

Shiga toxin (Stx)-producing Escherichia coli (STEC) causes foodborne outbreaks of bloody diarrhea. There are two major types of immunologically distinct Stxs: Stx1a and Stx2a.
KEYWORDS: Escherichia coli, O26:H11, Shiga toxin, Stx1, Stx2, hemolytic-uremic syndrome
ABSTRACT
Shiga toxin (Stx)-producing Escherichia coli (STEC) causes foodborne outbreaks of bloody diarrhea. There are two major types of immunologically distinct Stxs: Stx1a and Stx2a. Stx1a is more cytotoxic to Vero cells than Stx2a, but Stx2a has a lower 50% lethal dose (LD50) in mice. Epidemiological data suggest that infections by STEC strains that produce only Stx2a progress more often to a life-threatening sequela of infection called hemolytic-uremic syndrome (HUS) than isolates that make Stx1a only or produce both Stx1a and Stx2a. In this study, we found that an E. coli O26:H11 strain that produces both Stx1a and Stx2a was virulent in streptomycin- and ciprofloxacin-treated mice and that mice were protected by administration of an anti-Stx2 antibody. However, we discovered that in the absence of ciprofloxacin, neutralization of Stx1a enhanced the virulence of the strain, a result that corroborated our previous finding that Stx1a reduces the toxicity of Stx2a by the oral route. We further found that intraperitoneal administration of the purified Stx1a B subunit delayed the mean time to death of mice intoxicated with Stx2a and reduced the cytotoxic effect of Stx2a on Vero cells. Taken together, our data suggest that Stx1a reduces both the pathogenicity of Stx2 in vivo and cytotoxicity in vitro.
INTRODUCTION
Shiga toxin (Stx)-producing Escherichia coli (STEC) is estimated to cause more than 175,000 domestically acquired foodborne infections per year in the United States (1). STEC infection leads to diarrhea that is often grossly bloody. In some individuals, STEC infection may progress to the life-threatening manifestation hemolytic-uremic syndrome (HUS). HUS most often occurs in children and is characterized by thrombocytopenia, hemolytic anemia, and acute kidney failure (2). The O157:H7 serotype is most commonly associated with STEC infections and HUS in the United States (3), followed by serogroup O26 (usually O26:H11) (4–6).
Stx is an AB5 toxin that inhibits protein synthesis in sensitive eukaryotic cells (7). The A subunit of the toxin contains the enzymatic domain, which inactivates eukaryotic ribosomes and inhibits protein synthesis. The B pentamer binds to the cellular receptor globotriaosylceramide (Gb3) or, to a lesser extent, globotetraosylceramide (Gb4) (8–10). There are two antigenically distinct, but highly homologous, types of Stx that may be produced by E. coli, Stx1a and Stx2a (11). The stx1 and stx2 genes are carried on separate prophages. Toxin production and release are coordinated with bacteriophage induction and cell lysis (12). Antibiotics such as ciprofloxacin (Cip) can induce bacteriophages and thus stimulate Stx production (13). STEC can produce either toxin alone or can make both Stx1a and Stx2a (or subtypes of those prototype toxins). Epidemiological data suggest that STEC infection with a strain that expresses Stx2a only will more often progress to HUS than infection with a strain that makes Stx1a only or both Stx1a and Stx2a (14, 15). Analogously, in a gnotobiotic pig model of STEC infection, a strain that made both Stx1 and Stx2 caused less disease than the stx1 mutant of the isolate (16).
Although Stx1a and Stx2a share 60% sequence identity (17), they do exhibit some biological differences in addition to being antigenically distinct: Stx1a is 10 times more potent toward Vero cells than Stx2a, most likely because Stx1a has a higher affinity for Gb3, and the murine 50% lethal dose (LD50) for Stx2a administered parenterally is 100-fold lower than that for Stx1a (18–20). The cause of death of Stx-treated mice is necrosis of the distal tubular epithelial cells, which leads to impaired kidney function, electrolyte imbalance, and ultimately organ failure, represented by increased blood urea nitrogen and creatinine levels (21). In addition, by the oral route, the LD50 of purified Stx2a in mice is 2.8 μg, while Stx1a-intoxicated mice are unaffected at doses as high as 157 μg/mouse. This difference in oral potency between the Stxs occurs even though Stx1a reaches the kidney at levels similar to those of Stx2a (19). Unexpectedly, the same study found that cointoxication of mice with Stx1a and Stx2a results in increased survival and extends the mean time to death beyond that for animals intoxicated with Stx2a alone. The protection from Stx2a was also observed when Stx2a was coadministered with an Stx1a active-site toxoid, a finding that demonstrates that the protection is due to the binding capacity of Stx1a (21).
In this study, we found that Stx1a reduces the pathogenicity for mice of an O26:H11 STEC strain that makes both Stx1a and Stx2a. We also showed that intraperitoneal (i.p.) administration of the B subunit of Stx1a extended the mean time to death due to Stx2a intoxication in a mouse model. Finally, we discovered that the B subunit of Stx1a protected Vero cells from cytotoxicity induced by Stx2a.
RESULTS
Stx production by TW08571.
We selected an stx1a+ stx2a+ O26:H11 strain (TW08571) that was isolated from a patient with bloody diarrhea to evaluate the roles of individual Stxs in the pathogenicity of an STEC strain in a mouse model of infection. First, we assessed the ability of TW08571 to produce both Stx1a and Stx2a in vitro. Immuno-dot blots of cell-associated (CA) and supernatant (Sup) fractions from TW08571 after overnight growth showed that the strain produces both Stx1a and Stx2a (Fig. 1). Stx1 is most often found in the CA fraction, while Stx2 is found in the Sup. We measured very low levels of Stx1 in the Sup fraction and were unable to detect Stx2 in the CA fraction of TW08571. Conclusions about relative toxin production cannot be made from the immuno-dot blot, because the antibodies used have different affinities for the individual toxins they bind (22); however, the antibodies are specific for their designated toxin types (23, 24). To quantify the level of overall toxin production, cytotoxicity was measured on Vero cells after growth without or with Cip (to induce the phages that carry the stx genes). CA and Sup fractions from overnight cultures of TW08571 grown in the absence of Cip contained about 5.5 log 50% cytotoxic doses (CD50)/ml (Fig. 2), whereas similar Sup fractions from cultures grown with 15 ng/ml Cip (a sublethal dose [data not shown]) showed about 7 log CD50/ml. The amount of CA toxin remained about the same even after TW08571 was grown with Cip (Fig. 2). The increase of toxicity in the Sup fraction is expected for an STEC in which the stx genes are located on inducible bacteriophages.
FIG 1.

Immuno-dot blot of 2-fold serial dilutions of cell-associated (CA) or supernatant (Sup) fractions from overnight cultures of TW08571 probed with Stx1a (top) or Stx2a (bottom).
FIG 2.
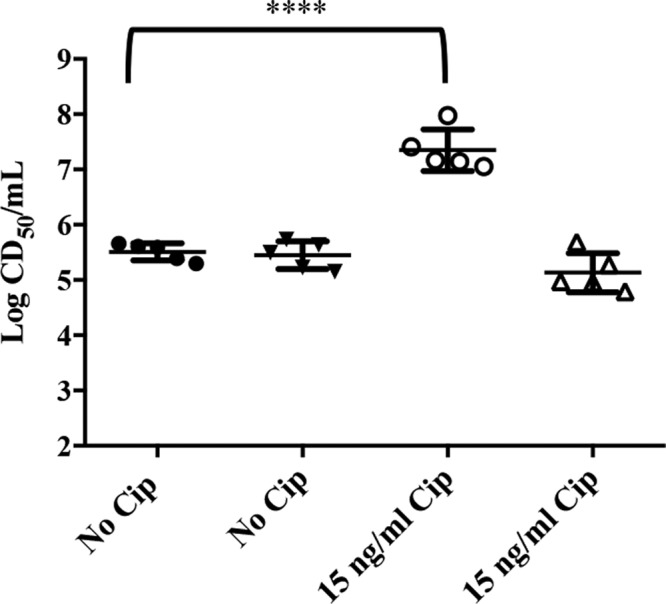
Cytotoxicity of supernatant (circles) and cell-associated (triangles) fractions of overnight cultures of TW08571 grown in the absence or presence of 15 ng/ml Cip. Each wide horizontal bar indicates the median CD50 (log) from five experiments, and error bars represent standard deviations. A significant difference was found between supernatant fractions with no Cip and those with 15 ng/ml Cip (P < 0.0001), while the difference between cell-associated fractions with no Cip and those with 15 ng/ml Cip was not significant, by Tukey’s multiple-comparison test.
Virulence capacity of TW08571 in mice.
Infection of streptomycin (Str)-treated mice with TW08571 caused no observable signs of morbidity and no mortality. However, when mice infected with TW08571 were injected daily with 15 μg of Cip starting on day 2 postinfection (D2PI), mice began to lose weight and succumb to infection (Fig. 3A). To determine which toxin was responsible for the virulence of TW08571 in Str- and Cip-treated mice, the infected mice were passively immunized with a Stx1- or Stx2-neutralizing antibody, or with anti-cholera toxin (CT) as a negative control. We found that only the Stx2-neutralizing antibody protected Cip-treated mice from infection with TW08571 (Fig. 3B), an observation that shows that Stx2a is responsible for the virulence of TW08571 in Str- and Cip-treated infected animals. Attempts to create an stx2 mutant of TW08571 to confirm the role of Stx2a in virulence were unsuccessful.
FIG 3.

Survival of TW08571-infected, Str-treated mice. (A) Percentages of survival of Str-treated mice infected with TW08571 and given daily injections, starting on D2PI, of PBS (black) or 15 μg Cip (red) (P, 0.0089 by the log rank [Mantel-Cox] test). (B) Percentages of survival of Str-treated mice infected with TW08571, given Cip starting on D2PI, and administered a tail vein injection of PBS (black), anti-CT (green), anti-Stx2 (blue), or anti-Stx1 (red) on D2PI. A significant difference was found between PBS and anti-Stx2 (P = 0.002) by the log rank (Mantel-Cox) test; the differences between all other treatments were nonsignificant.
TW08571-infected mice exhibit morbidity after treatment with anti-Stx1.
Unexpectedly, we observed an increase in morbidity in TW08571-infected, Str-treated mice when either monoclonal or polyclonal anti-Stx1 was administered on D2PI in the absence of Cip treatment (Fig. 4). The weight loss of these anti-Stx1-treated animals was significantly increased from D7PI through D11PI. This increase in morbidity was not seen when either a control (anti-ricin) or anti-Stx2a was administered. The finding of morbidity after the administration of either monoclonal or polyclonal anti-Stx1 to TW08571-infected, Str-treated mice suggests that Stx1a attenuates the virulence of TW08571.
FIG 4.

Percentages of weight change for TW08571-infected, Str-treated mice. Mice were given a tail vein injection of PBS (black), anti-ricin (green), anti-Stx2 (blue), monoclonal anti-Stx1 (red), or polyclonal anti-Stx1 (orange) on D2PI. Points on the lines represent the mean weights of 10 animals. Error bars represent standard deviations; asterisks indicate days on which the mean weights of PBS-treated mice demonstrated a significant difference from those of the experimental group by Dunnett’s multiple-comparison test (**, P ≤ 0.006; ****, P ≤ 0.0001).
The Stx1a B subunit delays the mean time to death of Stx2a-intoxicated mice.
To determine whether the B subunit of Stx1a is sufficient to reduce the toxic potential of Stx2a in animals after systemic delivery, we injected mice with 2× LD50 Stx2a (5 ng) with or without the Stx1a B subunit (50 ng) given 3, 12, or 24 h before the Stx2a challenge. In the absence of the Stx1a B subunit, 100% of the Stx2a-intoxicated mice died by 100 h postintoxication (Fig. 5). In contrast, Stx2a-intoxicated mice previously injected with the Stx1a B subunit exhibited a delay in the mean time to death. The most significant delay in the mean time to death occurred when the Stx1a B subunit was given 12 h prior to Stx2a intoxication: the mean time to death in those animals was as long as 156 h postintoxication. When the Stx1a B subunit was neutralized with anti-Stx1 prior to intoxication, the extension in the mean time to death was not observed. These results further support our hypothesis that the Stx1a B subunit competes with Stx2a at the level of the target organ responsible for the death of the mice, the kidney, and thus can attenuate the toxic effect of Stx2a.
FIG 5.
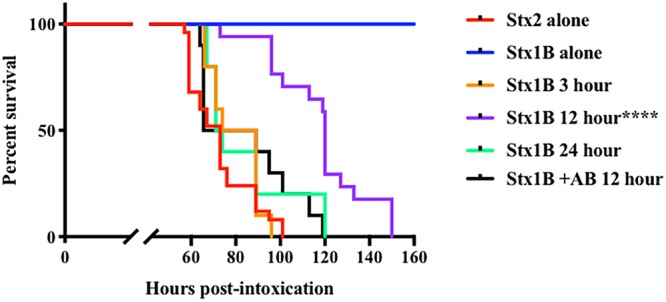
Survival curves of mice intoxicated with 2× LD50 of purified Stx2a (5 ng) alone or pretreated with 50 ng Stx1a B subunit at 3, 12 (with or without a Stx1 antibody [AB]), or 24 h before intoxication. Asterisks indicate a significant difference (****, P ≤ 0.0001) by the log rank (Mantel-Cox) test.
The Stx1a B subunit reduces the cytotoxicity of Stx2a for Vero cells.
We next used Vero cells to determine whether the Stx1 binding moiety could alter the function of Stx2a in vitro. Vero cells were incubated with 2 CD50 of Stx2a along with different concentrations of the Stx1a B subunit or Stx2a toxoid. Both Stx2a toxoid and the B subunit of Stx1a protected the Vero cells in a dose-dependent manner (Fig. 6). The addition of the Stx1a B subunit or the Stx2a toxoid alone at the concentrations shown in Fig. 6 did not affect Vero cell viability (data not shown). The reason for the use of Stx2a toxoid rather than the Stx2a B subunit is that the Stx2a B subunit was not in the correct conformation (since it existed primarily as a dimer [data not shown]) to properly bind to the receptor.
FIG 6.
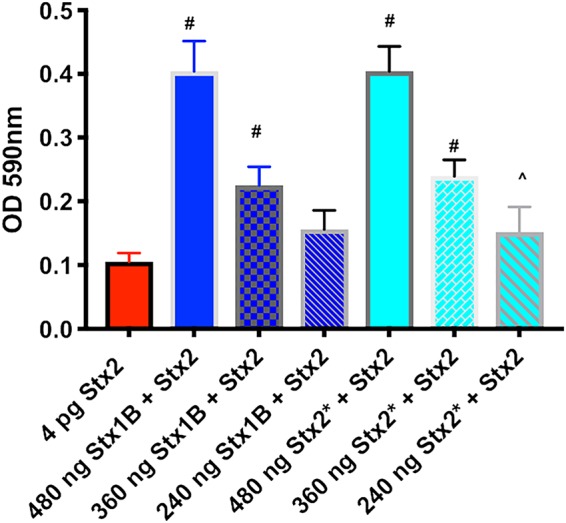
Stx2a cytotoxicity for Vero cells in the presence of the B subunit of Stx1a or Stx2a toxoid (Stx2*). Vero cells were intoxicated with a set concentration of Stx2a mixed with various concentrations of the Stx1a B subunit (dark blue bars) or Stx2* (light blue bars). Each bar represents the median for three separate experiments, and the error bar indicates 1 standard deviation. Symbols mark concentrations of Stx1B or Stx2* that resulted in an optical density at 590 nm (OD590) significantly different from that with Stx2a alone by use of Dunnett’s multiple-comparison test (^, P ≤ 0.01; #, P ≤ 0.001).
Stx1a and Stx2a bind to kidney tissue in similar patterns.
As mentioned above, we hypothesize that Stx1a might attenuate the virulence of TW08571 by competing with Stx2a for binding on target renal tissue. To test the feasibility of this theory, we sought to determine whether the two toxins bind to the same regions of the kidney. Therefore, we overlaid Stx1a or Stx2a onto kidney sections and assessed the location of toxin binding by immunofluorescence. We observed no discernible differences between the binding patterns for purified Stx1a and Stx2a (Fig. 7), a finding similar to that of Tesh et al., in which purified Stx1 and Stx2 were shown to bind tubular epithelial cells in the cortex and medulla (18). These results leave open the possibility that Stx1a could block Stx2a binding to the kidney. We attempted to measure decreased binding of Stx2a in the presence of Stx1a but were unable to observe blocking in experiments similar to those described above (data not shown). We suspect that the reason that Stx1 did not appear to block the binding of Stx2a to the kidney section is most likely the high number of Stx binding sites on the kidney sections and/or the inability of this method to detect slight differences in the amount of bound toxin.
FIG 7.

Patterns of binding of Stx1a and Stx2a to mouse kidney tissue. Purified Stx1a (top) and Stx2a (bottom) are imaged at magnifications of ×400 (left) and ×1,000 (right).
DISCUSSION
In this study, we used three complementary methods to extend our previous observation that orally delivered Stx1a (and an A subunit toxoid of Stx1a) protects mice orally intoxicated with Stx2a (21). First, we found that the presence of Stx1a in the O26:H11 strain TW08571 reduced the potential of that strain to cause morbidity in Str-treated mice. Second, we showed that the Stx1a B subunit decreased the toxicity of Stx2a in vivo (when both were administered parenterally). Third, we discovered that the Stx1a B subunit also reduced the cytotoxicity of Stx2a for Vero cells.
Infection with TW08571 alone in our Str-treated mouse model resulted in no morbidity or mortality. However, when mice were given a daily injection of Cip starting on D2PI, an established method of phage induction (13), mice began to rapidly lose weight and succumb to infection. Neutralization of Stx2a, but not Stx1a, resulted in full protection of Cip-treated mice infected with TW08571. The latter result agrees with a previous study from our lab that demonstrated that neutralization of Stx2a, but not Stx1a, protects Str-treated mice from infection with Stx1a+ Stx2a+ O157:H7 strain 933cu-rev. Similar results were also observed in gnotobiotic piglets: an STEC strain that produced Stx2 alone caused more neurologic symptoms and disease than the same strain when it made only Stx1 or both Stx1 and Stx2 (16).
When we injected Str-treated, TW08571-infected mice (in the absence of Cip) with anti-Stx1, we were initially surprised to find that the mice exhibited morbidity, and we repeated the experiment with a polyclonal antibody to Stx1, with the same result. However, those findings of morbidity confirm that Stx1a has an inhibitory effect on the ability of Stx2a to cause disease in mice and support our earlier finding that Stx1a protects mice from oral gavage with Stx2a (21). Furthermore, this study is the first demonstration that the presence of Stx1 can attenuate the virulence of an STEC strain during infection in mice.
Our previous work showed that orally gavaged Stx1a and Stx2a disseminated similarly to the kidney and that the Stx1a B subunit reduces Stx2a toxicity. In this study, we further tested whether the Stx1a B subunit could directly influence Stx2a intoxication by bypassing the gastrointestinal tract. When both Stx2a and the Stx1a B subunit were administered parenterally, and the Stx1a B subunit was administered prior to Stx2a, we observed an extension in the mean time to death due to Stx2a intoxication, with the maximal protective effect achieved by a 12-h pretreatment. We hypothesize that (i) the protective effect is due to a reduction of the binding of Stx2a to the kidney and (ii) the reason the Stx1a B subunit had to be given prior to Stx2a is that the Stx1a B pentamer has to reach the kidney before Stx2a to prevent Stx2a binding.
Although the administration of 1 μg lipopolysaccharide (LPS) 8 or 24 h prior to Stx2a intoxication can reduce the toxicity of Stx2a, likely though immunomodulatory effects (25), our preparations had <40 pg/μg toxin, so mice never received more than 2.2 ng LPS in our studies. Furthermore, preincubation of the Stx1a B subunit with the Stx1a monoclonal antibody that neutralizes the binding capacity of the B subunit eliminated protection from Stx2a by the Stx1 B subunit, a result that further supports our hypothesis that the protection we observe is not caused by LPS.
We cannot fully rule out the possibility that the Stx1 B subunit has immunomodulatory activity that can impact Stx2a toxicity. However, we believe that the low levels of protein used in our studies most likely do not elicit enough of an immune response to result in protection from Stx2a. In vitro studies with purified Stx1a and Stx2a show that although Stx2a causes an increase in cytokine and chemokine expression in THP-1 cells, the B subunit of either toxin alone is unable to induce such a cytokine response (26). Inflammasome-dependent caspase-1 activation and interleukin 1β (IL-1β) secretion from THP-1 cells also requires A subunit activity (27). However, in HCT-8 cells, which are of intestinal origin, the Stx1a B subunit transiently causes ERK1/2 activation (28), although 10 μg/ml Stx1 B subunit for 24 h was necessary for detection of an immune response. Our study that demonstrated protection from Stx2a by the Stx1 B subunit on Vero cells (of kidney origin) maximally used 480 ng of the B subunit for 5 h. In addition, the protection we observed in mice with the B subunit alone occurred after parenteral injection, a route that bypassed the intestine.
Overall, we provide a model of oral bacterial infection in which the presence of Stx1a reduces Stx2a toxicity. This attenuation was seen with full-length active Stx1a and with the B subunit of the toxin alone. We hypothesize that the protection observed may be attributed to a significant inhibition of Stx2a binding to Gb3 due to the higher affinity of Stx1a for this receptor (7, 29); however, a more detailed investigation of receptor binding would be necessary in order to characterize the competition between the Stx subtypes for receptor binding. Since the pathogenic potential of STEC is determined mainly by the amount of toxin binding to Gb3 at the kidney, strains that produce more Stx1a than Stx2a could exhibit lower pathogenicity than strains that produce more Stx2a than Stx1a. Additionally, the loss or acquisition of an Stx1a phage by E. coli strains that express Stx2a may change their pathogenic potential.
In conclusion, our data suggest that Stx1a reduces the pathogenic potential of Stx2a in vivo and in vitro by decreasing the binding of Stx2a to receptors on the kidney. It is also possible that Stx1a influences the internalization or trafficking of Stx2a bound to the receptor. However, our work did not address either of these possibilities. Further studies will be required to determine how Stx1a mediates the reduction of Stx2a cytotoxicity at the cellular level.
MATERIALS AND METHODS
Bacterial strains.
Escherichia coli O26:H11 strain TW08571 (stx1a+ stx2a+) was isolated in Germany from a patient who had bloody diarrhea in 2000 (deposited by L. Beutin in the STEC repository at Michigan State University). A spontaneous streptomycin (Str)-resistant mutant of TW08571 was isolated and used for mouse studies. Bacteria were inoculated onto Luria broth (LB) plates supplemented with Str (100 μg/ml). The plates were then incubated overnight at 37°C, and colonies were inoculated into LB with Str. Cultures were grown overnight at 37°C with shaking (225 rotations per min). For some experiments, ciprofloxacin was added to LB medium at various concentrations to increase Stx production. For animal infections, bacteria were harvested by centrifugation and were resuspended in phosphate-buffered saline (PBS) to a 50× final concentration.
Immuno-dot blots.
The relative expression levels of Stx1a and Stx2a from TW08571 were determined as described previously (21). Briefly, overnight cultures of STEC strains grown in LB were centrifuged at 15,871 relative centrifugal force (RCF) for 30 s. The supernatant fraction was collected, and the cells were resuspended in an equal volume of water to make the cell-associated fraction. Both fractions were freeze-thawed twice to lyse the cells. Samples were then centrifuged at 15,871 RCF for 30 s to pellet debris and were serially diluted 1:5 in Tris-buffered saline (TBS), and 400 μl of each dilution was applied to a 0.45-μm nitrocellulose membrane in a 96-well Minifold I Dot-Blot system (Whatman). The primary monoclonal antibody to Stx1 (13C4 [30]) or Stx2 (11E10 [31]) was diluted 1:300 in TBS-Tween (TBST). The secondary antibody was a horseradish peroxidase (HRP)-conjugated rabbit anti-mouse antibody (Bio-Rad) diluted 1:200.
Cell culture.
Vero cells (CCL-81; American Type Culture Collection [ATCC], Manassas, VA) were grown in Eagle’s minimal essential medium (EMEM) (Lonza, Inc., Walkersville, MD). Tissue culture media were supplemented with 10% fetal bovine serum (FBS), 10 U/ml penicillin, and 10 μg/ml Str.
Cytotoxicity assay.
The Vero cytotoxicity assay was carried out as published previously (32). Briefly, Vero cells were seeded into 96-well plates and were incubated at 37°C under 5% CO2 for 24 h. Serial dilutions of toxin samples were overlaid on cells, and the plates were incubated for an additional 48 h. The cells were then fixed in formalin and stained with crystal violet. The plates were then washed and air dried, and the absorbance of the plates at 590 nm was measured spectrophotometrically. The CD50 for each sample was determined by the inverse of the toxin dilution that caused 50% killing of cells relative to the number of untreated control cells.
Purification of Stx1a, the Stx1 B subunit, Stx2a, and Stx2a toxoid.
Proteins were purified from E. coli DH5α lysates that contained Stx1a (expressed from pLPSH3 [33]), the Stx1a B subunit [expressed from pMJS33::stxB1 in pBluescript II KS(–) as described in reference 34], the Stx2a B subunit [expressed from pMJS34::stxB2 in pBluescript II KS(–) as described in reference 34], Stx2a (expressed from pJES120 [35]), or Stx2a toxoid (expressed from pNR100 [23]). The proteins were purified by affinity chromatography over AminoLink Coupling Resin columns as described previously (19). LPS levels were quantified with a Limulus amebocyte assay (Thermo Fisher Scientific, Waltham, MA) and were found to be <40 pg/μg protein for each protein preparation.
Mice.
All mouse studies were conducted in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (36) and were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University. Six-week-old male BALB/c mice from Charles River Laboratories (Wilmington, MA) were used for all animal experiments. Mice were housed in filter-top cages with access to food and water ad libitum, unless otherwise stated.
Infection.
Mice were fasted and were given water supplemented with 5 g/liter Str for 18 h prior to infection. On the day of infection, overnight bacterial cultures were concentrated 50-fold by centrifugation. The mice were given 0.1 ml of the concentrated bacterial sample by intragastric administration. One hour postinfection, food and Str-supplemented water were returned to the mice. The mice were weighed daily and were observed for 14 days to monitor morbidity and mortality. For some experiments, mice were given daily i.p. injections of 15 μg Cip dissolved in PBS or PBS only (control mice) starting on day 2 postinfection (D2PI).
Passive immunization experiments.
For some experiments, mice were infected as described above and were then given a tail vein injection of monoclonal anti-Stx1 (cαStx1 [37]) or anti-Stx2 (11E10 [31]) or polyclonal anti-Stx1 (24) on D2PI. The humanized monoclonal antibody cαStx1 binds the B subunit of Stx1, and the mouse monoclonal antibody 11E10 recognizes the A subunit of Stx2. All antibodies also neutralize the cytotoxic and lethal activities of the Stx they recognize. Two nonspecific control antibodies were used: anti-ricin (Sigma, St. Louis, MO) or anti-cholera toxin (Novus Biologicals, Littleton, CO). Antibodies were diluted in PBS to a concentration of 600 μg/ml, and each mouse received 100 μl. Control mice were given an equal volume of PBS.
Intoxication experiments.
Purified Stx1a B subunit and Stx2a toxin were diluted in PBS and were administered to mice via i.p. injection on opposite sides of the abdomen at the desired concentrations. Mice were observed for signs of morbidity and mortality.
Toxin binding to mouse kidney sections.
Uninfected mice were euthanized, and kidneys were collected and flash frozen in liquid nitrogen. To examine toxin binding, frozen sections were incubated with purified toxin at a concentration of 10 ng/ml, rinsed, and then incubated with a 1:10 dilution of cαStx1 (13.7 mg/ml) to visualize Stx1a (37) or cαStx2 (10 mg/ml) to visualize Stx2a (37). The secondary antibody was a 1:50 dilution of an Alexa Fluor 488-conjugated goat anti-human antibody (Invitrogen, Eugene, OR). Control slides were also stained with the secondary antibody alone to ensure specific binding; no fluorescence was observed in these control slides (data not shown). The sections were counterstained with SlowFade Diamond Antifade Mountant with 4',6-diamidino-2-phenylindole (DAPI) (Invitrogen, Eugene, OR). All sections were stored at 4°C in the dark and were examined by fluorescence microscopy.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI020148 to A.D.O.
We thank Kieron Torres for technical assistance.
The opinions and assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense, the Department of the Navy, the Uniformed Services University of the Health Sciences, or the National Institutes of Health.
REFERENCES
- 1.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.091101p1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 3.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. 1999. Food-related illness and death in the United States. Emerg Infect Dis 5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks JT, Sowers EG, Wells JG, Greene KD, Griffin PM, Hoekstra RM, Strockbine NA. 2005. Non-O157 Shiga toxin-producing Escherichia coli infections in the United States, 1983–2002. J Infect Dis 192:1422–1429. doi: 10.1086/466536. [DOI] [PubMed] [Google Scholar]
- 5.Marder EP, Griffin PM, Cieslak PR, Dunn J, Hurd S, Jervis R, Lathrop S, Muse A, Ryan P, Smith K, Tobin-D'Angelo M, Vugia DJ, Holt KG, Wolpert BJ, Tauxe R, Geissler AL. 2018. Preliminary incidence and trends of infections with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 2006–2017. MMWR Morb Mortal Wkly Rep 67:324–328. doi: 10.15585/mmwr.mm6711a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogura Y, Gotoh Y, Itoh T, Sato MP, Seto K, Yoshino S, Isobe J, Etoh Y, Kurogi M, Kimata K, Maeda E, Pierard D, Kusumoto M, Akiba M, Tominaga K, Kirino Y, Kato Y, Shirahige K, Ooka T, Ishijima N, Lee KI, Iyoda S, Mainil JG, Hayashi T. 2017. Population structure of Escherichia coli O26:H11 with recent and repeated stx2 acquisition in multiple lineages. Microb Genom 3. doi: 10.1099/mgen.0.000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MN. 2004. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem 279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 8.Lindberg AA, Brown JE, Stromberg N, Westling-Ryd M, Schultz JE, Karlsson KA. 1987. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J Biol Chem 262:1779–1785. [PubMed] [Google Scholar]
- 9.Lingwood CA, Law H, Richardson S, Petric M, Brunton JL, De Grandis S, Karmali M. 1987. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J Biol Chem 262:8834–8839. [PubMed] [Google Scholar]
- 10.Ling H, Boodhoo A, Hazes B, Cummings MD, Armstrong GD, Brunton JL, Read RJ. 1998. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 37:1777–1788. doi: 10.1021/bi971806n. [DOI] [PubMed] [Google Scholar]
- 11.Scheutz F, Teel LD, Beutin L, Pierard D, Buvens G, Karch H, Mellmann A, Caprioli A, Tozzoli R, Morabito S, Strockbine NA, Melton-Celsa AR, Sanchez M, Persson S, O'Brien AD. 2012. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J Clin Microbiol 50:2951–2963. doi: 10.1128/JCM.00860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krüger A, Lucchesi PM. 2015. Shiga toxins and stx phages: highly diverse entities. Microbiology 161:451–462. doi: 10.1099/mic.0.000003. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. 2000. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J Infect Dis 181:664–670. doi: 10.1086/315239. [DOI] [PubMed] [Google Scholar]
- 14.Orth D, Grif K, Khan AB, Naim A, Dierich MP, Würzner R. 2007. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn Microbiol Infect Dis 59:235–242. doi: 10.1016/j.diagmicrobio.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. 1989. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J Infect Dis 160:994–998. doi: 10.1093/infdis/160.6.994. [DOI] [PubMed] [Google Scholar]
- 16.Donohue-Rolfe A, Kondova I, Oswald S, Hutto D, Tzipori S. 2000. Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J Infect Dis 181:1825–1829. doi: 10.1086/315421. [DOI] [PubMed] [Google Scholar]
- 17.Fuller CA, Pellino CA, Flagler MJ, Strasser JE, Weiss AA. 2011. Shiga toxin subtypes display dramatic differences in potency. Infect Immun 79:1329–1337. doi: 10.1128/IAI.01182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O’Brien AD, Samuel JE. 1993. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect Immun 61:3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo LM, Melton-Celsa AR, Smith MA, Smith MJ, O’Brien AD. 2014. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect Immun 82:1213–1221. doi: 10.1128/IAI.01264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith MJ, Teel LD, Carvalho HM, Melton-Celsa AR, O’Brien AD. 2006. Development of a hybrid Shiga holotoxoid vaccine to elicit heterologous protection against Shiga toxins types 1 and 2. Vaccine 24:4122–4129. doi: 10.1016/j.vaccine.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 21.Russo LM, Melton-Celsa AR, O’Brien AD. 2016. Shiga toxin (Stx) type 1a reduces the oral toxicity of Stx type 2a. J Infect Dis 213:1271–1279. doi: 10.1093/infdis/jiv557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards AC, Melton-Celsa AR, Arbuthnott K, Schmitt CK, Wong HC, O’Brien AD. 1998. Vero cell neutralization and mouse protective efficacy of humanized monoclonal antibodies against Escherichia coli toxins Stx1 and Stx2, p 388–392. In Kaper JB, O’Brien AD (ed), Escherichia coli O157:H7 and other Shiga-toxin-producing E. coli strains. ASM Press, Washington, DC. [Google Scholar]
- 23.Wen S, Teel L, Judge N, O’Brien A. 2006. Genetic toxoids of Shiga toxin types 1 and 2 protect mice against homologous but not heterologous toxin challenge. Vaccine 24:1142–1148. doi: 10.1016/j.vaccine.2005.08.094. [DOI] [PubMed] [Google Scholar]
- 24.Strockbine NA, Marques LR, Newland JW, Smith HW, Holmes RK, O’Brien AD. 1986. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect Immun 53:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palermo M, Alves-Rosa F, Rubel C, Fernandez G, Fernandez-Alonso G, Alberto F, Rivas M, Isturiz M. 2000. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1β exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin Exp Immunol 119:77–83. doi: 10.1046/j.1365-2249.2000.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandelli JR, Griener TP, Laing A, Mulvey G, Armstrong GD. 2015. The effects of Shiga toxin 1, 2 and their subunits on cytokine and chemokine expression by human macrophage-like THP-1 cells. Toxins 7:4054–4066. doi: 10.3390/toxins7104054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee MS, Kwon H, Lee EY, Kim DJ, Park JH, Tesh VL, Oh TK, Kim MH. 2016. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of proinflammatory cytokine interleukin-1β and apoptotic cell death. Infect Immun 84:172–186. doi: 10.1128/IAI.01095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jandhyala DM, Ahluwalia A, Schimmel JJ, Rogers AB, Leong JM, Thorpe CM. 2016. Activation of the classical mitogen-activated protein kinases is part of the Shiga toxin-induced ribotoxic stress response and may contribute to Shiga toxin-induced inflammation. Infect Immun 84:138–148. doi: 10.1128/IAI.00977-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda T, Yoshino K, Adachi E, Sato Y, Yamagata K. 1999. In vitro assessment of a chemically synthesized Shiga toxin receptor analog attached to chromosorb P (Synsorb Pk) as a specific absorbing agent of Shiga toxin 1 and 2. Microbiol Immunol 43:331–337. doi: 10.1111/j.1348-0421.1999.tb02413.x. [DOI] [PubMed] [Google Scholar]
- 30.Strockbine NA, Marques LR, Holmes RK, O’Brien AD. 1985. Characterization of monoclonal antibodies against Shiga-like toxin from Escherichia coli. Infect Immun 50:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perera LP, Marques LR, O’Brien AD. 1988. Isolation and characterization of monoclonal antibodies to Shiga-like toxin II of enterohemorrhagic Escherichia coli and use of the monoclonal antibodies in a colony enzyme-linked immunosorbent assay. J Clin Microbiol 26:2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentry MK, Dalrymple JM. 1980. Quantitative microtiter cytotoxicity assay for Shigella toxin. J Clin Microbiol 12:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tesh VL, Samoel JE, Perera LP, Sharefkin JB, O’Brien AD. 1991. Evaluation of the role of Shiga and Shiga-like toxins in mediating direct damage to human vascular endothelial cells. J Infect Dis 164:344–352. doi: 10.1093/infdis/164.2.344. [DOI] [PubMed] [Google Scholar]
- 34.Smith MJ, Carvalho HM, Melton-Celsa AR, O’Brien AD. 2006. The 13C4 monoclonal antibody that neutralizes Shiga toxin type 1 (Stx1) recognizes three regions on the Stx1 B subunit and prevents Stx1 from binding to its eukaryotic receptor globotriaosylceramide. Infect Immun 74:6992–6998. doi: 10.1128/IAI.01247-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindgren SW, Melton AR, O’Brien AD. 1993. Virulence of enterohemorrhagic Escherichia coli O91:H21 clinical isolates in an orally infected mouse model. Infect Immun 61:3832–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 37.Melton-Celsa AR, Carvalho HM, Thuning-Roberson C, O’Brien AD. 2015. Protective efficacy and pharmacokinetics of human/mouse chimeric anti-Stx1 and anti-Stx2 antibodies in mice. Clin Vaccine Immunol 22:448–455. doi: 10.1128/CVI.00022-15. [DOI] [PMC free article] [PubMed] [Google Scholar]