC Laboratory of Molecular Biology, Cambridge, UK) and dATF4.5′UTR > dsRed reporter (kind gift from K. Kang and M-J. Kang, University of Ulsan College of Medicine, Seoul, Republic of Korea). All experiments on adult flies were performed using only males.
RNA extraction and quantitative real-time RT-PCR
Total RNA was extracted using TRIzol (Ambion) and quantified by spectrophotometric analysis. Quantitative real-time PCR with reverse transcription (qRT-PCR) was performed on a real-time cycler (Applied Biosystems 7500 Fast Real-Time PCR Systems) using the SensiFAST SYBR Lo-ROX one-Step Kit (Bioline). Gene-specific primers were obtained from QIAGEN (QuantiTect Primer Assays) for the following genes: dMfn (QT00499205), Nmdmc (dm: QT00503153), Shmt2 (dm: QT00498904). Gene-specific primer rp49 (forward, TGTCCTTCCAGCTTCAAGATGACCATC; reverse, CTTGGGCTTGCGCCATTTGTG) was obtained from Sigma and used as a housekeeping gene.
Protein extraction and western blotting
Protein extracts from whole flies were prepared by grinding flies in lysis buffer (100 mM KCl, 20 mM Hepes at pH 7.5, 5% (v/v) glycerol, 10 mM EDTA, 0.1% (v/v) Triton X-100, 10 mM DTT, (1 mL leupeptin, 1 mL antipain, 1 mL chymostatin and 1 mL pepstatin). The suspensions were cleared by centrifugation at 21,000 × g for 10 min at 4 °C and protein concentrations of the supernatants were measured using the Bradford assay (Bio-Rad). All supernatants were mixed with 4 × LDS loading buffer. For SDS–PAGE, equivalent amounts of proteins were resolved on 10% Precast Gels (Invitrogen) and transferred onto PVDF membranes (Millipore). The membranes were blocked in TBS (0.15 M NaCl and 10 mM Tris-HCl, pH 7.5) containing 10% (w/v) dried non-fat milk for 1 h at room temperature, then probed with the primary antibodies (anti-α-tubulin, Sigma, T6074) or anti-dMfn (a gift from A. Whitworth, MRC, Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK), before being incubated with the appropriate HRP-conjugated secondary antibody. Antibody complexes were visualised by Pierce enhanced chemiluminescence (ECL).
Climbing assay
Climbing assays were performed as previously described22 using a counter-current apparatus equipped with six chambers. A total of 15–20 male 3-day-old flies were placed into the first chamber, tapped to the bottom, and then given 20 s to climb a distance of 10 cm. The flies that successfully climbed 10 cm or beyond within 20 s were then shifted to a new chamber, and both sets of flies were given another opportunity to climb the 10 cm distance. This procedure was repeated a total of five times. After five trials, the number of flies in each chamber was counted. A video demonstrating this technique can be found at https://youtu.be/vmR6s_WAXgc. The climbing index was measured using a weighted average approach with the following formula:
In this formula, n0 corresponds to the number of flies that failed the first trial, and n1 through n5 are the numbers of flies that successfully passed each successive trial. At least 100 flies were used for each genotype tested.
Lifespan analysis
Groups of 15 newly eclosed males of each genotype were placed into separate vials with food and maintained at 25 °C. The flies were transferred to vials containing fresh food every two to three days, and the number of dead flies was recorded. The data are presented as Kaplan–Meier survival distributions, and the significance was determined by the log-rank test.
Dietary supplements
Folic acid (Sigma, F7876) was incorporated into the fly food at a final concentration of 4 mM. The animals were treated with FA throughout development. Adult flies were maintained on FA-containing food throughout their lifespan, and they were transferred to vials with fresh food every two to three days.
Microscopy-based assessment of mitochondrial function and length
Measurement of Δψm in brains of 3-day-old flies was performed using tetramethylrhodamine (TMRM) as previously described17. Briefly, adult fly brains were loaded with 40 nM TMRM in loading buffer (10 mM HEPES, pH 7.35, 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2 and 10 mM glucose) for 40 min at room temperature, and the dye was present during the experiment. In this experiment, TMRM was used in the redistribution mode to assess Δψm, and therefore, a reduction in TMRM fluorescence represents mitochondrial depolarisation. Confocal images were obtained using a Zeiss 510 confocal microscope equipped with a 40× oil immersion objective. Illumination intensity was kept to a minimum (at 0.1–0.2% of laser output) to avoid phototoxicity, and the pinhole was set to give an optical slice of 2 μm. Fluorescence was quantified by exciting TMRM using the 565 nm laser and measured above 580 nm. Z-stacks of 5 fields of 300 μm2 each per brain were acquired, and the mean maximal fluorescence intensity was measured for each group.
Mitochondrial length was quantitated in mechanosensory axons in the ventral nerve cord (VNC) from third-instar larvae. The VNC was dissected in PBS, transferred to a drop of PBS as mounting medium on glass slides, covered with a coverslip and imaged on a Zeiss LSM510 confocal microscope. Mitochondrial length was calculated using the “segmented line” tools in ImageJ to measure the length of mitoGFP-positive mitochondria across their largest dimension. Z-projection of 10 μm-thick stacks was used to follow the mitochondrial 3D distribution and measure lengths more accurately.
Citrate synthase assay
Citrate synthase activity was measured using a protocol adapted from the Citrate Synthase Assay kit (CS070 SIGMA). Ten male flies (3-days-old) were homogenised in lysis buffer (100 mM KCl, 20 mM Hepes at pH 7.5, 5% (v/v) glycerol, 10 mM EDTA, 0.1% (v/v) Triton X-100, 10 mM DTT, 1 μg/mL leupeptin, 1 μg/mL antipain, 1 μg/mL chymostatin and 1 μg/mL pepstatin). The suspensions were cleared twice by centrifugation at 2,000 × g for 15 s at 4 °C, and the protein concentrations were determined by Bradford assay (Bio-Rad). Sample volume was resuspended in reaction buffer (75 mM Tris-HCl pH 8, 100 μM DTNB, 0.1% Triton, 350 μg/ml, 0.5 mM Oxalacetate) and absorbance was measured at 412 nm for 2 min using M200PRO plate reader (TECAN, Switzerland). Absorbance values were plotted against time (min) for each reaction. Changes in absorbance (ΔA412/minute) were used to calculate the citrate synthase activity using the following equation: units (mmole/ml/min) = (ΔA412/min × V(ml)/ εmM × L(cm) × Venz (ml). V(ml) = reaction volume, εmM = 13.6 mM−1 cm−1, Venz (ml) = volume of sample. Units of citrate synthase activity were normalised to protein concentration (mg/ml).
ATP assays
Five male flies (3-days-old) were homogenised in 100 μL of 6 M guanidine-HCl in extraction buffer (100 mM Tris and 4 mM EDTA, pH 7.8) to inhibit ATPases. Homogenised samples were subjected to rapid freezing in liquid nitrogen followed by boiling for 5 min. Samples were then cleared by centrifugation and the supernatant was diluted (1/50) with extraction buffer and mixed with luminescent solution (CellTiter-Glo Luminescent Cell Viability Assay, Promega). The luminescence was measured on an infinitive M200PRO plate reader (TECAN, Switzerland). The relative ATP levels were calculated by dividing the luminescence by the total protein concentration, which was determined by the Bradford method.
Analysis of ATF4 activation in Drosophila wing discs
To analyse ATF4 activation, ATF4.5′UTR > dsRed reporter larvae with the expression of GFP in the posterior compartment of wing discs under the control of hhGal4 were used. Wing discs were dissected in PBS and imaged on a Zeiss LSM510 confocal microscope. The 15 μm-thick stacks were acquired and maximum projection of DsRed signal was measured by using ImageJ software.
Measurement of mitochondrial ROS in Drosophila adult brain
Drosophila adult brains were dissected in PBS and incubated with 5 μM MitoSOXTM Red mitochondrial superoxide indicator (M36008, Molecular Probes) for 30 min. After incubation brains were washed with PBS for 10 min and imaged on a Zeiss LSM510 confocal microscope. The 100 μm-thick stacks were acquired and used to measure the MitoSOX signal using ImageJ software.
Analysis of mitochondrial speed
Axonal mitochondrial speeds were measured as previously described23,24. The 2–5-day-old flies expressing GFP under the dpr driver were anaesthetised with CO2 and enclosed in a custom-built chamber formed by cover glasses where the body of the fly was placed ventral side up and wings were positioned under small coverslips and covered with a drop of halocarbon oil. Imaging of the axonal wing nerve was performed using a Zeiss LSM510 confocal microscope with a 100× oil immersion objective. Mitochondrial movements were quantified using ImageJ. Z-projection was performed and anterograde and retrograde mitochondrial movements were scored following the direction of movement as previously reported. Anterograde runs towards the fly thorax and retrograde to the cell body of the neuron24. Transported mitochondria were manually tracked across the frames and speeds calculated dividing the total distance by the time. Data were plotted using Prism (GraphPad).
Electron microscopy
For transmission electron microscopy, adult fly brains were fixed overnight in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2% paraformaldehyde, 2.5% glutaraldehyde and 0.1% Tween-20. Then, the samples were post-fixed for 1 h at room temperature in a solution containing 1% osmium tetroxide and 1% potassium ferrocyanide. After fixation, the samples were stained en bloc with 5% aqueous uranyl acetate overnight at room temperature; then, they were dehydrated via a series of ethanol washes and embedded in TAAB epoxy resin (TAAB Laboratories Equipment Ltd., Aldermaston, UK). Semi-thin sections were stained with toluidine blue, and areas of the sections were selected for ultramicrotomy. Ultrathin sections were stained with lead citrate and imaged using a MegaView 3 digital camera and iTEM software (Olympus Soft Imaging Solutions GmbH, Münster, Germany) with a Jeol 100-CXII electron microscope (Jeol UK Ltd., Welwyn Garden City, UK).
Microarray acquisition and analysis
RNA was prepared from 3-day-old male adult flies (8 samples in total, 4 replicates for each genotype). The dMfn RNAi was driven by daGAL4. The RNA quality was confirmed using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Detailed experimental protocols and raw data were deposited in ArrayExpress under accession E-MTAB-6579. Differential expression was analysed using the Partek Genomics Suite (Partek Inc. Missouri, USA). Pathway enrichments were calculated using a pathway ANOVA statistical model in Partek Pathway (Partek Inc. Missouri, USA).
Respirometry
Mitochondrial respiration in 3-day-old flies was assayed at 37◦C by high-resolution respirometry as previously described25. The OROBOROS Oxygraph DatLab software package (OROBOROS, Innsbruck, Austria) was used for data acquisition (2 s time intervals) and analysis, including calculation of the time derivative of the oxygen concentration and signal deconvolution dependent on the response time of the oxygen sensor, with correction for instrumental background oxygen flux. Respiration was assessed by homogenising two flies using a pestle in MiR05 respiration buffer (20 mM HEPES, 10 mM KH2PO4, 110 mM sucrose, 20 mM taurine, 60 mM K-lactobionate, 0.5 mM EGTA, 3 mM MgCl2, and 1 g/l fatty acid-free BSA). Coupled state 3 respiration for complex I was assayed in MiR05 respiration buffer in the presence of 2 mM malate, 10 mM glutamate and 5 mM ADP.
Statistical analyses
Descriptive and inferential statistical analyses were performed using GraphPad Prism 8 (www.graphpad.com). The data are presented as the mean value, and the error bar indicates ± SD or ± SEM (as indicated). In the combined violin and box plots, the median is shown with a solid white line and the quartiles are represented by the dashed white lines. The number of biological replicates per experimental variable (n) is indicated in either the respective figure or figure legend. Significance is indicated as **** for p < 0.0001; *** for p < 0.001; ** for p < 0.01 and * for p < 0.05. The investigators gathering quantitative data on the biological samples were not blinded to the sample identities at the time of analysis. No specific randomisation strategies were employed when the biological replicates were assigned to the treatment groups.
Digital image processing
Western blot images were acquired as uncompressed, bitmapped digital images (TIFF format). The images were processed using Adobe Photoshop, employing established scientific imaging workflows26.
Supplementary information
Acknowledgements
We would like to thank A. Vagnoni and S. Bullock (MRC Laboratory of Molecular Biology, Cambridge, UK) for sharing reagents and unpublished experimental protocols, K. Kang and M-J. Kang (University of Ulsan College of Medicine, Seoul, Republic of Korea) for the generous gift of ATF4.5′UTR > dsRed reporter line, and T. Ashby and M. Patel for preparing the fly food. We also thank T. Smith and M. Martin for their assistance with electron microscopy. FUNDING This work is funded by the UK Medical Research Council, intramural project RG94521.
Author Contributions
J.G.M., I.C., A.C.C., S.L., S.H.Y.L. and L.M.M. conceived, designed and performed the experiments and analysed the data. S.H.Y.L. and L.M.M. wrote the paper and contributed equally as joint last authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Melino
Enhancing folic acid metabolism restores mitochondrial function and is neuroprotective following suppression of mitofusin.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Samantha H. Y. Loh, Phone: +44 116 223 1501, Email: shyl2@mrc-tox.cam.ac.uk
L. Miguel Martins, Phone: +44 116 252 5533, Email: martins.lmiguel@gmail.com.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41419-019-1496-2).
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Zuchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 3.de Castro IP, Martins LM, Tufi R. Mitochondrial quality control and neurological disease: an emerging connection. Expert. Rev. Mol. Med. 2010;12:e12. doi: 10.1017/S1462399410001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 5.Celardo I, et al. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016;7:e2271. doi: 10.1038/cddis.2016.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ugur B, Chen K, Bellen HJ. Drosophila tools and assays for the study of human diseases. Dis. Model Mech. 2016;9:235–244. doi: 10.1242/dmm.023762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu. Rev. Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorn GW, 2nd, et al. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debattisti V, Pendin D, Ziviani E, Daga A, Scorrano L. Reduction of endoplasmic reticulum stress attenuates the defects caused by Drosophila mitofusin depletion. J. Cell. Biol. 2014;204:303–312. doi: 10.1083/jcb.201306121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magwere T, et al. The effect of dietary restriction on mitochondrial protein density and flight muscle mitochondrial morphology in Drosophila. J. Gerontol. A. Biol. Sci. Med. Sci. 2006;61:36–47. doi: 10.1093/gerona/61.1.36. [DOI] [PubMed] [Google Scholar]
- 11.Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front. Biosci. 1996;1:d91–d102. doi: 10.2741/A118. [DOI] [PubMed] [Google Scholar]
- 12.Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012;32:4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Celardo I, Lehmann S, Costa AC, Loh SH. & Miguel Martins, L. dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ. 2017;24:638–648. doi: 10.1038/cdd.2016.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang K, Ryoo HD, Park JE, Yoon JH, Kang MJ. A Drosophila reporter for the translational activation of ATF4. PLoS One. 2015;10:e0126795. doi: 10.1371/journal.pone.0126795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly GS. Folates: supplemental forms and therapeutic applications. Altern. Med. Rev. 1998;3:208–220. [PubMed] [Google Scholar]
- 17.Tufi R, et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat. Cell Biol. 2014;16:157–166. doi: 10.1038/ncb2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis. 2018;9:330. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawalec M, et al. Mitofusin 2 deficiency affects energy metabolism and mitochondrial biogenesis in MEF cells. PLoS One. 2015;10:e0134162. doi: 10.1371/journal.pone.0134162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rana A, et al. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017;8:448. doi: 10.1038/s41467-017-00525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franco A, et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540:74–79. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greene JC, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vagnoni A, Bullock SL. A simple method for imaging axonal transport in aging neurons using the adult Drosophila wing. Nat. Protoc. 2016;11:1711–1723. doi: 10.1038/nprot.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vagnoni A, Hoffmann PC, Bullock SL. Reducing Lissencephaly-1 levels augments mitochondrial transport and has a protective effect in adult Drosophila neurons. J. Cell. Sci. 2016;129:178–190. doi: 10.1242/jcs.179184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costa A, Loh S, Martins LM. Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis. 2013;4:e467. doi: 10.1038/cddis.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wexler, E. J. Photoshop CS3 Extended for Biomedical Research [DVD-ROM and online course]. (Lynda.com, Ventura, 2008).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.