

Adaptation of Staphylococcus aureus to host microenvironments during chronic infection involves spontaneous mutations, yet changes underlying adaptive phenotypes remain incompletely explored. Here, we employed artificial selection and whole-genome sequencing to better characterize spontaneous chromosomal mutations that alter two pathogenicity phenotypes relevant to chronic infection in S. aureus: intracellular invasiveness and intracellular cytotoxicity.
KEYWORDS: microbial genetics, microbial pathogenesis, Staphylococcus aureus, evolution, genome analysis, molecular genetics, virulence
ABSTRACT
Adaptation of Staphylococcus aureus to host microenvironments during chronic infection involves spontaneous mutations, yet changes underlying adaptive phenotypes remain incompletely explored. Here, we employed artificial selection and whole-genome sequencing to better characterize spontaneous chromosomal mutations that alter two pathogenicity phenotypes relevant to chronic infection in S. aureus: intracellular invasiveness and intracellular cytotoxicity. We identified 23 genes whose alteration coincided with enhanced virulence, 11 that were previously known and 12 (52%) that had no previously described role in S. aureus pathogenicity. Using precision genome editing, transposon mutants, and gene complementation, we empirically assessed the contributions of individual genes to the two virulence phenotypes. We functionally validated 14 of 21 genes tested as measurably influencing invasion and/or cytotoxicity, including 8 newly implicated by this study. We identified inactivating mutations (murA, ndhC, and a hypothetical membrane protein) and gain-of-function mutations (aroE Thr182Ile, yhcF Thr74Ile, and Asp486Glu in a hypothetical peptidase) in previously unrecognized S. aureus virulence genes that enhance pathogenesis when introduced into a clean genetic background, as well as a novel activating mutation in the known virulence regulator gene saeS (Ala106Thr). Investigation of potentially epistatic interactions identified a tufA mutation (Ala271Val) that enhances virulence only in the context of purine operon repressor gene (purR) inactivation. This project reveals a functionally diverse range of genes affected by gain- or loss-of-function mutations that contribute to S. aureus adaptive virulence phenotypes. More generally, the work establishes artificial selection as a means to determine the genetic mechanisms underlying complex bacterial phenotypes relevant to adaptation during infection.
INTRODUCTION
Staphylococcus aureus is a prevalent opportunistic pathogen that is associated with a diverse spectrum of disease (1–4) and is responsible for a high burden of patient morbidity and mortality worldwide (1, 2, 5–7). S. aureus infections, especially noncutaneous diseases, are often chronic or relapsing (8–11) and are difficult to eradicate even with appropriate antibiotic treatment. The organism employs a diverse array of strategies and virulence factors that contribute to its pathogenesis and persistence (5, 12), including invasion of host cells and survival within the phagosome or cytoplasm (13–15).
Although the genetic composition of S. aureus strains can influence their inherent virulence phenotypes (16–19), the host-pathogen interaction is dynamic and must change over time in order for the organism to establish and maintain infection (20–22). For example, during acute phases, S. aureus must evade the immune system and destroy host cells to enter deep tissue structures, whereas in chronic infection, nutrient deprivation and lysosomal degradation pose challenges (20). Regulation of virulence gene expression is thought to facilitate adaptation to host microenvironments over the short term, as evidenced by rapidly reversible changes associated with persistence as early as 3 days postinfection (23), but as infection becomes more established, spontaneous chromosomal mutations can fix beneficial regulatory patterns or alter the coding sequences of effector genes (21, 22). Consequently, identifying spontaneous mutations that go to fixation during infection can illuminate not only genes that are important in establishing chronic disease, but also those that may be differentially regulated in more acute phases.
Most studies of the adaptive changes in S. aureus occurring during chronic infection have focused on specific, previously identified genetic or phenotypic alterations (24–27). Comparatively little work has examined spontaneous mutations associated with disease progression on a genome-wide scale. Transposon mutant screens have been performed to identify genes that influence virulence under various experimental conditions or in model organisms (28–31), although this approach is unable to identify gain-of-function mutations and is also subject to artifacts arising from polar effects (32). More recently, whole-genome sequencing of isolates collected longitudinally during the course of infection has been conducted (10, 21, 27, 33–35). Although illuminating, genomic analyses to date have characterized only limited numbers of strains from patients with particular infection types and do not identify how mutations impact specific pathogenic processes during infection.
To better characterize the spectrum of spontaneous mutations capable of promoting S. aureus infection and persistence, here, we have employed artificial selection for virulence phenotypes under defined laboratory conditions, followed by whole-genome sequencing. We have used this approach to explore chromosomal changes enhancing S. aureus capacity for intracellular cytotoxicity (18) and intracellular invasion (13–15), phenotypes relevant to chronic infective states (12, 36). Prior work has identified multiple staphylococcal factors involved in invasion, including fibronectin-binding proteins (37, 38), autolysin Atl (39), the lipoprotein-like gene cluster and other cyclomodulins (40, 41), and manipulation of host autophagy mechanisms (42). The mechanisms underlying intracellular cytotoxicity are less well understood but may involve the same cytolytic toxins expressed under the control of the agr, sar, and arl systems (43, 44) that mediate extracellular tissue destruction. The spontaneous mutations that refine, adapt, augment, and enhance the actions of these factors during the course of infection are currently unknown. Our work implicates a combination of loss-of-function and gain-of-function mutations that occur in both known and novel virulence genes and that involve a variety of cellular and metabolic pathways.
RESULTS
In vitro selection increases S. aureus phenotypes relevant to infection.
We designed separate experiments to enrich spontaneous mutants having increased capacity for either intracellular invasion or intracellular cytotoxicity (Fig. 1). In brief, bacteria were serially passaged in the presence of HeLa cells using modified gentamicin protection assays (15, 45, 46), which specifically kill bacteria that are not internalized by eukaryotic cells. For both assays, HeLa cells were incubated with S. aureus for 1 h to allow invasion, followed by application of antibiotics to kill extracellular organisms. When selecting for intracellular invasion, bacteria were released from HeLa cells by isotonic lysis 3 h after antibiotic exposure. For intracellular-cytotoxicity selection, incubation was extended for 1 to 2 days in antibiotic-free medium that was unable to sustain staphylococcal growth (47) (see Fig. S1 in the supplemental material), after which bacteria that had been released into the culture supernatant were collected. The harvested bacterial populations were briefly expanded by overnight culture in supplemented medium (48) that supported growth of auxotrophic strains (22, 49–51), washed to remove soluble toxins, and then used to infect fresh HeLa cells. At the midpoint and endpoint of passaging, two isolates were picked from each replicate for a total of 24 isolates per selection, and their activity for the desired phenotype was quantitatively assessed (Fig. 2; see Table S1 in the supplemental material).
FIG 1.

Schema of in vitro selection for enhanced-pathogenicity phenotypes. (A) Washed S. aureus bacteria are combined with cultured HeLa cells in antibiotic- and serum-free medium. (B) Brief incubation (1 h) allows a fraction of the bacteria to invade the HeLa cells. (C) Extracellular S. aureus bacteria are killed by the addition of antibiotics, which are unable to penetrate eukaryotic-cell membranes. (D) For cytotoxicity selection, the medium is subsequently replaced with antibiotic-free medium and cultures are incubated for 1 to 2 days, allowing intracellular bacteria to replicate and exert cytotoxic effects. (E) Cytotoxic bacteria rupture host cell membranes and are released into the culture supernatant. The extracellular bacteria are unable to replicate in the culture medium. (F) Bacteria in the culture supernatant are collected, leaving behind intracellular organisms. (G) For invasion selection, HeLa cells are subjected to isotonic lysis 3 h after the application of antibiotics. (H) Bacteria released from host cells are collected. (I) For both selections, harvested bacteria are expanded by overnight culture in nutritionally supplemented medium that prevents reversion of small-colony variants. The cells are washed to remove soluble toxins prior to the next round of selection.
FIG 2.

Virulence phenotypes and growth rates of evolved S. aureus isolates. (A) Average invasiveness of isolates selected for enhanced-virulence phenotypes. (B) Average cytotoxicities of isolates selected for enhanced-virulence phenotypes. (C) Average doubling times of isolates selected for enhanced-virulence phenotypes. (A to C) Measurements were normalized to the parental strain, and isolates were aggregated by selection type and time of passaging. The reference activity of the wild-type strain (set to 1) is highlighted on each y axis. The data points correspond to individual strains, the horizontal lines indicate medians for aggregated groups of strains, and the error bars indicate the SEM for the groups. Note the log2 scale in panel A. Populations that are significantly different (Z test) from the parental strain are indicated by asterisks: *, P < 0.05; **, P < 0.001.
By 14 passages under conditions designed to select for enhanced intracellular invasion, significant (P < 0.012; 2-tailed t test) increases in invasiveness were observed for 11 of the 12 evolved isolates and, at 24 passages, for all 12 evolved isolates. Average quantitative measures of invasiveness also increased between midpoint and endpoint passaging (Fig. 2A), and at a maximum, the invasiveness of selected replicates had activity exceeding that of the parental strain by a factor of 700.
Selection for intracellular cytotoxicity similarly increased isolates’ activity with passaging. After 10 passages, all 12 isolates demonstrated statistically significant (P < 0.018) increases in quantitative measurements of cytotoxicity. For unclear reasons, this number dropped to 8 of 12 isolates by 20 passages; however, overall quantitative measurements of HeLa cell death continued to increase (Fig. 2B). Isolates with the maximum cytotoxicity activity had activity that exceeded that of the parental strain by a factor of ∼1.5.
These selected phenotypes are not necessarily mutually exclusive, as intracellular cytotoxicity is dependent on prior invasion of host cells by bacteria. To assess this, we subjected evolved isolates to both virulence assays (see Table S1). Consistent with phenotypic overlap, evolved isolates tended to demonstrate increased capabilities for both cytotoxicity and invasion relative to the parental strain regardless of the selection conditions used (Fig. 2A and B). Nineteen of 24 isolates from cytotoxicity selections showed significant (P < 0.012; 2-tailed t test) gains in invasiveness, while 20 of 24 isolates selected for invasiveness had elevated intracellular-cytotoxicity activity at the same significance level. Isolates selected for invasion showed the greatest magnitude of activity for both of the assayed virulence phenotypes. This finding could exclusively reflect increased bacterial entry into host cells: indeed, there was a positive correlation between the intracellular cytotoxicity of individual isolates selected for invasion and their measured invasiveness (Pearson correlation coefficient = 0.63), but not for isolates selected for cytotoxicity (Pearson correlation coefficient = −0.084). Despite similarities in virulence phenotypes, we conclude that the properties and mechanisms of virulence activity are distinct when comparing isolates from the two selections.
Slow growth is a consequence of evolved invasion, but not cytotoxicity, phenotypes.
The S. aureus small-colony variant (SCV) is a phenotype that is associated with persistent infection and is characterized by slow growth on most laboratory media (22, 49–51). SCVs often emerge during chronic infection and have a number of adaptive traits that increase their persistence in the host (22, 49, 50). To determine whether selected isolates displayed growth deficiencies, we quantified the doubling times of evolved isolates in rich medium (52) (Fig. 2C; see Table S2 in the supplemental material). All the strains selected for invasiveness had increased doubling times compared to the unpassaged parental strain (1.65- ± 0.18-fold change [average ± standard deviation]), representing a significant difference for that group (P = 2.9 × 10−10; 2-tailed t test). There was no correlation between the degree of intracellular invasion and an isolate’s doubling time (Pearson correlation coefficient = 0.30), indicating that enhanced invasion is not a direct consequence of slow growth. There was also no significant difference in the doubling times seen for isolates taken from midpoint and endpoint selection (P = 0.58), showing that growth deficiencies did not increase proportionally to passaging. Strikingly, strains passaged under conditions selecting for cytotoxicity did not generally display differences in doubling times compared to the unpassaged progenitor (1.08- ± 0.16-fold change; P = 0.26 for the population; 2-tailed t test).
Although both populations of selected bacteria showed enhanced abilities to invade host cells (Fig. 2A), these results indicate that a slow-growth phenotype accompanies selection for intracellular invasion, but not selection for intracellular cytotoxicity.
Evolved isolates display differences in colony growth and pigmentation.
To directly determine whether any evolved strains displayed an SCV phenotype, we evaluated them for growth deficiency on blood agar, which is a hallmark of that colony morphology (51). Consistent with SCVs, 7 of the 48 evolved isolates demonstrated greatly reduced growth on blood agar relative to the parental strain (see Fig. S2A in the supplemental material). Two lineages selected for cytotoxicity were affected: both endpoint passaging isolates from one lineage and both midpoint and endpoint isolates from the other displayed the phenotype. The final isolate was from endpoint passage of a replicate selected for invasiveness, whereas the other isolate collected at the time point was not affected.
We separately assessed the evolved strains for differences in pigment production, a phenotype more broadly dependent on sigB regulation (53, 54) that contributes to S. aureus pathogenesis in vivo through promoting bacterial fitness and escape from neutrophil killing (53, 55), reasoning that the phenotype could be influenced by mutations involved in other pathways relevant to our selection conditions (56). The pigmentation of most evolved isolates (41 of 48) matched that of the progenitor, but phenotypic heterogeneity was observed in seven of the strains (Fig. S2B). Four isolates selected for intracellular cytotoxicity displayed a high level of pigmentation, all of which were derived from the same replicate lineage at different time points. An intermediate level of pigment production was seen in an additional seven isolates derived from four replicates selected for intracellular survival. In all but one case, both isolates picked from a replicate had the same intermediate pigmentation phenotype.
We conclude that selection for narrowly defined virulence activities can more broadly enhance accessory phenotypes that increase S. aureus fitness in a human host.
sigB, but not agrA, expression is selected in evolved isolates.
sigB and agr represent two interrelated global regulatory factors that control phenotypes relevant to acute infection and intracellular persistence (20, 57–59), where sigB expression is antagonistic to that of agr (60). Expression of sigB promotes intracellular replication of S. aureus (57) and accessory phenotypes relevant to persistence during chronic infection (20). In contrast, the agr locus is thought to be more active during acute infection (20) and regulates the production of specific cytotoxins (58) and genes participating in cell internalization (61). To explore a potential role of these regulators in our evolved isolates, we quantitated their expression using real-time PCR (Fig. 3; see Table S3 in the supplemental material).
FIG 3.

Expression of sigB and agrA in evolved S. aureus isolates. Real-time PCR quantitation of sigB (A) and agrA (B) was normalized to the parental strain, with isolates aggregated by selection type and time of passaging. The reference activity of the wild-type strain (set to 1) is highlighted on each y axis. The data points correspond to individual strains, the horizontal lines indicate medians for aggregated groups of strains, and the error bars indicate SEM for the groups. Note the log2 scale in panel B. Populations that are significantly different (Z test) from the parental strain are indicated by asterisks: *, P < 0.05; **, P < 0.001.
Relative to the parental strain, sigB (Fig. 3A) expression was significantly (P ≤ 0.046; 2-tailed t test) elevated in 20 of 24 isolates selected for cytotoxicity (average 1.65-fold increase; standard error of the mean [SEM], 0.46). In contrast, sigB expression was much more variable in isolates selected for invasiveness, with only 8 of 24 showing increased expression at the same significance level (average 1.14-fold increase; SEM, 0.073). Cytotoxic selection isolates expressed significantly (P = 9.0 × 10−5; 2-tailed t test) more sigB than those selected for invasiveness. In contrast, expression of the response regulator gene agrA (62) was more heterogeneous (Fig. 3B), with a wider range of gene expression levels observed across each discrete population. No statistically significant differences were observed when comparing agrA expression across separately selected populations or comparing time points within individual selected populations. Interestingly, there was also no relationship between expression levels of agrA and sigB (Pearson correlation coefficient = 0.172).
Increased sigB expression in intracellular-cytotoxicity isolates suggests selection for enhanced intracellular persistence (57), whereas there is no evidence for correlation of agr expression with either selection scheme. As our experiments were designed to select and recover spontaneous chromosomal mutants with increased pathogenicity phenotypes, it was not unexpected that canonical gene-regulatory patterns might be disrupted by direct mutation of effector genes or subsequent perturbations to regulatory feedback loops. Consequently, mutated factors regulated by sigB or agrA may contribute to pathogenicity phenotypes independently of the expression of higher-level regulatory factors.
Whole-genome sequencing of selected S. aureus isolates identifies chromosomal mutations.
Diversity in quantitative virulence phenotypes among different selected replicates suggests contributions of multiple, nonredundant genes that additively confer those traits. In order to define the range of spontaneous coding mutations that contribute to virulence phenotypes, we performed whole-genome sequencing of evolved isolates.
An average of 2.7 coding mutations (range, 1 to 5) were identified per strain (see Table S4 in the supplemental material). A total of 23 genes were mutated across evolved isolates compared to the parent (Table 1). Sixteen genes were mutated exclusively during selection for intracellular invasion, 9 were found during selection for intracellular cytotoxicity, and 3 were mutated under both conditions. Thus, despite superficial similarities in the virulence phenotypes of the evolved isolates (Fig. 2A and B), the two selections resulted in largely nonoverlapping mutations.
TABLE 1.
Mutations recovered after selection for intracellular invasion and/or intracellular cytotoxicity
| Common gene name | Reported virulence factor in S. aureus (reference)? | Protein accession no. | Gene function | Protein changea | Mutation type | Occurrence in selection isolates (n = 24) |
|
|---|---|---|---|---|---|---|---|
| Intracellular cytotoxicity | Intracellular invasion | ||||||
| pheT | No | AHM68762.1 | Phenylalanyl-tRNA synthase subunit beta | Leu110Ile | Missense | 0 | 1 |
| ctaB | Yes (56) | AHM68781.1 | Protoheme IX farnesyltransferase | Thr222Ile | Missense | 1 | 0 |
| qoxD | Yes (81, 83) | AHM68836.1 | Quinol oxidase subunit 4 | Gly83Arg | Missense | 0 | 2 |
| menD | Yes (77) | AHM68850.1 | 2-Succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase | Ile91fs | Frameshift | 0 | 1 |
| Ala83del | In-frame deletion | 6 | 0 | ||||
| menF | Yes (33) | AHM68851.1 | Isochorismate synthase | Ser31* | Stop gain | 0 | 1 |
| ndhC | No | AHM68945.1 | NADH dehydrogenase | Ala386Val | Missense | 0 | 1 |
| None (unnamedA) | No | AHM68983.1 | Hypothetical peptidase | Asp486Glu | Missense | 0 | 2 |
| saeS | Yes (72) | AHM69145.1 | Histidine kinase | Ala106Thr | Missense | 0 | 16 |
| nupC | No | AHM69205.1 | Nucleoside permease | Pro189Leu | Missense | 0 | 2 |
| mnhA | Yes (111) | AHM69226.1 | Monovalent cation/H+ antiporter subunit A | Ala722Thr | Missense | 0 | 2 |
| sdrC | Yes (84, 85) | AHM69286.1 | Hydrolase | Glu469Gly | Missense | 2 | 0 |
| tufA | No | AHM69299.1 | Elongation factor Tu | Ala271Val | Missense | 0 | 4 |
| purR | Yes (29, 30, 68) | AHM69351.1 | Purine operon repressor | Gly210fs | Frameshift | 6 | 0 |
| Ala208fs | Frameshift | 6 | 0 | ||||
| Ser189Leu | Missense | 0 | 4 | ||||
| Gly119Asp | Missense | 0 | 2 | ||||
| Gly67Arg | Missense | 0 | 2 | ||||
| Lys37Thr | Missense | 2 | 2 | ||||
| Val30fs | Frameshift | 0 | 2 | ||||
| epsC | No | AHM69661.1 | Polysaccharide biosynthesis protein | Asn333fs | Frameshift | 6 | 0 |
| ypfH | No | AHM69943.1 | Carboxylesterase | Ser104* | Stop gain | 0 | 3 |
| None (unnamedB) | No | AHM70189.1 | Hypothetical membrane protein | Phe41fs | Frameshift | 0 | 4 |
| murA | No | AHM70338.1 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | Ala378del | In-frame deletion | 4 | 0 |
| yhcF | No | AHM70475.1 | GntR family transcriptional regulator | Thr74Ile | Missense | 0 | 1 |
| aroE | No | AHM70790.1 | Shikimate dehydrogenase | Thr182Ile | Missense | 1 | 0 |
| Tyr164fs | Frameshift | 10 | 0 | ||||
| srrA | Yes (92) | AHM70893.1 | Transcriptional regulator | Arg197fs | Frameshift | 2 | 0 |
| Glu232* | Stop gain | 2 | 0 | ||||
| ubiE | No | AHM70914.1 | Ubiquinone biosynthesis methyltransferase | Lys21fs | Frameshift | 4 | 0 |
| ebh | Yes (78) | AHM70948.1 | Matrix-binding protein | Glu6622Lys | Missense | 0 | 4 |
| brnQ2 | Yes (112) | AHM70974.1 | Branched-chain amino acid ABC transporter | Asp198Tyr | Missense | 0 | 1 |
An asterisk indicates a stop gain mutation at the amino acid position listed.
Of the genes recovered, 11 (48%) have documented roles in S. aureus virulence and pathogenesis, whereas the remaining 12 (52%) are newly implicated by our selection. Two genes have no previously documented function and are only predicted: for the purposes of this study, we have arbitrarily designated these genes unnamedA and unnamedB.
Directed inactivating lesions identify loss-of-function mutations contributing to pathogenicity phenotypes.
It has been reported that the majority of mutations arising during in-host evolution in S. aureus inactivate protein function (34). To assess genes identified by our selection, we therefore initially generated isogenic single-gene nonpolar deletions using recombineering and CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9-mediated counterselection (63) or obtained bursa aurealis transposon mutants constructed in a different strain background (JE2) (64) when this strategy was unsuccessful. Two genes (mnhA and tufA) could not be constructed as nonpolar deletions, but strains were engineered to carry the specific missense mutations resulting from selection. Integrating these strategies, 21 of the 23 genes implicated by selection could be further studied, with only epsC and pheT recalcitrant to modification. We assessed the virulence phenotypes of each single-gene mutant strain compared to the unmodified parental strain (Fig. 4; see Table S5 in the supplemental material).
FIG 4.

Virulence phenotypes of single-gene mutant strains. Intracellular cytotoxicity (left axis) and intracellular invasion (right axis) activities of single-gene mutants relative to the parental strain are displayed. The reference activity of the parental strain (set to 1) is highlighted separately on each y axis. Measured values that are significantly different (by 2-tailed t test) than those of the parental strain are indicated by asterisks: *, P < 0.03; **, P < 0.001. The asterisks are colored to correspond to the phenotype being assessed. The error bars indicate SEM. Information about the selection conditions that identified specific gene mutations, the types of mutations identified, and the background and design of engineered mutant strains is color coded beneath the chart.
When disrupted, eight genes (qoxD, menD, murA, ndhC, purR, ebh, sdrC, and unnamedB) conferred a statistically significant increase in the virulence phenotype under which the mutation had been selected and, in some cases, both of the assayed virulence phenotypes. Because intracellular cytotoxicity is dependent on prior cellular entry by bacteria, it is expected that a gene’s increasing invasiveness could additionally enhance cytotoxicity: this pattern was observed for five of the eight mutants (qoxD, ndhC, purR, sdrC, and unnamedB). Evolved isolates carried a variety of missense and nonsense mutations in these genes, suggesting that the phenotypic changes associated with those mutations result from loss of function.
Despite being recovered as nonsense mutations in evolved isolates, disruption of three genes (menF, srrA, and ypfH) failed to confer an increase in either assayed virulence phenotype. Five other genes affected by missense mutations did not increase virulence when constructed as nonpolar single-gene deletions (aroE, saeS, nupC, yhcF, and unnamedA). Isogenic missense mutation of tufA (Ala271Val) and mnhA (Ala722Thr) also failed to increase either virulence phenotype.
Three gene disruption mutants (brnQ2, ctaB, and ubiE) did not increase the expected virulence phenotypes but unexpectedly showed enhancement of the virulence phenotype for which strains bearing the gene mutations had not been selected.
The change in virulence activity seen with any singular isogenic mutant was considerably less than the maximal levels observed for evolved strains (Fig. 2), particularly for the cytotoxicity phenotype, suggesting that contributory mutations exert a synergistic effect.
Complementation studies identify gain-of-function mutations contributing to pathogenicity phenotypes.
Genes targeted for disruption that failed to confer an increase in the expected virulence phenotype and that accumulated missense mutations during selection (ctaB, unnamedA, saeS, nupC, yhcF, aroE, and brnQ2) may exert effects through gain of function. To test this possibility, we performed complementation studies after mobilizing wild-type and mutant genes onto plasmids under the control of their native promoters. The plasmids were reintroduced into the appropriate deletion strains, and their virulence activities were measured.
Testing did not identify any mutant-complemented strains that showed increased cytotoxicity relative to those complemented with the wild-type allele, including the two mutations selected during cytotoxicity passaging (ctaB and aroE). However, invasion assays revealed that five mutations (ctaB Thr222Ile, aroE Thr182Ile, saeS Ala106Thr, unnamedA Asp486Glu, and yhcF Thr74Ile) conferred significant increases in pathogenicity compared to matched controls (Fig. 5; see Table S6 in the supplemental material). For ctaB, increased invasiveness was also seen for the corresponding nonpolar deletion mutant, indicating that the allele carries a loss-of-function mutation. Nonpolar deletion of the other four genes resulted in reduced invasiveness, showing that these mutant alleles confer gain-of-function effects that promote invasiveness.
FIG 5.

Invasiveness of complemented mutant strains. Shown are intracellular invasion activities of single-gene deletion strains complemented with the specified mutant genes relative to the values seen for the same strains complemented with wild-type genes. The reference activity of the wild-type-complemented strain (set to 1) is highlighted on the y axis. The error bars indicate SEM. Measured values that are significantly different (by 2-tailed t test) than those of the wild-type-complemented strain are indicated by asterisks: *, P < 0.04; **, P < 0.001.
Potential for epistatic interactions governing virulence phenotypes.
Some mutations that failed to yield measurable phenotypes when introduced as single changes may reflect epistatic interactions (65). To explore this possibility, we selected one such mutation, tufA Ala271Val, for further analysis. tufA Ala271Val was observed in four isolates from two separate replicates selected for invasiveness, but in each case, it occurred in the context of an inactivating purR mutation. We therefore constructed an isogenic double mutant containing both of the mutations (tufA Ala271Val and ΔpurR) and tested its invasiveness relative to mutants carrying each of the mutations in isolation.
Consistent with our earlier results (Fig. 4), the tufA mutant showed reduced invasiveness relative to the wild-type strain, whereas the ΔpurR strain had an elevated capacity (Fig. 6; see Table S7 in the supplemental material). Remarkably, when both mutations were present in the same background, invasiveness was significantly increased over that of the wild-type strain and the ΔpurR strain alone. These findings indicate that the tufA Ala271Val mutation functions as a virulence-enhancing mutation but that it is epistatic to purR inactivation.
FIG 6.
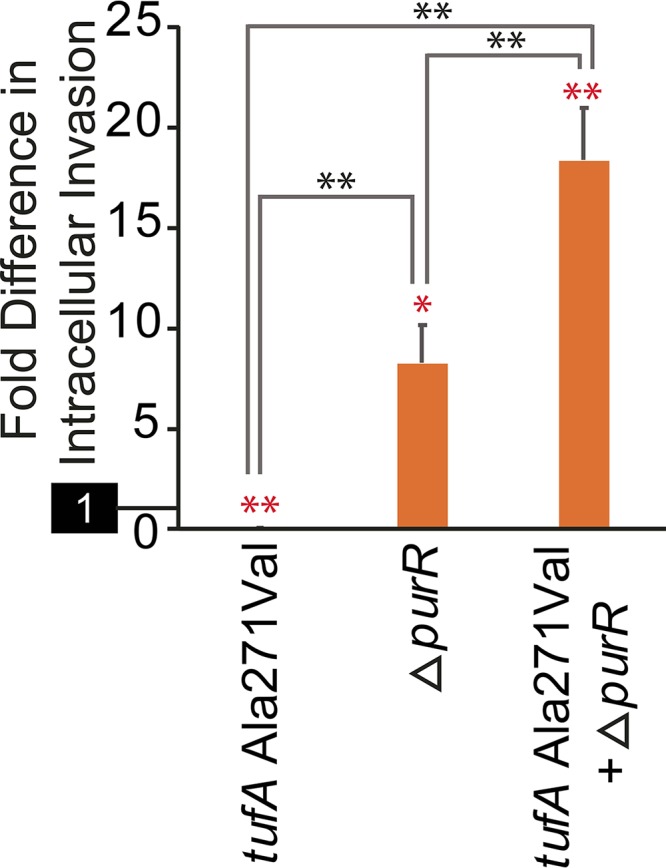
Epistatic interactions influence virulence phenotypes. Intracellular invasion of single- and double- mutant strains of ATCC 29213 relative to the parental strain are displayed. The reference activity of the wild-type parent (set to 1) is highlighted separately on the y axis. The error bars indicate SEM. The statistical significance of comparisons is shown by asterisks: *, P < 0.03; **, P < 0.001 (2-tailed t test). The red asterisks indicate statistical comparison to the parental strain; the black asterisks correspond to comparisons between different mutant strains.
DISCUSSION
Despite the importance of genome evolution in promoting chronic S. aureus infection and persistence, knowledge of the adaptive changes that the organism acquires during pathogenesis in the host remains critically lacking in multiple respects (16, 20). To provide a well-controlled framework for investigating the range and scope of these genomic changes, here, we employed a strategy of in vitro selection and whole-genome characterization to identify and subsequently functionally validate genes responsible for two virulence phenotypes, mammalian-cell invasion and intracellular cytotoxicity, that are relevant to chronic infection.
In addition to enhanced-virulence phenotypes (Fig. 2A and B), evolved isolates showed marked differences in growth rates (Fig. 2C) and pigmentation (see Fig. S1B), revealing that secondary consequences relevant to pathogenicity also resulted from selection. Of note, seven isolates recovered across the selections (15%) had evolved an SCV phenotype (see Fig. S2A), all of which carried mutations in menD (12) (see Table S4), showing that passaging recapitulated an important S. aureus colony phenotype arising during infection in vivo (20, 49, 66, 67). Despite some phenotypic overlap in the virulence phenotypes of strains resulting from different selections, the correlation between invasiveness and cytotoxicity was markedly different among isolates resulting from the two selections, as were the majority of gene mutations that they carried, indicating that separate features were advantageous during passaging.
Whole-genome sequencing implicated 23 genes whose mutation correlated with increased virulence in selected strains (Table 1), only 11 of which (48%) have previously been reported as having roles in S. aureus virulence. The virulence functions of six of these known genes could be validated through genetic studies (Fig. 4 and 5), but even so, our findings reveal new modes of action and virulence effects.
Disruption of purR, the repressor for the purine biosynthetic operon, was selected under both conditions (Table 1) and resulted in enhanced invasion and intracellular cytotoxicity as an isogenic deletion mutant (see Table S5). Transposon mutagenesis experiments have indicated that purine biosynthesis is necessary to achieve full virulence in S. aureus (29, 30, 68) and other organisms (69–71). Our results support a complementary model, in that upregulated purine biosynthetic capacity actually enhances S. aureus virulence.
saeS is a well-characterized sensor histidine kinase gene involved in the regulation of many virulence factors present in the S. aureus genome, and substitution mutations in the gene are known to alter larger gene expression patterns (72). Intriguingly, 16 of the 24 strains selected for infectivity in our study carried identical, previously unreported nonsynonymous mutations in saeS (Ala106Thr), making this the most common recurrent point mutation identified in our study. Complementation experiments revealed that saeS Ala106Thr significantly enhanced invasiveness relative to the wild-type allele, thereby confirming a gain-of-function effect. There are three previously reported variants of saeS (72), at least one of which contains a constitutively activating mutation (Leu18Pro) that has been found to increase invasiveness (73, 74). The mutation identified by our work affects the HAMP domain, which is a motif often involved in signal transduction (75) and is a different region of the protein than those altered in previously reported mutants.
menD is a key gene in the menaquinone biosynthesis pathway, and mutations in menD are often observed in clinical SCV isolates (33, 51). Nonsense mutations of menD in our study resulted from intracellular-cytotoxicity selection, while one isolate from invasiveness selections uniquely carried a missense mutation in the gene. Directed menD deletion mutants have been reported as having variable effects on virulence, with prior work showing unaltered infectivity in a rabbit endocarditis model (76) but reduced virulence in a Caenorhabditis elegans infection model (77). Here, menD transposon mutagenesis measurably increased intracellular cytotoxicity without impacting invasiveness, more consistent with rabbit models (76).
ebh encodes a large (∼1,000-residue) membrane protein (78). Stable SCVs significantly upregulate expression of the gene (79), and mutation of ebh has previously been shown to reduce complement resistance and virulence in mice (78). Here, disruption of ebh increased both invasiveness and cytotoxicity, suggesting that in vivo attenuation of ebh mutants may reflect higher rates of complement-induced bacterial death (78) rather than reduced pathogenicity per se. Mutations in ebh may consequently promote resistance in specific host microenvironments.
S. aureus mutants with deficiencies in heme biosynthesis demonstrate an SCV phenotype with increased intracellular persistence (80), and two genes involving heme usage and having known impact on virulence were similarly identified as loss-of-function mutations in our work. First, the protoheme IX farnesyltransferase gene ctaB is involved in heme biosynthesis and is utilized by S. aureus when it is unable to scavenge the metabolite from a host (56). Prior work has demonstrated that ctaB deletion attenuates growth and virulence in mouse infection models but enhances the growth of persister cells and the production of pigment (56). In our study, inactivating ctaB mutation resulted from passaging for cytotoxicity, but loss of the gene increased invasiveness without affecting the selected phenotype, suggesting that ctaB mutation indirectly contributes to cytotoxicity phenotypes through increased intracellular invasion. Second, qoxD (cyoD) encodes a subunit of quinol oxidase, one of two oxidase systems present in S. aureus that support aerobic respiration (81) and also appear to protect against heme toxicity (82). qox is itself heme dependent and incorporates heme A generated by the action of ctaB (56). Although inactivating qoxD mutations have not been previously investigated, it is expected that their functional effects will mimic loss of other qox subunits in that they all should destabilize the protein complex. It is known that loss of qoxB results in reduced growth under aerobic conditions, as well as organ-specific reduction of colonization in murine systemic-infection models (81, 83). Mutation of qoxD in our study occurred during selection for invasion; however, nonpolar deletion of the gene resulted in enhanced invasiveness and intracellular cytotoxicity. Given their attenuated phenotypes in vivo (56, 81, 83), it was somewhat unexpected that nonpolar deletion of ctaB and qoxD produced mutants with enhanced, rather than reduced, pathogenicity phenotypes. Our findings suggest that prior in vivo results (56, 81, 83) likely reflect decreased overall fitness and survival in the host and that mutations of these genes could nevertheless promote virulence in particular host microenvironments.
Lastly, the adhesin gene sdrC is thought to be involved in host colonization (84). The gene is upregulated during invasive S. aureus infection, and its disruption attenuates disease in vivo (84, 85). However, like qoxD and ebh, disruption of sdrC in our study paradoxically resulted in increased invasiveness and intracellular cytotoxicity, suggesting again that in vivo attenuation results from a secondary effect unrelated to these virulence traits.
Beyond these known virulence contributors, our study has newly implicated 12 genes whose mutation accompanies enhanced pathogenicity phenotypes. We have empirically determined that mutation of eight such genes carries a quantifiable contribution to defined virulence traits in S. aureus (Fig. 4 and 6):
The shikimate dehydrogenase gene aroE is considered a housekeeping gene in S. aureus (86). Several different pathogenic organisms with deficiencies in shikimic acid biosynthesis are known to be attenuated, likely reflecting auxotrophy (87, 88), and isogenic nonpolar deletion of aroE similarly decreased S. aureus invasiveness in our study. Nevertheless, complementation utilizing the aroE Thr182Ile mutant significantly increased invasiveness, indicating that the gene not only is required for full virulence in wild-type S. aureus, but can also act as a virulence-enhancing factor through gain-of-function mutation, possibly by increasing the availability of limiting metabolites (87, 88). Mutation of aroE was selected during passaging for intracellular cytotoxicity but affected only invasiveness, suggesting that it indirectly contributes to cytotoxicity by increasing the burden of intracellular bacteria.
ndhC encodes an NADH-menaquinone oxidoreductase (89, 90) that was found in our study to enhance invasion and decrease cytotoxicity when inactivated. Chemically reduced menaquinone increases the action of srrAB (91), with consequent reduction of virulence gene expression and other metabolic changes (91, 92). The augmented-virulence phenotypes observed with loss of ndhC may therefore be mediated indirectly through the amount of reduced menaquinone interacting with the known virulence factor genes srrAB.
The S-adenosylmethionine:2-DMK methyltransferase gene ubiE is responsible for catalyzing the final step of menaquinone biosynthesis (82), and nonpolar deletion of the gene in our study imparted increased invasiveness. Deletion of ubiE has previously been shown to impact neither colony growth rates nor resistance to heme toxicity resulting from superoxide production (82), making its contribution to virulence in this work unclear. We note that intermediates of menaquinone biosynthesis are required for intracellular pathogenesis of Listeria monocytogenes (93), and it is possible that ubiE mutation results in a buildup of these metabolites and thereby promotes intracellular pathogenesis of S. aureus.
A gain-of-function mutation in the GntR family transcriptional regulator gene yhcF increased strain invasiveness, potentially by affecting diverse transcriptional changes within S. aureus. yhcF is known to influence antibiotic resistance in Bacillus subtilis (94, 95), but its regulatory role in S. aureus has not yet been explored and appears to extend across other traits.
murA encodes one of two functionally redundant UDP-N-acetylglucosamine enolpyruvyl transferase genes present in Gram-positive organisms, which catalyze the initial step of cell wall biosynthesis (96). Its deletion has been shown to significantly reduce cell peptidoglycan content (96). In L. monocytogenes, murA is required for full virulence in vivo, while its loss has not been found to impact infection rates in cell culture models (97, 98). The role of murA in S. aureus pathogenesis appears to be somewhat different. Mutation of the gene was selected under conditions promoting cytotoxicity, and its nonpolar deletion elevates intracellular cytotoxicity while negatively impacting invasiveness in a cell culture model. The mechanism by which this effect occurs is presently unclear.
The elongation factor tufA is considered a housekeeping gene, but screens of transposon libraries have previously shown that the gene is necessary for maximal host cell invasion in Pseudomonas aeruginosa (99). In our work, selection for invasion recovered a point mutation in tufA (Ala271Val) that, when engineered in an isogenic strain, reduced invasiveness. In exploring possible epistasis effects, we found that tufA Ala271Val was able to enhance invasiveness in a background where purR had been inactivated (Fig. 6). The functional mechanism of this mutation and its relationship to purine metabolism are unclear.
Excitingly, two genes uncovered by our study that were functionally validated have no previously defined functions. We have arbitrarily designated these genes unnamedA and unnamedB. The unnamedA gene encodes a hypothetical peptidase that increased invasiveness when carrying an Asp486Glu gain-of-function mutation. The unnamedB gene, encoding a hypothetical membrane protein, was subjected to nonsense mutations during selection for invasion and increased both invasiveness and cytotoxicity phenotypes when constructed as an isogenic nonpolar deletion. By BLAST (100) search, both the unnamedA and unnamedB genes can be identified in a large number of S. aureus genomes, suggesting that they are broadly distributed across S. aureus lineages. These genes are worthy of further characterization in future studies.
Six other mutated genes (ypfH, srrA, menF, brnQ2, mnhA, and nupC) did not measurably increase the expected pathogenesis phenotype when produced as isogenic mutants or in complementation experiments using specific missense mutations (where such testing was appropriate). This does not discount mutations in these genes as contributors to virulence. Indeed, several encode known virulence factors (Table 1), and our data show that their intact function is necessary for full virulence in S. aureus (Fig. 4). The failure of these genes to be validated in our experiments may indicate dependence on concordant cellular signaling events or genetic epistasis (65), as we have demonstrated for the tufA Ala271Val mutation (Fig. 6). Dependence on accessory factors is also supported by the identification of gene mutations that specifically enhanced a virulence effect for which those mutations had not been selected (Fig. 4). In light of these observations, the interrelatedness of virulence-modifying genes and mutations may prove to be extensive and their contributions to pathogenesis to be multifactorial (65). Alternatively, it has been reported that gentamicin is able to undergo limited diffusion across eukaryotic membranes under certain conditions (101, 102), and it is possible that the action of the antibiotic on internalized bacteria could have impacted our results by selecting for increased resistance or by inducing SCV formation (103). We view this possibility as less likely, however, given both the limited temporal exposure of cell culture models to antibiotics and the finding that most individual genes implicated by our study could be experimentally validated as virulence factors conferring relevant phenotypes.
In summary, our study utilized artificial selection as a means to explore genetic mechanisms underlying defined virulence activities relevant to adaptation during infection of the host. We found that even an innocuous laboratory S. aureus strain had the potential to greatly enhance relative virulence activity through the accumulation of spontaneous chromosomal mutations. Analysis of these mutations identified known and previously undescribed genes that contribute to pathogenic phenotypes in S. aureus, with effector mutations spanning both loss-of-function and gain-of-function effects. In addition to enhancing understanding of the mechanisms by which these mutations exert an impact on virulence, future work will seek to further define epistatic interactions that may govern complex virulence phenotypes in S. aureus.
MATERIALS AND METHODS
Bacterial strains and plasmids.
S. aureus strain ATCC 29213 was from the American Type Culture Collection. Strain JE2 and JE2 transposon mutants (menD, NR-47887; ndhC, NR-48426; sdrC, NR-46975; and ebh, NR-46544) and plasmids pCN50 (104) and pCN33 (104) were from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). Plasmids for conditional recombineering and CRISPR/Cas9 counterselection have been previously described, as have nonpolar deletion mutants of aroE, saeS (i.e., baeS), qoxD (i.e., cyoD), nupC, purR, ypfH, and yhcF in ATCC 29213, created using that system (63).
Artificial selection for virulence phenotypes.
For both selections, bacteria were expanded overnight in sup-MH broth (48) and washed twice in Dulbecco's phosphate-buffered saline (DPBS) (Difco) to remove soluble toxins, and cell density was assessed by optical density at 600 nm (OD600). Six replicates were independently passaged in parallel for each of the two selections. Selections were performed at 37°C with 5% atmospheric CO2.
Selection for invasiveness was based on previously described gentamicin protection assays (18, 46). Briefly, bacteria were combined with cultured HeLa cells at a multiplicity of infection (MOI) of 100 in serum-free minimal essential medium (MEM) without glucose (Difco) for 1 h. The medium was then replaced with MEM containing 100 μg/ml gentamicin (Gibco) and 10 μg/ml lysostaphin (Sigma) for 30 min, after which MEM containing 100 μg/ml gentamicin alone was applied for 3 h. The cells were then washed with DPBS and lysed with water containing 0.025% Triton X-100 (Sigma), and the lysate was transferred to sup-MH for overnight expansion prior to the next passage.
Selection for cytotoxicity followed the invasiveness selection protocol with several modifications. After removal of gentamicin/lysostaphin medium, the medium was replaced with serum-free MEM without glucose, and the cells were incubated for an additional 24 h (for an MOI of 10) to 48 h (for an MOI of 100). After incubation, the culture supernatant was collected, washed in DPBS, and inoculated into sup-MH for overnight expansion prior to the next passage. As the bacteria became more cytotoxic with increased passaging, it was necessary to reduce the MOI over time to prevent all the host cells from being lysed during infection while maintaining selective pressure for mutants with increased cytotoxic capability. Consequently, for the first 10 passages, an MOI of 100 was used, after which the MOI was reduced to 10.
Real-time PCR.
RNA was extracted from mid-log-phase cultures using the Qiagen RNeasy Protect Bacteria Mini kit protocols 4 and 7 with the substitution of 100 μg/ml lysostaphin instead of lysozyme. RNA was then converted to cDNA using the Promega ImProm II reverse transcription system. Primers for amplification of the 16S rRNA gene (105), agrA (106), and sigB (106) have been previously described (see Table S8 in the supplemental material). All real-time PCRs were performed on a ViiA7 system (Thermo Fisher) at least in triplicate, with specimens internally normalized to 16S rRNA expression. Relative quantitation of each factor was determined with respect to the parental strain.
Genome sequencing and analysis.
Whole-genome sequencing of evolved isolates (named “Cyto” for cytotoxicity selections and “Intra” for invasiveness selections) was performed as described previously (107). Sequence reads were mapped to the S. aureus strain 502 A reference genome (GenBank accession no. CP007454.1), which we determined to be the reference genome most closely matched to our parental strain, as before (107). Variant calling was performed as described previously (108). Common gene names were established by performing BLAST-P (100) searches restricted to S. aureus sequences in order to distinguish named genes from other strains having identical protein sequences.
Measurements of isolate virulence phenotypes and growth rates.
Quantitative invasiveness assays were performed following the protocols used for selection, with the exception that serial dilutions of the initial cell inoculum and the cell lysate were plated onto sup-MH medium to quantitate the fraction of successfully internalized cells. Invasiveness was calculated as the ratio of internalized bacteria to total inoculated bacteria.
Quantitative cytotoxicity assays were also performed following the protocols employed for selection, except that gentamicin/lysostaphin medium was not removed in order to kill any bacterial cells released into the medium and to therefore prevent reinfection. After 24 h of incubation, the number of viable cells was determined using a Cell Counting Kit-8 (Dojindo Molecular Technologies). Intracellular cytotoxicity was expressed as the fraction of lysed cells relative to that measured for the parental strain.
To account for variability inherent in cell culture invasion assays (109), all quantitative virulence measurements were performed at least in triplicate and normalized to paired controls run at the same time utilizing the parental strain.
Growth rates were monitored by absorbance at OD600 during incubation at 37°C in brain heart infusion (BHI) broth (Difco) (52), with experiments performed at least in triplicate.
Qualitative pigmentation and blood agar growth phenotypes were assessed by spotting 1-μl overnight cultures diluted to ∼1.3 × 106 CFU/ml onto BBL TSA II 5% sheep blood agar (Becton, Dickinson).
Construction of isogenic mutant strains.
Isogenic single and double mutants were constructed using conditional recombineering and CRISPR/Cas9-mediated counterselection, as previously described (63). Mutagenic recombineering and counterselection oligonucleotides are listed in Table S8 and were synthesized by IDT. All strains were cured of genomic-engineering plasmids prior to use (63).
Complementation experiments.
The cloning primers (IDT, Coralville, IA) are summarized in Table S8. The transcriptional terminator of pCN50 was amplified and cloned into pCN33, which can be maintained stably in the absence of antibiotic selection (104), to make the vector pCN33TT. Wild-type and mutant genes were subsequently amplified and cloned into this vector. The entire sae operon (110) was cloned in order to evaluate saeS mutant function, but for other genes, ∼1 kb of upstream flanking sequence was included to capture the native regulatory elements. Complementation experiments were performed in ATCC 29213 mutants carrying the appropriate single-gene deletion. Vectors were introduced into recipient S. aureus strains by electroporation as previously described (63).
Data availability.
Sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA) under accession no. SRP157935.
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00884-18.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Wertheim HFL, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JAJW, van Keulen PHJ, Vandenbroucke-Grauls CMJE, Meester MHM, Verbrugh HA. 2004. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet Lond Engl 364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak JM, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis 52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 4.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 5.Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, Fridkin S, Emerging Infections Program—Active Bacterial Core Surveillance MRSA Surveillance Investigators. 2013. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern Med 173:1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gould IM. 2007. MRSA bacteraemia. Int J Antimicrob Agents 30:S66–S70. doi: 10.1016/j.ijantimicag.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 8.Conlon BP. 2014. Staphylococcus aureus chronic and relapsing infections: evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. BioEssays 36:991–996. doi: 10.1002/bies.201400080. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed MI, Mukherjee S. 2016. Treatment for chronic methicillin-sensitive Staphylococcus aureus pulmonary infection in people with cystic fibrosis. Cochrane Database Syst Rev 3:CD011581. doi: 10.1002/14651858.CD011581.pub2. [DOI] [PubMed] [Google Scholar]
- 10.McAdam PR, Holmes A, Templeton KE, Fitzgerald JR. 2011. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS One 6:e24301. doi: 10.1371/journal.pone.0024301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah PL, Mawdsley S, Nash K, Cullinan P, Cole PJ, Wilson R. 1999. Determinants of chronic infection with Staphylococcus aureus in patients with bronchiectasis. Eur Respir J 14:1340–1344. doi: 10.1183/09031936.99.14613409. [DOI] [PubMed] [Google Scholar]
- 12.Dastgheyb SS, Otto M. 2015. Staphylococcal adaptation to diverse physiologic niches: an overview of transcriptomic and phenotypic changes in different biological environments. Future Microbiol 10:1981–1995. doi: 10.2217/fmb.15.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraunholz M, Sinha B. 2012. Intracellular Staphylococcus aureus: live-in and let die. Front Cell Infect Microbiol 2:43. doi: 10.3389/fcimb.2012.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowy FD. 2000. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol 8:341–343. doi: 10.1016/S0966-842X(00)01803-5. [DOI] [PubMed] [Google Scholar]
- 15.Garzoni C, Kelley WL. 2009. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol 17:59–65. doi: 10.1016/j.tim.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald JR. 2014. Evolution of Staphylococcus aureus during human colonization and infection. Infect Genet Evol 21:542–547. doi: 10.1016/j.meegid.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 17.Recker M, Laabei M, Toleman MS, Reuter S, Saunderson RB, Blane B, Török ME, Ouadi K, Stevens E, Yokoyama M, Steventon J, Thompson L, Milne G, Bayliss S, Bacon L, Peacock SJ, Massey RC. 2017. Clonal differences in Staphylococcus aureus bacteraemia-associated mortality. Nat Microbiol 2:1381–1388. doi: 10.1038/s41564-017-0001-x. [DOI] [PubMed] [Google Scholar]
- 18.Krut O, Utermöhlen O, Schlossherr X, Krönke M. 2003. Strain-specific association of cytotoxic activity and virulence of clinical Staphylococcus aureus isolates. Infect Immun 71:2716–2723. doi: 10.1128/IAI.71.5.2716-2723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spaulding AR, Satterwhite EA, Lin Y-C, Chuang-Smith ON, Frank KL, Merriman JA, Schaefers MM, Yarwood JM, Peterson ML, Schlievert PM. 2012. Comparison of Staphylococcus aureus strains for ability to cause infective endocarditis and lethal sepsis in rabbits. Front Cell Infect Microbiol 2:18. doi: 10.3389/fcimb.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuchscherr L, Bischoff M, Lattar SM, Noto Llana M, Pförtner H, Niemann S, Geraci J, Van de Vyver H, Fraunholz MJ, Cheung AL, Herrmann M, Völker U, Sordelli DO, Peters G, Löffler B. 2015. Sigma factor SigB is crucial to mediate Staphylococcus aureus adaptation during chronic infections. PLoS Pathog 11:e1004870. doi: 10.1371/journal.ppat.1004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suligoy CM, Lattar SM, Noto Llana M, González CD, Alvarez LP, Robinson DA, Gómez MI, Buzzola FR, Sordelli DO. 2018. Mutation of Agr is associated with the adaptation of Staphylococcus aureus o the host during chronic osteomyelitis. Front Cell Infect Microbiol 8:18. doi: 10.3389/fcimb.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuchscherr L, Heitmann V, Hussain M, Viemann D, Roth J, von Eiff C, Peters G, Becker K, Löffler B. 2010. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J Infect Dis 202:1031–1040. doi: 10.1086/656047. [DOI] [PubMed] [Google Scholar]
- 23.Tuchscherr L, Medina E, Hussain M, Völker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Löffler B. 2011. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goerke C, Wolz C. 2004. Regulatory and genomic plasticity of Staphylococcus aureus during persistent colonization and infection. Int J Med Microbiol 294:195–202. doi: 10.1016/j.ijmm.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Goerke C, Wolz C. 2010. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int J Med Microbiol 300:520–525. doi: 10.1016/j.ijmm.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 26.Cullen L, McClean S. 2015. Bacterial adaptation during chronic respiratory infections. Pathogens 4:66–89. doi: 10.3390/pathogens4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windmüller N, Witten A, Block D, Bunk B, Spröer C, Kahl BC, Mellmann A. 2015. Transcriptional adaptations during long-term persistence of Staphylococcus aureus in the airways of a cystic fibrosis patient. Int J Med Microbiol 305:38–46. doi: 10.1016/j.ijmm.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A 101:12312–12317. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Chen J, Chen G, Du X, Cui P, Wu J, Zhao J, Wu N, Zhang W, Li M, Zhang Y. 2015. Transposon mutagenesis identifies novel genes associated with Staphylococcus aureus persister formation. Front Microbiol 6:1437. doi: 10.3389/fmicb.2015.01437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mei JM, Nourbakhsh F, Ford CW, Holden DW. 1997. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature-tagged mutagenesis. Mol Microbiol 26:399–407. doi: 10.1046/j.1365-2958.1997.5911966.x. [DOI] [PubMed] [Google Scholar]
- 31.Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, Buysse JM. 2004. Large-scale identification of genes required for full virulence of Staphylococcus aureus. J Bacteriol 186:8478–8489. doi: 10.1128/JB.186.24.8478-8489.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg DE, Weiss A, Crossland L. 1980. Polarity of Tn5 insertion mutations in Escherichia coli. J Bacteriol 142:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dean MA, Olsen RJ, Long SW, Rosato AE, Musser JM. 2014. Identification of point mutations in clinical Staphylococcus aureus strains that produce small-colony variants auxotrophic for menadione. Infect Immun 82:1600–1605. doi: 10.1128/IAI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young BC, Golubchik T, Batty EM, Fung R, Larner-Svensson H, Votintseva AA, Miller RR, Godwin H, Knox K, Everitt RG, Iqbal Z, Rimmer AJ, Cule M, Ip CLC, Didelot X, Harding RM, Donnelly P, Peto TE, Crook DW, Bowden R, Wilson DJ. 2012. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc Natl Acad Sci U S A 109:4550–4555. doi: 10.1073/pnas.1113219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benoit JB, Frank DN, Bessesen MT. 2018. Genomic evolution of Staphylococcus aureus isolates colonizing the nares and progressing to bacteremia. PLoS One 13:e0195860. doi: 10.1371/journal.pone.0195860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuchscherr L, Löffler B. 2015. Staphylococcus aureus dynamically adapts global regulators and virulence factor expression in the course from acute to chronic infection. Curr Genet doi: 10.1007/s00294-015-0503-0. [DOI] [PubMed] [Google Scholar]
- 37.Sinha B, Francois P, Que YA, Hussain M, Heilmann C, Moreillon P, Lew D, Krause KH, Peters G, Herrmann M. 2000. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect Immun 68:6871–6878. doi: 10.1128/IAI.68.12.6871-6878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha B, François PP, Nüsse O, Foti M, Hartford OM, Vaudaux P, Foster TJ, Lew DP, Herrmann M, Krause KH. 1999. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol 1:101–117. doi: 10.1046/j.1462-5822.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 39.Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol 12:1746–1764. doi: 10.1111/j.1462-5822.2010.01506.x. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen MT, Kraft B, Yu W, Demircioglu DD, Demicrioglu DD, Hertlein T, Burian M, Schmaler M, Boller K, Bekeredjian-Ding I, Ohlsen K, Schittek B, Götz F. 2015. The νSaα specific lipoprotein like cluster (lpl) of S. aureus USA300 contributes to immune stimulation and invasion in human cells. PLoS Pathog 11:e1004984. doi: 10.1371/journal.ppat.1004984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen M-T, Deplanche M, Nega M, Le Loir Y, Peisl L, Götz F, Berkova N. 2016. Staphylococcus aureus Lpl lipoproteins delay G2/M phase transition in HeLa cells. Front Cell Infect Microbiol 6:201. doi: 10.3389/fcimb.2016.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauthe M, Yu W, Krut O, Krönke M, Götz F, Robenek H, Proikas-Cezanne T. 2012. WIPI-1 positive autophagosome-like vesicles entrap pathogenic Staphylococcus aureus for lysosomal degradation. Int J Cell Biol 2012:179207. doi: 10.1155/2012/179207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dinges MM, Orwin PM, Schlievert PM. 2000. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev 13:16–34. doi: 10.1128/CMR.13.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seidl K, Leemann M, Zinkernagel AS. 2018. The ArlRS two-component system is a regulator of Staphylococcus aureus-induced endothelial cell damage. Eur J Clin Microbiol Infect Dis 37:289–292. doi: 10.1007/s10096-017-3130-5. [DOI] [PubMed] [Google Scholar]
- 45.Das S, Lindemann C, Young BC, Muller J, Österreich B, Ternette N, Winkler A-C, Paprotka K, Reinhardt R, Förstner KU, Allen E, Flaxman A, Yamaguchi Y, Rollier CS, van Diemen P, Blättner S, Remmele CW, Selle M, Dittrich M, Müller T, Vogel J, Ohlsen K, Crook DW, Massey R, Wilson DJ, Rudel T, Wyllie DH, Fraunholz MJ. 2016. Natural mutations in a Staphylococcus aureus virulence regulator attenuate cytotoxicity but permit bacteremia and abscess formation. Proc Natl Acad Sci U S A 113:E3101–E3110. doi: 10.1073/pnas.1520255113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheung AL, Bayles KW. 2007. Tissue culture assays used to analyze invasion by Staphylococcus aureus. Curr Protoc Microbiol Chapter 9:Unit 9C.4. [DOI] [PubMed] [Google Scholar]
- 47.Lone AG, Atci E, Renslow R, Beyenal H, Noh S, Fransson B, Abu-Lail N, Park J-J, Gang DR, Call DR. 2015. Staphylococcus aureus induces hypoxia and cellular damage in porcine dermal explants. Infect Immun 83:2531–2541. doi: 10.1128/IAI.03075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Precit MR, Wolter DJ, Griffith A, Emerson J, Burns JL, Hoffman LR. 2016. optimized in vitro antibiotic susceptibility testing method for small-colony variant Staphylococcus aureus. Antimicrob Agents Chemother 60:1725–1735. doi: 10.1128/AAC.02330-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 50.Proctor RA, Balwit JM, Vesga O. 1994. Variant subpopulations of Staphylococcus aureus as cause of persistent and recurrent infections. Infect Agents Dis 3:302–312. [PubMed] [Google Scholar]
- 51.Melter O, Radojevič B. 2010. Small colony variants of Staphylococcus aureus—review. Folia Microbiol 55:548–558. doi: 10.1007/s12223-010-0089-3. [DOI] [PubMed] [Google Scholar]
- 52.Kahl BC, Belling G, Becker P, Chatterjee I, Wardecki K, Hilgert K, Cheung AL, Peters G, Herrmann M. 2005. Thymidine-dependent Staphylococcus aureus small-colony variants are associated with extensive alterations in regulator and virulence gene expression profiles. Infect Immun 73:4119–4126. doi: 10.1128/IAI.73.7.4119-4126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clauditz A, Resch A, Wieland K-P, Peschel A, Götz F. 2006. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect Immun 74:4950–4953. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelz A, Wieland K-P, Putzbach K, Hentschel P, Albert K, Götz F. 2005. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem 280:32493–32498. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 55.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. 2005. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu T, Han J, Zhang J, Chen J, Wu N, Zhang W, Zhang Y. 2016. Absence of protoheme IX farnesyltransferase CtaB causes virulence attenuation but enhances pigment production and persister survival in MRSA. Front Microbiol 7:1625. doi: 10.3389/fmicb.2016.01625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell G, Fugère A, Pépin Gaudreau K, Brouillette E, Frost EH, Cantin AM, Malouin F. 2013. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS One 8:e65018. doi: 10.1371/journal.pone.0065018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong C, Neoh H, Nathan S. 2016. Targeting Staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins 8:E72. doi: 10.3390/toxins8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenkman B, Rubinstein E, Cheung AL, Brill GE, Dardik R, Tamarin I, Savion N, Varon D. 2001. Adherence properties of Staphylococcus aureus under static and flow conditions: roles of agr and sar loci, platelets, and plasma ligands. Infect Immun 69:4473–4478. doi: 10.1128/IAI.69.7.4473-4478.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bischoff M, Entenza JM, Giachino P. 2001. Influence of a functional sigB operon on the global regulators Sar and Agr in Staphylococcus aureus. J Bacteriol 183:5171–5179. doi: 10.1128/JB.183.17.5171-5179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wesson CA, Liou LE, Todd KM, Bohach GA, Trumble WR, Bayles KW. 1998. Staphylococcus aureus Agr and Sar global regulators influence internalization and induction of apoptosis. Infect Immun 66:5238–5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koenig RL, Ray JL, Maleki SJ, Smeltzer MS, Hurlburt BK. 2004. Staphylococcus aureus AgrA binding to the RNAIII-agr regulatory region. J Bacteriol 186:7549–7555. doi: 10.1128/JB.186.22.7549-7555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Penewit K, Holmes EA, McLean K, Ren M, Waalkes A, Salipante SJ. 2018. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 9:e00067-18. doi: 10.1128/mBio.00067-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laabei M, Recker M, Rudkin JK, Aldeljawi M, Gulay Z, Sloan TJ, Williams P, Endres JL, Bayles KW, Fey PD, Yajjala VK, Widhelm T, Hawkins E, Lewis K, Parfett S, Scowen L, Peacock SJ, Holden M, Wilson D, Read TD, van den Elsen J, Priest NK, Feil EJ, Hurst LD, Josefsson E, Massey RC. 2014. Predicting the virulence of MRSA from its genome sequence. Genome Res 24:839–849. doi: 10.1101/gr.165415.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garcia LG, Lemaire S, Kahl BC, Becker K, Proctor RA, Denis O, Tulkens PM, Van Bambeke F. 2013. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J Antimicrob Chemother 68:1455–1464. doi: 10.1093/jac/dkt072. [DOI] [PubMed] [Google Scholar]
- 67.Proctor RA, Kahl B, von Eiff C, Vaudaux PE, Lew DP, Peters G. 1998. Staphylococcal small colony variants have novel mechanisms for antibiotic resistance. Clin Infect Dis 27:S68–S74. doi: 10.1086/514906. [DOI] [PubMed] [Google Scholar]
- 68.Connolly J, Boldock E, Prince LR, Renshaw SA, Whyte MK, Foster SJ. 2017. Identification of Staphylococcus aureus factors required for pathogenicity and growth in human blood. Infect Immun 85:e00337-17. doi: 10.1128/IAI.00337-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jenkins A, Cote C, Twenhafel N, Merkel T, Bozue J, Welkos S. 2011. Role of purine biosynthesis in Bacillus anthracis pathogenesis and virulence. Infect Immun 79:153–166. doi: 10.1128/IAI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alcantara RB, Read RDA, Valderas MW, Brown TD, Roop RM. 2004. Intact purine biosynthesis pathways are required for wild-type virulence of Brucella abortus 2308 in the BALB/c mouse model. Infect Immun 72:4911–4917. doi: 10.1128/IAI.72.8.4911-4917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cersini A, Martino MC, Martini I, Rossi G, Bernardini ML. 2003. Analysis of virulence and inflammatory potential of Shigella flexneri purine biosynthesis mutants. Infect Immun 71:7002–7013. doi: 10.1128/IAI.71.12.7002-7013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu Q, Yeo W-S, Bae T. 2016. The SaeRS two-component system of Staphylococcus aureus. Genes 7:E81. [Google Scholar]
- 73.Adhikari RP, Novick RP. 2008. Regulatory organization of the staphylococcal sae locus. Microbiology 154:949–959. doi: 10.1099/mic.0.2007/012245-0. [DOI] [PubMed] [Google Scholar]
- 74.Schäfer D, Lâm T-T, Geiger T, Mainiero M, Engelmann S, Hussain M, Bosserhoff A, Frosch M, Bischoff M, Wolz C, Reidl J, Sinha B. 2009. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J Bacteriol 191:7306–7314. doi: 10.1128/JB.00630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matamouros S, Hager KR, Miller SI. 2015. HAMP domain rotation and tilting movements associated with signal transduction in the PhoQ sensor kinase. mBio 6:e00616–15. doi: 10.1128/mBio.00616-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bates DM, von Eiff C, McNamara PJ, Peters G, Yeaman MR, Bayer AS, Proctor RA. 2003. Staphylococcus aureus menD and hemB mutants are as infective as the parent strains, but the menadione biosynthetic mutant persists within the kidney. J Infect Dis 187:1654–1661. doi: 10.1086/374642. [DOI] [PubMed] [Google Scholar]
- 77.Sifri CD, Baresch-Bernal A, Calderwood SB, von Eiff C. 2006. Virulence of Staphylococcus aureus small colony variants in the Caenorhabditis elegans infection model. Infect Immun 74:1091–1096. doi: 10.1128/IAI.74.2.1091-1096.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng AG, Missiakas D, Schneewind O. 2014. The giant protein Ebh is a determinant of Staphylococcus aureus cell size and complement resistance. J Bacteriol 196:971–981. doi: 10.1128/JB.01366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bui LMG, Hoffmann P, Turnidge JD, Zilm PS, Kidd SP. 2015. Prolonged growth of a clinical Staphylococcus aureus strain selects for a stable small-colony-variant cell type. Infect Immun 83:470–481. doi: 10.1128/IAI.02702-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.von Eiff C, Heilmann C, Proctor RA, Woltz C, Peters G, Götz F. 1997. A site-directed Staphylococcus aureus hemB mutant is a small-colony variant which persists intracellularly. J Bacteriol 179:4706–4712. doi: 10.1128/jb.179.15.4706-4712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hammer ND, Reniere ML, Cassat JE, Zhang Y, Hirsch AO, Indriati Hood M, Skaar EP. 2013. Two heme-dependent terminal oxidases power Staphylococcus aureus organ-specific colonization of the vertebrate host. mBio 4:e00241-13. doi: 10.1128/mBio.00241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wakeman CA, Hammer ND, Stauff DL, Attia AS, Anzaldi LL, Dikalov SI, Calcutt MW, Skaar EP. 2012. Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus. Mol Microbiol 86:1376–1392. doi: 10.1111/mmi.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lan L, Cheng A, Dunman PM, Missiakas D, He C. 2010. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J Bacteriol 192:3068–3077. doi: 10.1128/JB.00928-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, Stover CK, Sellman BR. 2015. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. mBio 6:e02272-14. doi: 10.1128/mBio.02272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miyazaki S, Matsumoto Y, Sekimizu K, Kaito C. 2012. Evaluation of Staphylococcus aureus virulence factors using a silkworm model. FEMS Microbiol Lett 326:116–124. doi: 10.1111/j.1574-6968.2011.02439.x. [DOI] [PubMed] [Google Scholar]
- 86.Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Izhar M, DeSilva L, Joysey HS, Hormaeche CE. 1990. Moderate immunodeficiency does not increase susceptibility to Salmonella typhimurium aroA live vaccines in mice. Infect Immun 58:2258–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stritzker J, Janda J, Schoen C, Taupp M, Pilgrim S, Gentschev I, Schreier P, Geginat G, Goebel W. 2004. Growth, virulence, and immunogenicity of Listeria monocytogenes aro mutants. Infect Immun 72:5622–5629. doi: 10.1128/IAI.72.10.5622-5629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schurig-Briccio LA, Yano T, Rubin H, Gennis RB. 2014. Characterization of the type 2 NADH:menaquinone oxidoreductases from Staphylococcus aureus and the bactericidal action of phenothiazines. Biochim Biophys Acta 1837:954–963. doi: 10.1016/j.bbabio.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mayer S, Steffen W, Steuber J, Götz F. 2015. The Staphylococcus aureus NuoL-like protein MpsA contributes to the generation of membrane potential. J Bacteriol 197:794–806. doi: 10.1128/JB.02127-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mashruwala AA, A van de G, Boyd JM. 2017. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. Elife 6:e23845. doi: 10.7554/eLife.23845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kinkel TL, Roux CM, Dunman PM, Fang FC. 2013. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. mBio 4:e00696-13. doi: 10.1128/mBio.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen GY, McDougal CE, D’Antonio MA, Portman JL, Sauer J-D. 2017. A genetic screen reveals that synthesis of 1,4-dihydroxy-2-naphthoate (DHNA), but not full-length menaquinone, is required for Listeria monocytogenes cytosolic survival. mBio 8:e00119-17. doi: 10.1128/mBio.00119-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leyn SA, Kazanov MD, Sernova NV, Ermakova EO, Novichkov PS, Rodionov DA. 2013. Genomic reconstruction of the transcriptional regulatory network in Bacillus subtilis. J Bacteriol 195:2463–2473. doi: 10.1128/JB.00140-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Serizawa M, Sekizuka T, Okutani A, Banno S, Sata T, Inoue S, Kuroda M. 2010. Genomewide screening for novel genetic variations associated with ciprofloxacin resistance in Bacillus anthracis. Antimicrob Agents Chemother 54:2787–2792. doi: 10.1128/AAC.01405-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blake KL, O'Neill AJ, Mengin-Lecreulx D, Henderson PJF, Bostock JM, Dunsmore CJ, Simmons KJ, Fishwick CWG, Leeds JA, Chopra I. 2009. The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol Microbiol 72:335–343. doi: 10.1111/j.1365-2958.2009.06648.x. [DOI] [PubMed] [Google Scholar]
- 97.Machata S, Hain T, Rohde M, Chakraborty T. 2005. Simultaneous deficiency of both MurA and p60 proteins generates a rough phenotype in Listeria monocytogenes. J Bacteriol 187:8385–8394. doi: 10.1128/JB.187.24.8385-8394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lenz LL, Mohammadi S, Geissler A, Portnoy DA. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci U S A 100:12432–12437. doi: 10.1073/pnas.2133653100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dubern J-F, Cigana C, De Simone M, Lazenby J, Juhas M, Schwager S, Bianconi I, Döring G, Eberl L, Williams P, Bragonzi A, Cámara M. 2015. Integrated whole-genome screening for Pseudomonas aeruginosa virulence genes using multiple disease models reveals that pathogenicity is host specific. Environ Microbiol 17:4379–4393. doi: 10.1111/1462-2920.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 101.VanCleave TT, Pulsifer AR, Connor MG, Warawa JM, Lawrenz MB. 2017. Impact of gentamicin concentration and exposure time on intracellular Yersinia pestis. Front Cell Infect Microbiol 7:505. doi: 10.3389/fcimb.2017.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eze MO, Yuan L, Crawford RM, Paranavitana CM, Hadfield TL, Bhattacharjee AK, Warren RL, Hoover DL. 2000. Effects of opsonization and gamma interferon on growth of Brucella melitensis 16M in mouse peritoneal macrophages in vitro. Infect Immun 68:257–263. doi: 10.1128/IAI.68.1.257-263.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tuchscherr L, Kreis CA, Hoerr V, Flint L, Hachmeister M, Geraci J, Bremer-Streck S, Kiehntopf M, Medina E, Kribus M, Raschke M, Pletz M, Peters G, Löffler B. 2016. Staphylococcus aureus develops increased resistance to antibiotics by forming dynamic small colony variants during chronic osteomyelitis. J Antimicrob Chemother 71:438–448. doi: 10.1093/jac/dkv371. [DOI] [PubMed] [Google Scholar]
- 104.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tiwari KB, Gatto C, Wilkinson BJ. 2018. Interrelationships between fatty acid composition, staphyloxanthin content, fluidity, and carbon flow in the Staphylococcus aureus membrane. Molecules 23:E1201. doi: 10.3390/molecules23051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee J-H, Cho HS, Kim Y, Kim J-A, Banskota S, Cho MH, Lee J. 2013. Indole and 7-benzyloxyindole attenuate the virulence of Staphylococcus aureus. Appl Microbiol Biotechnol 97:4543–4552. doi: 10.1007/s00253-012-4674-z. [DOI] [PubMed] [Google Scholar]
- 107.Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, Shendure J, Salipante SJ. 2015. A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet 11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol 53:1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Elsinghorst EA. 1994. Measurement of invasion by gentamicin resistance. Methods Enzymol 236:405–420. doi: 10.1016/0076-6879(94)36030-8. [DOI] [PubMed] [Google Scholar]
- 110.Steinhuber A, Goerke C, Bayer MG, Döring G, Wolz C. 2003. Molecular architecture of the regulatory locus sae of Staphylococcus aureus and its impact on expression of virulence factors. J Bacteriol 185:6278–6286. doi: 10.1128/JB.185.21.6278-6286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vaish M, Price-Whelan A, Reyes-Robles T, Liu J, Jereen A, Christie S, Alonzo F, Benson MA, Torres VJ, Krulwich TA. 2018. Roles of Staphylococcus aureus Mnh1 and Mnh2 antiporters in salt tolerance, alkali tolerance, and pathogenesis. J Bacteriol 200:e00611-17. doi: 10.1128/JB.00611-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaiser JC, Omer S, Sheldon JR, Welch I, Heinrichs DE. 2015. Role of BrnQ1 and BrnQ2 in branched-chain amino acid transport and virulence in Staphylococcus aureus. Infect Immun 83:1019–1029. doi: 10.1128/IAI.02542-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA) under accession no. SRP157935.