

The enteric bacterium and intracellular human pathogen Shigella causes hundreds of millions of cases of the diarrheal disease shigellosis per year worldwide. Shigella is acquired by ingestion of contaminated food or water; upon reaching the colon, the bacteria invade colonic epithelial cells, replicate intracellularly, spread to adjacent cells, and provoke an intense inflammatory response.
KEYWORDS: human intestinal enteroid, Shigella flexneri, host-pathogen interactions, intracellular pathogen
ABSTRACT
The enteric bacterium and intracellular human pathogen Shigella causes hundreds of millions of cases of the diarrheal disease shigellosis per year worldwide. Shigella is acquired by ingestion of contaminated food or water; upon reaching the colon, the bacteria invade colonic epithelial cells, replicate intracellularly, spread to adjacent cells, and provoke an intense inflammatory response. There is no animal model that faithfully recapitulates human disease; thus, cultured cells have been used to model Shigella pathogenesis. However, the use of transformed cells in culture does not provide the same environment to the bacteria as the normal human intestinal epithelium. Recent advances in tissue culture now enable the cultivation of human intestinal enteroids (HIEs), which are derived from human intestinal stem cells, grown ex vivo, and then differentiated into “mini-intestines.” Here, we demonstrate that HIEs can be used to model Shigella pathogenesis. We show that Shigella flexneri invades polarized HIE monolayers preferentially via the basolateral surface. After S. flexneri invades HIE monolayers, S. flexneri replicates within HIE cells and forms actin tails. S. flexneri also increases the expression of HIE proinflammatory signals and the amino acid transporter SLC7A5. Finally, we demonstrate that disruption of HIE tight junctions enables S. flexneri invasion via the apical surface.
INTRODUCTION
Shigellosis is an acute human diarrheal disease caused by the Gram-negative bacterium Shigella and is associated with significant morbidity and mortality worldwide, particularly among children in developing nations (1, 2). Shigella is an intracellular pathogen that replicates within the cytosol of human colon epithelial cells. Studies using rabbit ligated ileal loops indicate that Shigella does not invade colon cells via the apical face of the epithelium but rather exploits microfold (M) cells to transcytose the bacteria from the colon lumen to the basolateral face of the epithelium (3). Here, Shigella uses a type III secretion system (T3SS) to induce epithelial cell uptake (4–6), and once inside the cell, Shigella escapes from the engulfment vacuole and replicates in the host cell cytosol (7). Shigella then exploits host actin polymerization using the bacterial protein IcsA to recruit host N-WASP, catalyzing actin polymerization and propelling the bacterium into adjacent cells (8–10).
The incidence of drug-resistant Shigella strains is rising, and there is currently no vaccine for the treatment or prevention of Shigella infection (11–13). Complicating Shigella drug development, there is no small-animal model that completely and faithfully recapitulates human disease. Although intestinal infections of guinea pigs, rabbits, piglets, and zebrafish have revealed some aspects of Shigella interactions with the host (14–17), these model systems have distinct physiological differences from humans, and Shigella does not naturally infect these animals. While Shigella infection of nonhuman primates has been demonstrated, this model system is difficult and expensive to implement, and the infectious dose of Shigella in the nonhuman primate model is many orders of magnitude higher than that in humans, indicating Shigella human host specificity (18–23). Therefore, new models of Shigella infection are needed.
Cultured cells are routinely used to study Shigella pathogenesis, but these models are typically homogenous cell lines, such as HeLa (24), Henle-407 (25), or Caco-2 (26). While transformed lines have been and continue to be a fundamental tool to examine Shigella-host interactions, this model system diverges from a natural infection in several important ways. Transformed cell lines are subject to the Warburg effect, where cellular glycolysis is increased for energy production, leaving their metabolism fundamentally altered (27); this is particularly relevant during Shigella pathogenesis, as Shigella is a cytosolic intracellular pathogen that metabolizes host carbon during infection (28–30). Furthermore, transformed cell lines, such as HeLa, have altered cytosolic defense and immune response pathways (31). These differences make this model system less than ideal for studies involving metabolism or the immune response and limit the capacity of this model system for drug development. Human colon explants have also been used to characterize Shigella pathogenesis, but this model is not tractable or easily reproduced (32).
New advances in human tissue culture have led to the cultivation of human intestinal enteroids (HIEs), which are intestinal stem cells derived from human biopsy specimens and subsequently differentiated and grown ex vivo in tissue culture (33, 34). HIEs are composed of heterogeneous cell types and exhibit morphological characteristics of the human intestine, including polarization, mucus production, and three-dimensional (3D) structure, including crypt and villus formation, which is believed to mimic the natural intestinal environment (35). Importantly, HIEs can be expanded, passaged, cryogenically stored, and grown in monolayers, making this a sustainable and reproducible model system for gastrointestinal pathogenesis. Indeed, HIEs have been used to examine other gastrointestinal pathogens, such as Vibrio cholerae, enterohemorrhagic and enteroaggregative Escherichia coli, Salmonella, rotavirus, and others (33–39).
Here, we provide a technical report of proof-of-concept experiments that demonstrate that HIEs are a viable model system of Shigella pathogenesis. We show that Shigella flexneri invades HIEs preferentially via the basolateral face of polarized enteroid monolayers, replicates within enteroid cells, and forms actin tails. We believe that HIEs can be used in future studies to elucidate undiscovered aspects of Shigella pathogenesis that had not previously been described using established transformed cell lines, and HIEs offer the opportunity to study interactions of the bacteria with mucin, host immune cells, and innate immune responses.
RESULTS
S. flexneri apical and basolateral invasion of colonoid monolayers.
Previous studies indicate that S. flexneri cannot invade polarized epithelial cells grown in culture via the apical face but can access polarized cells with access to the basolateral face (26). We sought to determine if S. flexneri is capable of invading polarized human enteroid monolayers and whether S. flexneri invasion is specific to the basolateral surface. To this end, we cultivated from a single patient intestinal enteroid monolayers derived from the colon tissue (colonoids), where inflammatory lesions form in the gastrointestinal tract during human infection (40). Monolayers were grown on transwells with an 8-μm pore size, allowing ample colonoid cell access to S. flexneri when introduced to the basolateral surface. Enteroid monolayers were differentiated, and polarization was verified using transepithelial electrical resistance (TEER) measurements, as described previously (41, 42). S. flexneri was then introduced to either the apical or basolateral surfaces of the colonoid monolayers. As a control for invasion, we included an avirulent S. flexneri strain lacking the virulence plasmid (CFS100). After invasion, the monolayers were treated with gentamicin to eliminate any extracellular bacteria. At 3 h postinfection (p.i.), monolayers were lysed, and the intracellular bacteria were enumerated by plating.
When the bacteria were introduced to colonoid monolayers apically, the number of recovered bacteria was low, and we observed no significant difference in the recovery of S. flexneri wild-type (WT) and CFS100 strains. The few bacteria recovered likely represent adherent extracellular bacteria that had survived the gentamicin treatment. In contrast, we observed a significant increase in the number of S. flexneri WT bacteria recovered from colonoid monolayers compared to the CFS100 strain when the bacteria were introduced basolaterally (Fig. 1A). To confirm S. flexneri invasion, S. flexneri-infected monolayers were Giemsa stained and examined by light microscopy. We observed ample S. flexneri bacteria associated with colonoid cells in monolayers invaded with the WT strain via the basolateral surface, whereas WT S. flexneri was very rarely observed in colonoid monolayers invaded apically; no bacteria were observed in colonoid monolayers infected with the CFS100 strain either apically or basolaterally (Fig. 1B). We conclude that S. flexneri is able to invade colonoid monolayers, preferentially via the basolateral surface.
FIG 1.

Shigella demonstrates basolateral invasion preference in human colon enteroid monolayers. (A) S. flexneri WT and CFS100 strains were introduced apically or basolaterally to colon monolayers grown on transwells, and bacteria were enumerated at 3 h p.i. Statistical significance was determined by ANOVA with a Bonferroni posttest (n = 9; *, P < 0.05), with data pooled from three independent experiments. (B) Representative Giemsa stain of colonoid monolayers infected apically or basolaterally with WT S. flexneri or the noninvasive CFS100 strain.
S. flexneri can invade human enteroids derived from the duodenum, ileum, and colon.
A hallmark of shigellosis is acute colitis associated with inflammatory lesions. Shigella infection is typically localized in the colon and observed at a significantly increased frequency in the distal colon compared to the proximal colon (40, 43). Although Shigella infections may be associated with nonbloody diarrhea originating from the small intestine, there is little or no evidence of invasive lesions in the small intestine or changes to the human small bowel mucosa (44, 45), indicating that the colon is the primary site of Shigella invasion; however, little is known about which host or bacterial factors contribute to this S. flexneri intestinal tropism. We hypothesized that S. flexneri invasion is specific to human colon cells. To test this hypothesis, we cultivated intestinal enteroids derived from the colon and the duodenum and ileum of the small intestine of the same patient. Polarized enteroid monolayers were formed on transwells and differentiated, and basolateral S. flexneri invasion of these monolayers was then quantified by plating. We found no significant difference in S. flexneri invasion of enteroid monolayers derived from the duodenum, ileum, and colon (Fig. 2). As expected, the recovery of the avirulent S. flexneri strain CFS100 was minimal in all of the tissue types. These data indicate that S. flexneri can invade enteroids derived from small intestinal tissues as well as those derived from the colon. Thus, the preponderance of lesions in the colon during human infection is likely influenced by additional environmental factors not yet incorporated into HIEs.
FIG 2.
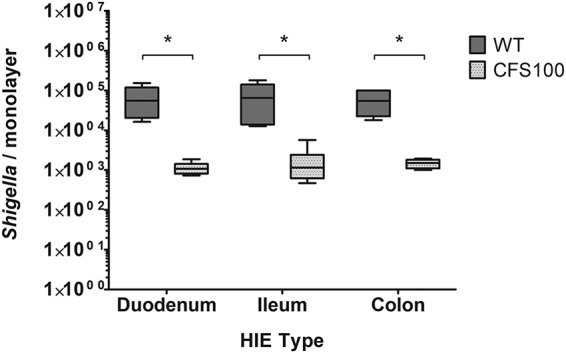
Shigella invades human enteroid monolayers derived from different segments. S. flexneri WT and CFS100 strains were introduced basolaterally to enteroid monolayers grown on transwells, and bacteria were enumerated at 3 h p.i. WT S. flexneri recovery was significantly higher than CFS100 recovery in all cell lines, as determined by ANOVA with a Bonferroni posttest (data from two independent experiments were pooled; n = 6; *, P < 0.05), while no statistically significant differences were observed among enteroid segment lines (P > 0.05 by ANOVA with a Bonferroni posttest).
Patient-patient variability of S. flexneri invasion.
Studies indicate that human shigellosis susceptibility is variable and is affected by demographic factors, including gender, age, and race/ethnicity (46, 47). To function as an S. flexneri virulence model system, it is important that S. flexneri be able to invade enteroids derived from different patients with similar efficiencies. Therefore, we next sought to determine if S. flexneri infection could be recapitulated in enteroid lines derived from normal colon tissues from different patients. Colonoids from three patients were cultured, and these colonoid lines were used to form polarized monolayers on transwells and then differentiated and basolaterally infected with S. flexneri WT and CFS100 strains. Consistent with the above-described experiments, the WT strain invaded all 3 patient colonoid lines, whereas only basal levels of CFS100 were detected (Fig. 3). Interestingly, while we observed modest differences in S. flexneri invasion between the different patient colonoid lines, these differences were not statistically significant.
FIG 3.
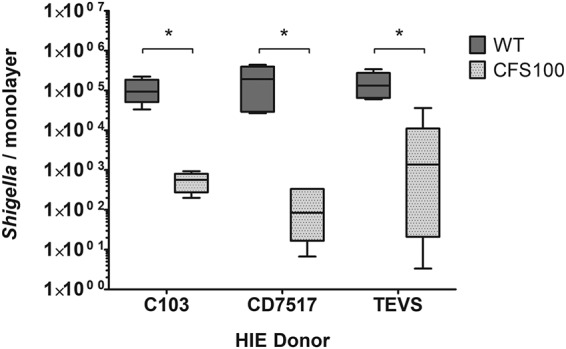
Shigella invades human colonoid monolayers derived from different patients. S. flexneri WT and CFS100 strains were introduced basolaterally to colonoid monolayers grown on transwells, and bacteria were enumerated at 3 h p.i. WT recovery was greater than CFS100 recovery in all colonoid lines as determined by ANOVA with a Bonferroni posttest (data from two independent experiments were pooled; n = 6; *, P < 0.05), while no statistically significant differences were observed comparing colonoid patient lines (P > 0.05 by ANOVA with a Bonferroni posttest).
S. flexneri replication in colonoid monolayers.
After S. flexneri invades a host cell, it escapes the engulfment vacuole and replicates within the host cell cytosol (48). To serve as a widely applicable model system of Shigella pathogenesis, it is important that S. flexneri is able to replicate within colonoid cells. To this end, colonoid monolayers were grown on transwells and basolaterally infected with S. flexneri. Colonoid monolayers were then lysed over time, and S. flexneri bacteria were enumerated by plating. We observed a significant increase in the number of S. flexneri bacteria recovered from monolayers over time, indicating that the bacteria are replicating within the colonoid monolayer (Fig. 4). We calculated the doubling time of S. flexneri to be 47.1 ± 6.7 min, which is slightly higher than doubling times reported for S. flexneri grown in metastatic Henle-407 or HeLa cells (28, 49, 50).
FIG 4.
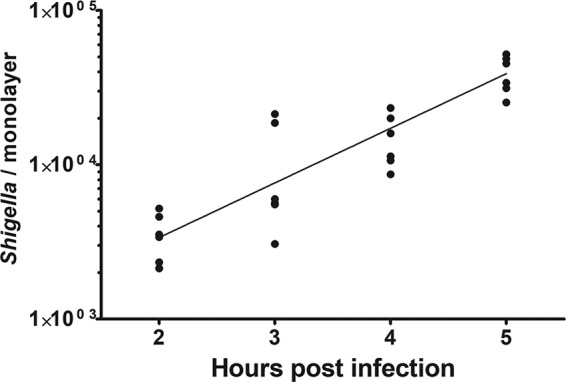
Shigella replicates within human colon enteroid monolayers. S. flexneri WT strain was introduced basolaterally to enteroid monolayers grown on transwells, and bacteria were enumerated over a time course. Line represents a nonlinear regression using the exponential growth model (R2, 0.80). The mean doubling time is calculated to be 47.1 ± 6.7 min, from two independent experiments.
S. flexneri forms actin tails in colonoid monolayers.
To spread from cell to cell, S. flexneri relies on host actin polymerization, mediated by the recruitment of host N-WASP by the polarly localized bacterial protein IcsA. Host actin polymerization by S. flexneri can be visualized using fluorescence microscopy. To determine if S. flexneri is motile in the colonoid cell cytosol, polarized colonoid monolayers were basolaterally infected with the S. flexneri WT or CFS100 strain harboring the plasmid pKP401 and expressing cfp, and at 3 h p.i., cells were fixed with paraformaldehyde and stained with Alexa Fluor 488 conjugated to phalloidin, which binds to actin. Confocal microscopy was used to capture z-stack micrographs. In the WT-infected monolayers, we observed bacterial cells within individual colonoid cells, and several of these cells were associated with actin tails, indicating motile bacteria (Fig. 5). Conversely, no intracellular bacteria were observed in colonoid monolayers infected with the CFS100 strain.
FIG 5.
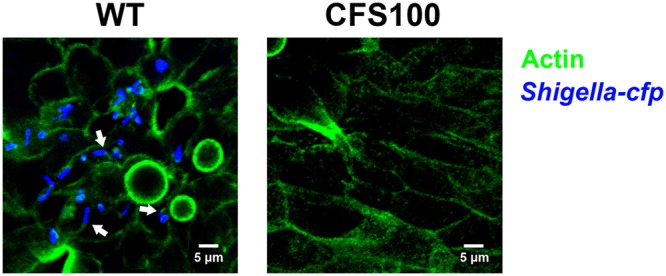
Shigella forms actin tails within human colon enteroid cells. S. flexneri WT and CFS100 strains expressing cyan fluorescent protein (blue) were introduced basolaterally to enteroid monolayers grown on transwells and visualized using confocal microscopy. Actin was stained with labeled phalloidin (green). Micrographs are average-intensity projections of a z-stack. Arrows indicate intracellular Shigella bacteria associated with actin tails.
Disruption of enteroid tight junctions facilitates S. flexneri apical invasion.
S. flexneri is able to apically invade polarized Caco-2 monolayers after cellular tight junctions have been disrupted (26). To determine if tight junction disruption enables S. flexneri apical invasion in human enteroid monolayers, colonoid monolayers were prepared and treated with 20 μM EGTA immediately prior to invasion, which was previously shown to disrupt tight junctions in polarized cells (26). These monolayers were then apically infected with WT S. flexneri. Consistent with the above-described experiments, the recovery of WT S. flexneri bacteria was minimal upon apical infection of untreated polarized colonoids; however, we observed a significant increase in the number of S. flexneri bacteria recovered from EGTA-treated monolayers compared to untreated monolayers (Fig. 6A). We measured the effect of EGTA on invasion of nonpolarized Henle-407 cells and found that EGTA pretreatment had no significant impact on WT S. flexneri invasion (mock, 76.1% ± 5.6%; EGTA, 67.2% ± 6.4% [P > 0.05 by a t test]), and no Henle-407 cells were observed to be invaded by the CFS100 strain with either treatment. To confirm S. flexneri invasion of EGTA-treated colonoid monolayers, we used confocal microscopy to visualize S. flexneri-invaded colonoid monolayers. Monolayers appeared to be confluent, and S. flexneri associated with actin tails was observed in the infected monolayers (Fig. 6B). No intracellular bacteria were observed in monolayers treated with phosphate-buffered saline (PBS) in the absence of EGTA and apically infected with S. flexneri. We conclude that tight junction disruption allows S. flexneri apical invasion of polarized monolayers.
FIG 6.

Disrupting colon enteroid tight junctions enables Shigella apical access to the host cytosol. (A) Colon enteroid monolayers were treated with EGTA, and S. flexneri bacteria were then introduced apically and enumerated at 3 h p.i. Mock indicates monolayers not treated with EGTA (PBS only). * indicates statistical significance, determined by a t test (n = 3; P < 0.05). (B) Confocal microscopy was used to visualize colon enteroid monolayers treated with EGTA and apically invaded by S. flexneri. Monolayers were stained with DAPI (blue) and phalloidin (green). Micrographs are average-intensity projections of a z-stack. An enlarged portion of the micrograph from EGTA-treated colonoids, annotated by a red box, highlights a Shigella cell associated with an actin tail.
S. flexneri infection alters colonoid host gene expression.
Host epithelial cells utilize several antimicrobial responses to limit S. flexneri infection. Extracellular bacteria are recognized by Toll-like receptor 2 (TLR2) or TLR4, which initiates an innate immune response (51, 52). Intracellular bacteria are recognized by STING, which binds bacterial cyclic dinucleotides and cytosolic DNA (53), or by peptidoglycan binding of NOD1/2 (54, 55), both of which initiate a type I interferon response. Cytosolic S. flexneri is also recognized through the TIFA cytosolic surveillance pathway, which stimulates NF-κB-mediated inflammation (56). Previous studies have shown that S. flexneri infection results in changes in host gene expression, specifically the upregulation of several genes related to type I interferons, inflammation, apoptosis, and autophagy (57). Because colonoids are primary cell lines and thus have unaltered metabolism and defense regulatory networks (31), we wanted to characterize the impact of S. flexneri infection on colonoid gene expression. Polarized colonoid monolayers were disrupted by EGTA, and infection by S. flexneri was done via the apical surface. At 3 h p.i., RNA was extracted from the colonoid monolayers, and reverse transcription-quantitative PCR (RT-qPCR) was used to determine the differential expression of host genes. We analyzed the expression of genes associated with the type I interferon response (beta interferon [IFN-β]), mitogen-activated protein kinase (MAPK), and NF-κB-mediated inflammation (c-Jun, interleukin-8 [IL-8], tumor necrosis factor alpha [TNF-α], and TNFAIP3) and the downstream prosurvival gene BCL-2, which are upregulated during S. flexneri infection of HeLa or Caco-2 cells (58, 59).
We observed significant increases in IL-8, TNF-α, IFN-β, and TNFAIP3 expression upon infection with WT S. flexneri compared to the mock-treated colonoids (Fig. 7). We also observed significant increases in TNFAIP3 expression in CFS100-infected colonoid monolayers, indicating that noninvasive extracellular bacteria can stimulate changes in the host gene response as well. TNF-α and TNFAIP3 levels were significantly higher in the WT-infected colonoid monolayers than in the CFS100-infected monolayers, indicating that intracellular S. flexneri bacteria are more potent stimulators of immunity-related genes.
FIG 7.
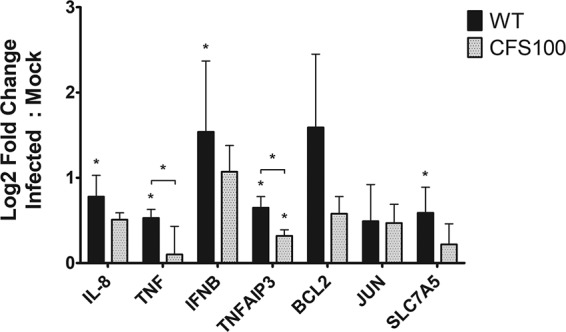
Shigella infection alters host gene expression. Colon enteroid monolayers were treated with EGTA, S. flexneri WT or CFS100 bacteria were then introduced apically, and host gene expression was quantified at 3 h p.i. using RT-qPCR. Gene expression was normalized to that of actB, and log2-fold change values are relative to those of the mock-infected control. Statistical significance was determined by a t test, comparing ΔCT values (n = 3; *, P < 0.05).
During infection, S. flexneri induces an amino acid starvation response associated with the GCN2/eIF2α (α subunit of eukaryotic initiation factor 2)/ATF4/ATF3 integrated stress response and reduces the levels of the cytosolic branched-chain amino acids leucine and isoleucine, together promoting mTOR-mediated xenophagy (60, 61). Importantly, mTOR-associated xenophagy is limited by the uptake of essential amino acids such as leucine (62); because amino acid transportation is limited by glutamine, and glutamine metabolism is altered in metastatic cell lines such as HeLa, Shigella studies of amino acid starvation and xenophagy are constrained by physiologically relevant model systems. In generalized amino acid starvation studies, autophagy is governed by the ATF4-mediated regulation of SLC7A5, a heterodimeric antiporter which couples the influx of leucine/isoleucine with the efflux of glutamine (63, 64). Because S. flexneri infection depletes host leucine and isoleucine and activates ATF3, likely through the ATF4 stress response (60), we hypothesized that SLC7A5 is upregulated during S. flexneri infection of colonoid monolayers. We observed a significant increase in SLC7A5 expression in S. flexneri-infected colonoid monolayers, compared to mock-treated colonoids, and this increase in SLC7A5 expression was not observed in CFS100-infected monolayers (Fig. 7).
DISCUSSION
HIEs are a powerful new tool to study gastrointestinal disease; here, we demonstrate that enteroids can serve as a model of Shigella pathogenesis. We show that S. flexneri is capable of invading human intestinal monolayers and replicating within host cells and exhibits characteristic behaviors in these cells, including host actin polymerization and stimulating a host innate immune response. HIEs are advantageous to use over other tissue culture model systems because they are a primary human-derived cell line with genetically intact signaling pathways and metabolism. Additionally, HIEs can be expanded, genetically manipulated, and cryogenically preserved (33), making them an easily reproducible model system of Shigella pathogenesis. Furthermore, HIEs can be sourced from various tissues and patients, making the model system genetically and phenotypically diverse. We demonstrate here that S. flexneri is able to invade HIEs derived from 3 different patients with similar efficiencies. Future studies examining an expanded pool of patient HIE lines will elucidate host factors that contribute to S. flexneri pathogenesis. Further underscoring the utility and reproducibility of HIEs for the study of Shigella pathogenesis is the work of Ranganathan et al. (65) that was done in parallel with our study. Although there are some slight differences in protocols, both studies show invasion and replication of Shigella flexneri in HIEs, and each study provides new information on the host response to S. flexneri invasion.
The interplay between Shigella and its human host is complex, and many aspects of the host-pathogen relationship are not captured using a homogenous tissue culture model. Shigella encounters different types of cells during infection of the colonic mucosa, and these differentiated cell types can contribute to S. flexneri pathogenesis in the host. For example, depletion of Paneth cells makes newborn mice susceptible to S. flexneri infection (66), and mucins produced by goblet cells are linked to TNF-α expression during S. flexneri infection (67, 68). Additionally, defensins secreted by the colonic epithelium have been shown to contribute to Shigella pathogenesis (69). Differentiated HIEs, which contain both Paneth and goblet cells, offer a promising holistic system to examine the impact of these and other factors on Shigella invasion and intracellular proliferation and to determine which Shigella factors contribute to overcoming obstacles of pathogenesis. In addition, Shigella interacts differently with epithelial cells and immune cells, such as macrophages or B lymphocytes (70, 71). In epithelial cells, S. flexneri induces slow necrosis while it replicates in the host cytosol; in contrast, when S. flexneri is phagocytosed by macrophages, it induces rapid pyroptosis (72). New developments have facilitated the coculture of human intestinal enteroids and macrophages (73); this system can be used to observe the role of macrophage pyroptosis in the context of the colon epithelium. Similar to polarized Caco-2 cells, polarized enteroid monolayers can be disrupted using EGTA, which enables S. flexneri apical invasion (26). Importantly, tight junction disruption by EGTA allows uniform invasion of colonoid monolayers, as opposed to basolateral invasion, which is limited to cells overlapping transwell pores.
The interaction between Shigella and host metabolism is inherently difficult to discern. Despite being able to replicate to a high cell density in the HeLa cell cytosol, host metabolism remains relatively stable, and host cells retain their energy charge during infection (7, 28). And while overall amino acid levels remain steady in infected HeLa cells, HeLa cells exhibit signs of amino acid starvation during Shigella infection (60). Confounding these studies is the Warburg effect, which alters, among other things, host glutamine metabolism (27, 74). Due to their intact metabolic pathways, human intestinal enteroids are an alternative model system to examine host-pathogen metabolic interactions. We find here that host colonoids upregulate the branched-chain-amino-acid transporter SLC7A5 during Shigella infection (Fig. 7), possibly in response to Shigella-induced amino acid depletion. SLC7A5 has been implicated in the pathogenesis of other intracellular pathogens, such as Salmonella and Legionella (75–78). Future studies will interrogate the role of amino acid transport in Shigella pathogenesis and how S. flexneri replication impacts host metabolism in human enteroids.
MATERIALS AND METHODS
Strains, cell lines, and media.
S. flexneri serotype 2a strain 2457T was used for this study (Table 1). S. flexneri was cultured on tryptic soy broth agar plates with 0.01% (wt/vol) Congo red (TSBA-CR) at 37°C, and red colonies were selected to ensure the presence of the S. flexneri virulence plasmid. Bacterial cultures were grown overnight in Luria-Bertani (LB) broth at 30°C, subcultured 1:100, and grown at 37°C to an optical density at 650 nm (OD650) of 0.5 to 1.0 (mid-log phase) prior to infection. Deoxycholate was supplemented at 0.1% (wt/vol) where indicated to increase the efficiency of invasion (79). S. flexneri harboring the plasmid pKP401 was used for microscopy where indicated; this plasmid is derived from pUC18 and contains the cfp allele.
TABLE 1.
Strains and cell lines used in this study
| Strain or cell line | Description or intestinal segment | Source or reference |
|---|---|---|
| S. flexneri strains | ||
| WT | S. flexneri serotype 2a strain 2457T | Walter Reed Institute of Research |
| CFS100 | 2457T lacking the virulence plasmid | 81 |
| Human intestinal enteroid lines | ||
| C103 | Colon | Baylor College of Medicine |
| D103 | Duodenum | Baylor College of Medicine |
| I103 | Ileum | Baylor College of Medicine |
| CD7517 | Colon | H. J. Kim |
| TEVS | Colon | N. F. Shroyer |
HIEs derived from either the small bowel or colon (colonoids) were sourced from the Gastrointestinal Experimental Model Systems (GEMS) Core in the Digestive Disease Center at Baylor College of Medicine, the CD7517 line was provided by Hyun Jung Kim at the University of Texas at Austin, and the TEVS line was provided by Noah Shroyer at Baylor College of Medicine (Table 1). HIEs were derived from adult intestinal biopsy specimens from routine endoscopy procedures or bariatric surgeries, and all biopsy specimens were assessed by physicians to identify healthy tissue.
Complete medium without growth factor (CMGF−) is advanced Dulbecco’s modified Eagle medium (catalog number 12634028; Thermo Fisher) supplemented with GlutaMAX (Thermo Fisher) and 10 mM HEPES (41). Complete growth medium with growth factor (CMGF+) is CMGF− supplemented with Wnt3A-conditioned medium (Wnt3A expressed in mouse fibroblasts), R-spondin-conditioned medium (R-spondin expressed in HEK293T cells), noggin-conditioned medium (noggin expressed in HEK293 cells), B27, N2, N-acetylcysteine, mouse epidermal growth factor (EGF), [Leu15]-gastrin I, nicotinamide, A83-01, and SB202190 (41). HighWNT is a 50:50 mixture of CMGF+ and Wnt3A-conditioned medium. Differentiation medium is CMGF+ without the addition of Wnt3A-conditioned medium, nicotinamide, or SB202190 and with half-concentrations of R-spondin- and noggin-conditioned media (41).
Enteroid cultivation.
3D HIEs were cultured in Matrigel (BD Biosciences) and grown in HighWNT as previously described (38, 41). Undifferentiated HIEs were washed with 0.5 mM EDTA in ice-cold PBS, pelleted for 5 min at 200 × g, and then trypsinized. Trypsin was then inactivated by adding medium with 10% fetal bovine serum (FBS). Cells were then gently pipetted several times with a P1000 pipette before passing cells through a 40-μm nylon filter to produce a single-cell solution, and approximately 2 × 105 to 5 × 105 cells were seeded onto polycarbonate transwell inserts (catalog number 3422; Corning) coated with collagen IV (Sigma) to form monolayers. The monolayers were incubated with 10 μM ROCK inhibitor Y-27632 (Stemcell Technologies) for 24 h and then grown in differentiation medium (38, 41) for 4 days. Differentiation medium was changed every other day prior to infection. Monolayer confluence was confirmed by light microscopy, and polarization was verified by using transepithelial electrical resistance (TEER) measurements, as described previously (41, 42); a monolayer with a value of >700 Ω was considered polarized.
Enteroid invasion.
S. flexneri invasion of HIE monolayers was performed as previously described (42). S. flexneri was grown in LB medium plus deoxycholate (DOC) and then diluted to 5 × 108 CFU/ml (multiplicity of infection [MOI] of ∼100) in CMGF−. For apical infections, HIE monolayers on transwells were transferred to an empty well of a 24-well plate, and the medium was removed from the inner chamber of the transwell. Subsequently, 10 μl of S. flexneri bacteria was added to the inner chamber of the transwell. For basolateral infections, HIE monolayers on transwells were transferred to a 12-well plate upside down, and 10 μl of S. flexneri bacteria was then added to the upward-facing transwell membrane (basolateral side of the cells). Following either apical or basolateral infections, monolayers were incubated for 1 h at 37°C with 5% CO2, and the membranes were then washed four times with PBS. The medium was replaced with CMGF− with either 20 μg/ml gentamicin (for invasion assays and microscopy) or 4 μg/ml gentamicin (for intracellular growth and RT-qPCR), and the monolayers were incubated for an additional 2 h (3 h p.i. total).
For EGTA disruption, colonoid monolayers were prepared as described above, and the monolayers were then washed once with PBS and incubated with PBS plus 20 μM EGTA (or PBS alone for the mock treatment) at 37°C for 15 min prior to infection. PBS-EGTA was then removed, and monolayer disruption was confirmed by microscopy. One hundred microliters of S. flexneri bacteria diluted to 5 × 108 CFU/ml in CMGF− was then added to the apical side of the monolayers. Henle-407 cell invasion assays were performed as previously described (42, 49) and incorporated the same EGTA treatment as the one described above for colonoid monolayers.
S. flexneri intracellular growth assay.
To determine bacterial doubling times, colonoid monolayers were basolaterally infected with S. flexneri as described above at an MOI of 100. At 1 h p.i., monolayers were washed four times with PBS, and the medium was then changed to CMGF− with 4 μg/ml gentamicin. At 2 h.p.i., and every 60 min thereafter, one monolayer was lysed in 5% saponin in PBS at 37°C, diluted, and spot plated on TSBA-CR. The doubling time (r) was determined using the formula t × log2/(log y2 − log y1), where t is time (180 min), y1 is Shigella at 2 h p.i., and y2 is Shigella at 5 h p.i. (n = 3).
Microscopy.
For Giemsa stains, enteroid monolayers on transwells were basolaterally infected as described above, and at 3 h p.i., monolayers were washed twice with PBS, and 100 μl Giemsa stain was then added (Camco Quik stain II; Wright-Giemsa) for 2 min. Monolayers were then washed twice with water, excised using a razor blade, and mounted on a glass slide. Micrographs were captured using an Olympus BX41 microscope (100× objective) with a DP73 digital camera (Olympus) and processed using cellSens software (Olympus) and ImageJ (80).
For fluorescence microscopy, enteroid monolayers on transwells were infected as indicated, and at 3 h p.i., the cells were washed with PBS and then fixed with formaldehyde. Cells were then permeabilized with 0.5% Triton X-100 and blocked with 5% bovine serum albumin (BSA) in PBS. The cells were then incubated with phalloidin-Alexa Fluor 488 (excitation at 490 nm and emission at 525 nm) in PBS with 2.5% BSA and DAPI (4′,6-diamidino-2-phenylindole), where indicated, for approximately 2 h and washed with 0.05% Tween 20 in PBS. Enteroid monolayers were then excised using a razor blade and mounted on slides with Prolong diamond antifade mountant (Thermo Fisher). z-stack micrographs were acquired using a Zeiss LSM 710 confocal microscope (63× objective) at the University of Texas Microscopy and Imaging Facility.
RNA extraction and RT-qPCR.
For RNA extractions of S. flexneri-infected HIE cells, HIE monolayers were infected, as described above, with S. flexneri WT bacteria grown in deoxycholate and added at an MOI of 100. At 3 h p.i., monolayers were washed once with PBS, and 1 ml of cold RNA-Bee (TelTest) was pipetted over the monolayer. The cell lysates from HIE monolayers were transferred to new tubes, and 200 μl chloroform was added. The mixture was vortexed and, after 5 min on ice, centrifuged at 13,000 × g, and the upper aqueous phase was transferred to a new tube. RNA was then precipitated with isopropanol, treated with DNase I (Invitrogen), and solubilized in water.
RNA was quantified using an ND-1000 spectrophotometer (Nanodrop), and a total of 500 ng RNA was used with a high-capacity cDNA reverse transcription kit (Applied Biosystems). Two microliters of cDNA was then used as the template for SYBR green qPCR (Applied Biosystems) using a ViiA 7 real-time PCR system (Applied Biosystems) at the University of Texas at Austin Genomic Sequencing and Analysis Facility. Human gene expression was normalized to actB. All primers used for RT-qPCR are listed in Table S1 in the supplemental material. Statistical significance was determined using Student’s t test of ΔCT values comparing mock-treated samples to S. flexneri WT-infected HIE cells or S. flexneri CFS100 (noninvasive strain)-infected HIE cells.
Statistics.
Student’s t test or analysis of variance (ANOVA) was used, as indicated, to determine statistical significance (GraphPad Prism), and replicates from independent experiments were pooled to determine significance. For ANOVA, differences between specific groups were determined using a Bonferroni posttest. Asterisks in the figures indicate statistical significance (P < 0.05). Error bars represent standard deviations from the means. Bacterial growth data were fitted to an exponential growth model using nonlinear regression (GraphPad Prism).
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Shin and H. J. Kim for providing the CD7517 enteroid line and D. Omotajo and N. F. Shroyer for the TEVS enteroid line. We also thank the Baylor College of Medicine Digestive Disease Center for their assistance, and we thank M. Whiteley for providing the pKP401 plasmid.
This work was funded by Public Health Service grant AI131497 from the National Institutes of Health to S.M.P.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00733-18.
REFERENCES
- 1.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. 1999. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bull World Health Organ 77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 2.Kosek M, Yori PP, Olortegui MP. 2010. Shigellosis update: advancing antibiotic resistance, investment empowered vaccine development, and green bananas. Curr Opin Infect Dis 23:475–480. doi: 10.1097/QCO.0b013e32833da204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Killackey SA, Sorbara MT, Girardin SE. 2016. Cellular aspects of Shigella pathogenesis: focus on the manipulation of host cell processes. Front Cell Infect Microbiol 6:38. doi: 10.3389/fcimb.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ménard R, Sansonetti PJ, Parsot C. 1993. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol 175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marquart ME, Picking WL, Picking WD. 1996. Soluble invasion plasmid antigen C (IpaC) from Shigella flexneri elicits epithelial cell responses related to pathogen invasion. Infect Immun 64:4182–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P. 1999. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol 147:683–693. doi: 10.1083/jcb.147.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantis N, Prévost MC, Sansonetti P. 1996. Analysis of epithelial cell stress response during infection by Shigella flexneri. Infect Immun 64:2474–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci U S A 92:6572–6576. doi: 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. 1999. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol 146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.CDC. 2011. National Antimicrobial Resistance Monitoring System for Enteric Bacteria (NARMS): human isolates final report. CDC, Atlanta, GA. [Google Scholar]
- 12.CDC. 2013. Antibiotic resistance threats in the United States. CDC, Atlanta, GA. [Google Scholar]
- 13.WHO. 2017. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. WHO, Geneva, Switzerland. [Google Scholar]
- 14.Shim DH, Suzuki T, Chang SY, Park SM, Sansonetti PJ, Sasakawa C, Kweon MN. 2007. New animal model of shigellosis in the guinea pig: its usefulness for protective efficacy studies. J Immunol 178:2476–2482. doi: 10.4049/jimmunol.178.4.2476. [DOI] [PubMed] [Google Scholar]
- 15.West NP, Sansonetti P, Mounier J, Exley RM, Parsot C, Guadagnini S, Prevost MC, Prochnicka-Chalufour A, Delepierre M, Tanguy M, Tang CM. 2005. Optimization of virulence functions through glucosylation of Shigella LPS. Science 307:1313–1317. doi: 10.1126/science.1108472. [DOI] [PubMed] [Google Scholar]
- 16.Jeong KI, Zhang Q, Nunnari J, Tzipori S. 2010. A piglet model of acute gastroenteritis induced by Shigella dysenteriae type 1. J Infect Dis 201:903–911. doi: 10.1086/650995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mostowy S, Boucontet L, Moya MJM, Sirianni A, Boudinot P, Hollinshead M, Cossart P, Herbomel P, Levraud J-P, Colucci-Guyon E. 2013. The zebrafish as a new model for the in vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PLoS Pathog 9:e1003588. doi: 10.1371/journal.ppat.1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kent TH, Formal SB, LaBrec EH, Sprinz H, Maenza RM. 1967. Gastric shigellosis in rhesus monkeys. Am J Pathol 51:259–267. [PMC free article] [PubMed] [Google Scholar]
- 19.Honjo S, Takasaka M, Fujiwara T, Nakagawa M, Andoo K, Ogawa H, Takahashi R, Imaizumi K. 1964. Shigellosis in cynomolgus monkeys (Macaco irus). II. Experimental infection with Shigella flexneri 2A with special references to clinical and bacteriological findings. Jpn J Med Sci Biol 17:307–319. doi: 10.7883/yoken1952.17.307. [DOI] [PubMed] [Google Scholar]
- 20.Shipley ST, Panda A, Khan AQ, Kriel EH, Maciel M, Livio S, Nataro JP, Levine MM, Sztein MB, DeTolla LJ. 2010. A challenge model for Shigella dysenteriae 1 in cynomolgus monkeys (Macaca fascicularis). Comp Med 60:54–61. [PMC free article] [PubMed] [Google Scholar]
- 21.Higgins R, Sauvageau R, Bonin P. 1985. Shigella flexneri type 2 infection in captive nonhuman primates. Can Vet J 26:402–403. [PMC free article] [PubMed] [Google Scholar]
- 22.Sansonetti PJ, Arondel J, Fontaine A, d’Hauteville H, Bernardini ML. 1991. ompB (osmo-regulation) and icsA (cell-to-cell spread) mutants of Shigella flexneri: vaccine candidates and probes to study the pathogenesis of shigellosis. Vaccine 9:416–422. doi: 10.1016/0264-410X(91)90128-S. [DOI] [PubMed] [Google Scholar]
- 23.DuPont HL, Hornick RB, Dawkins AT, Snyder MJ, Formal SB. 1969. The response of man to virulent Shigella flexneri 2a. J Infect Dis 119:296–299. doi: 10.1093/infdis/119.3.296. [DOI] [PubMed] [Google Scholar]
- 24.Pal T, Hale TL. 1989. Plasmid-associated adherence of Shigella flexneri in a HeLa cell model. Infect Immun 57:2580–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale TL, Morris RE, Bonventre PF. 1979. Shigella infection of Henle intestinal epithelial cells: role of the host cell. Infect Immun 24:887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. 1992. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun 60:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu PP, Sabatini DM. 2008. Cancer cell metabolism: Warburg and beyond. Cell 134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 28.Kentner D, Martano G, Callon M, Chiquet P, Brodmann M, Burton O, Wahlander A, Nanni P, Delmotte N, Grossmann J, Limenitakis J, Schlapbach R, Kiefer P, Vorholt JA, Hiller S, Bumann D. 2014. Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc Natl Acad Sci U S A 111:9929–9934. doi: 10.1073/pnas.1406694111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waligora EA, Fisher CR, Hanovice NJ, Rodou A, Wyckoff EE, Payne SM. 2014. Role of intracellular carbon metabolism pathways in Shigella flexneri virulence. Infect Immun 82:2746–2755. doi: 10.1128/IAI.01575-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pieper R, Fisher CR, Suh M-J, Huang S-T, Parmar P, Payne SM. 2013. Analysis of the proteome of intracellular Shigella flexneri reveals pathways important for intracellular growth. Infect Immun 81:4635–4648. doi: 10.1128/IAI.00975-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stutz AM, Jauch A, Aiyar RS, Pau G, Delhomme N, Gagneur J, Korbel JO, Huber W, Steinmetz LM. 2013. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda) 3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coron E, Flamant M, Aubert P, Wedel T, Pedron T, Letessier E, Galmiche JP, Sansonetti PJ, Neunlist M. 2009. Characterisation of early mucosal and neuronal lesions following Shigella flexneri infection in human colon. PLoS One 4:e4713. doi: 10.1371/journal.pone.0004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Ettayebi K, Blutt SE, Hyser JM, Zeng XL, Crawford SE, Broughman JR, Estes MK, Donowitz M. 2014. Human enteroids as an ex-vivo model of host-pathogen interactions in the gastrointestinal tract. Exp Biol Med (Maywood) 239:1124–1134. doi: 10.1177/1535370214529398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovbasnjuk O, Zachos NC, In J, Foulke-Abel J, Ettayebi K, Hyser JM, Broughman JR, Zeng XL, Middendorp S, de Jonge HR, Estes MK, Donowitz M. 2013. Human enteroids: preclinical models of non-inflammatory diarrhea. Stem Cell Res Ther 4:S3. doi: 10.1186/scrt364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin Y, Zhou D. 2018. Organoid and enteroid modeling of Salmonella infection. Front Cell Infect Microbiol 8:102. doi: 10.3389/fcimb.2018.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, Graham DY, Qureshi W, Sherman V, Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Donowitz M, Estes MK. 2016. Human intestinal enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol 90:43–56. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poole NM, Green SI, Rajan A, Vela LE, Zeng X-L, Estes MK, Maresso AW. 2017. Role for FimH in extraintestinal pathogenic Escherichia coli invasion and translocation through the intestinal epithelium. Infect Immun 85:e00581-17. doi: 10.1128/IAI.00581-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajan A, Vela L, Zeng X-L, Yu X, Shroyer N, Blutt SE, Poole NM, Carlin LG, Nataro JP, Estes MK, Okhuysen PC, Maresso AW. 2018. Novel segment- and host-specific patterns of enteroaggregative Escherichia coli adherence to human intestinal enteroids. mBio 9:e02419-17. doi: 10.1128/mBio.02419-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forbester JL, Goulding D, Vallier L, Hannan N, Hale C, Pickard D, Mukhopadhyay S, Dougan G. 2015. Interaction of Salmonella enterica serovar Typhimurium with intestinal organoids derived from human induced pluripotent stem cells. Infect Immun 83:2926–2934. doi: 10.1128/IAI.00161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Speelman P, Kabir I, Islam M. 1984. Distribution and spread of colonic lesions in shigellosis: a colonoscopic study. J Infect Dis 150:899–903. doi: 10.1093/infdis/150.6.899. [DOI] [PubMed] [Google Scholar]
- 41.Poole NM, Rajan A, Maresso AW. 2018. Human intestinal enteroids for the study of bacterial adherence, invasion, and translocation. Curr Protoc Microbiol 50:e55. doi: 10.1002/cpmc.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koestler BJ, Ward CM, Payne SM. 2018. Shigella pathogenesis modeling with tissue culture assays. Curr Protoc Microbiol 49:e57. doi: 10.1002/cpmc.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Islam MM, Azad AK, Bardhan PK, Raqib R, Islam D. 1994. Pathology of shigellosis and its complications. Histopathology 24:65–71. doi: 10.1111/j.1365-2559.1994.tb01272.x. [DOI] [PubMed] [Google Scholar]
- 44.Levine MM, DuPont HL, Formal SB, Hornick RB, Takeuchi A, Gangarosa EJ, Snyder MJ, Libonati JP. 1973. Pathogenesis of Shigella dysenteriae 1 (Shiga) dysentery. J Infect Dis 127:261–270. doi: 10.1093/infdis/127.3.261. [DOI] [PubMed] [Google Scholar]
- 45.Zalev AH, Warren RE. 1989. Shigella colitis with radiological and endoscopic correlation: case report. Can Assoc Radiol J 40:328–330. [PubMed] [Google Scholar]
- 46.Shiferaw B, Shallow S, Marcus R, Segler S, Soderlund D, Hardnett FP, Van Gilder T. 2004. Trends in population-based active surveillance for shigellosis and demographic variability in FoodNet sites, 1996–1999. Clin Infect Dis 38:S175–S180. doi: 10.1086/381584. [DOI] [PubMed] [Google Scholar]
- 47.Chang M, Groseclose SL, Zaidi AA, Braden CR. 2009. An ecological analysis of sociodemographic factors associated with the incidence of salmonellosis, shigellosis, and E. coli O157:H7 infections in US counties. Epidemiol Infect 137:810–820. doi: 10.1017/S0950268808001477. [DOI] [PubMed] [Google Scholar]
- 48.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7:333–340. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 49.Koestler BJ, Fisher CR, Payne SM. 2018. Formate promotes Shigella intercellular spread and virulence gene expression. mBio 9:e01777-18. doi: 10.1128/mBio.01777-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossi RM, Yum L, Agaisse H, Payne SM. 2017. Cardiolipin synthesis and outer membrane localization are required for Shigella flexneri virulence. mBio 8:e01199-17. doi: 10.1128/mBio.01199-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pore D, Mahata N, Pal A, Chakrabarti MK. 2011. Outer membrane protein A (OmpA) of Shigella flexneri 2a, induces protective immune response in a mouse model. PLoS One 6:e22663. doi: 10.1371/journal.pone.0022663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paciello I, Silipo A, Lembo-Fazio L, Curcurù L, Zumsteg A, Noël G, Ciancarella V, Sturiale L, Molinaro A, Bernardini ML. 2013. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci U S A 110:E4345–E4354. doi: 10.1073/pnas.1303641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girardin SE, Tournebize R, Mavris M, Page A-L, Li X, Stark GR, Bertin J, DiStefano PS, Yaniv M, Sansonetti PJ, Philpott DJ. 2001. CARD4/Nod1 mediates NF‐κB and JNK activation by invasive Shigella flexneri. EMBO Rep 2:736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Girardin SE, Boneca IG, Carneiro LAM, Antignac A, Jéhanno M, Viala J, Tedin K, Taha M-K, Labigne A, Zäthringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ. 2003. Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 56.Gaudet RG, Guo CX, Molinaro R, Kottwitz H, Rohde JR, Dangeard A-S, Arrieumerlou C, Girardin SE, Gray-Owen SD. 2017. Innate recognition of intracellular bacterial growth is driven by the TIFA-dependent cytosolic surveillance pathway. Cell Rep 19:1418–1430. doi: 10.1016/j.celrep.2017.04.063. [DOI] [PubMed] [Google Scholar]
- 57.Krokowski S, Mostowy S. 2016. Interactions between Shigella flexneri and the autophagy machinery. Front Cell Infect Microbiol 6:17. doi: 10.3389/fcimb.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pédron T, Thibault C, Sansonetti PJ. 2003. The invasive phenotype of Shigella flexneri directs a distinct gene expression pattern in the human intestinal epithelial cell line Caco-2. J Biol Chem 278:33878–33886. doi: 10.1074/jbc.M303749200. [DOI] [PubMed] [Google Scholar]
- 59.Faherty CS, Merrell DS, Semino-Mora C, Dubois A, Ramaswamy AV, Maurelli AT. 2010. Microarray analysis of Shigella flexneri-infected epithelial cells identifies host factors important for apoptosis inhibition. BMC Genomics 11:272. doi: 10.1186/1471-2164-11-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, Yang C, Emili A, Philpott DJ, Girardin SE. 2012. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 61.Tsalikis J, Tattoli I, Ling A, Sorbara MT, Croitoru DO, Philpott DJ, Girardin SE. 2015. Intracellular bacterial pathogens trigger the formation of U small nuclear RNA bodies (U bodies) through metabolic stress induction. J Biol Chem 290:20904–20918. doi: 10.1074/jbc.M115.659466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. 2009. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scalise M, Galluccio M, Console L, Pochini L, Indiveri C. 2018. The human SLC7A5 (LAT1): the intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front Chem 6:243. doi: 10.3389/fchem.2018.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, Hao Q, Li R, Yu L. 2014. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol 206:173–182. doi: 10.1083/jcb.201403009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ranganathan S, Doucet M, Grassel CL, Delaine-Elias B, Zachos NC, Barry EM. 2019. Evaluating Shigella flexneri pathogenesis in the human enteroid model. Infect Immun 87:e00740-18. doi: 10.1128/IAI.00740-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez M-I, Regnault B, Mulet C, Tanguy M, Jay P, Sansonetti PJ, Pédron T. 2008. Maturation of Paneth cells induces the refractory state of newborn mice to Shigella infection. J Immunol 180:4924–4930. doi: 10.4049/jimmunol.180.7.4924. [DOI] [PubMed] [Google Scholar]
- 67.Sperandio B, Fischer N, Chevalier-Curt MJ, Rossez Y, Roux P, Robbe Masselot C, Sansonetti PJ. 2013. Virulent Shigella flexneri affects secretion, expression, and glycosylation of gel-forming mucins in mucus-producing cells. Infect Immun 81:3632–3643. doi: 10.1128/IAI.00551-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nutten S, Sansonetti P, Huet G, Bourdon-Bisiaux C, Meresse B, Colombel J-F, Desreumaux P. 2002. Epithelial inflammation response induced by Shigella flexneri depends on mucin gene expression. Microbes Infect 4:1121–1124. doi: 10.1016/S1286-4579(02)01636-2. [DOI] [PubMed] [Google Scholar]
- 69.Xu D, Liao C, Zhang B, Tolbert WD, He W, Dai Z, Zhang W, Yuan W, Pazgier M, Liu J, Yu J, Sansonetti PJ, Bevins CL, Shao Y, Lu W. 2018. Human enteric α-defensin 5 promotes Shigella infection by enhancing bacterial adhesion and invasion. Immunity 48:1233.e7–1244.e7. doi: 10.1016/j.immuni.2018.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nothelfer K, Arena ET, Pinaud L, Neunlist M, Mozeleski B, Belotserkovsky I, Parsot C, Dinadayala P, Burger-Kentischer A, Raqib R, Sansonetti PJ, Phalipon A. 2014. B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection. J Exp Med 211:1215–1229. doi: 10.1084/jem.20130914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lucchini S, Liu H, Jin Q, Hinton JCD, Yu J. 2005. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect Immun 73:88–102. doi: 10.1128/IAI.73.1.88-102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashida H, Kim M, Sasakawa C. 2014. Manipulation of the host cell death pathway by Shigella. Cell Microbiol 16:1757–1766. doi: 10.1111/cmi.12367. [DOI] [PubMed] [Google Scholar]
- 73.Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, Zachos NC. 2017. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep 7:45270. doi: 10.1038/srep45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu W, Pelicano H, Huang P. 2010. Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell 18:199–200. doi: 10.1016/j.ccr.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tattoli I, Philpott DJ, Girardin SE. 2012. The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole. Biol Open 1:1215–1225. doi: 10.1242/bio.20122840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schuster AT, Homer CR, Kemp JR, Nickerson KP, Deutschman E, Kim Y, West G, Sadler T, Stylianou E, Krokowski D, Hatzoglou M, de la Motte C, Rubin BP, Fiocchi C, McDonald C, Longworth MS. 2015. CAP-D3 promotes bacterial clearance in human intestinal epithelial cells by repressing expression of amino acid transporters. Gastroenterology 148:1405.e3–1416.e3. doi: 10.1053/j.gastro.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bruckert WM, Abu Kwaik Y. 2015. Complete and ubiquitinated proteome of the Legionella-containing vacuole within human macrophages. J Proteome Res 14:236–248. doi: 10.1021/pr500765x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Price CTD, Kwaik YA. 2014. The transcriptome of Legionella pneumophila-infected human monocyte-derived macrophages. PLoS One 9:e114914. doi: 10.1371/journal.pone.0114914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pope LM, Reed KE, Payne SM. 1995. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect Immun 63:3642–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rasband WS. 1997. ImageJ. US National Institutes of Health, Bethesda, MD. [Google Scholar]
- 81.Marman HE, Mey AR, Payne SM. 2014. Elongation factor P and modifying enzyme PoxA are necessary for virulence of Shigella flexneri. Infect Immun 82:3612–3621. doi: 10.1128/IAI.01532-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.