

Summary
Intracellular bacterial pathogens often rely on their hosts for essential nutrients. Host cells, in turn, attempt to limit nutrient availability, using starvation as a mechanism of innate immunity. Here we discuss both host mechanisms of amino acid starvation and the diverse adaptations of pathogens to their nutrient-deprived environments. These processes provide both key insights into immune subversion and new targets for drug development.
Introduction
Successful bacterial pathogens exploit their hosts to support their own survival. Pathogens rely on their hosts for nutrients necessary for survival, injuring the hosts in the process. For their part, the hosts are hardly gracious. In fact, the ability to keep would-be pathogens away from the nutrients turns out to be a key part of host defence. This interplay exists for all essential nutrients, including carbon, nitrogen and transitional metals (Eisenreich et al., 2010; Skaar, 2010; Rohmer et al, 2011; Hood and Skaar, 2012).
For many potential pathogens, amino acids are critical nutrients. While many organisms can synthesize their own amino acids, others must scavenge them in order to make proteins. Additionally, many bacterial pathogens are able to use amino acids as a carbon source, some even as their principle carbon source (Molofsky and Swanson, 2004; Wieland et al, 2005; Eylert et al, 2010; Venugopal et al., 2011). Accordingly, mammalian hosts have evolved mechanisms to starve bacteria of amino acids. And, to counter this, bacteria have a diversity of means to respond to this stress.
Here we will review the evidence for and mechanisms of host-mediated amino acid starvation and the bacterial response to amino acid depletion. We have chosen three intracellular pathogens to illustrate the diversity of bacterial responses. The first, Chlamydia trachomatis, is a natural auxotroph for many amino acids and responds to amino acid starvation primarily by differentiating to a viable but non-replicating form. The second, Mycobacterium tuberculosis, has evolved to become independent of host amino acid availability and constitutively synthesizes its own amino acids. The third, Legionella pneumophila, is able to extract amino acids from an otherwise stingy host by exploiting host machinery – including amino acid transporters and the host protea-some – to make amino acids available in the pathogen’s intracellular niche.
Host cells are able to starve intracellular pathogens of amino acids
The evidence
The exact intracellular niches of bacterial pathogens are difficult to isolate; thus their metabolite contents are difficult to measure directly. Pathogen adaptation to the intracellular environment, however, suggests that host cells are able to limit amino acid availability. The most compelling fact is that amino acid auxotroph strains of many bacterial pathogens are often attenuated for intracellular growth and during host infection. For example, M. tuberculosis requires the biosynthesis pathways for at least four amino acids to survive during a model infection of mice and within isolated mouse and human macrophages. Leucine, proline and lysine auxotrophs are attenuated in vivo, the latter by so much that they have been tested as live-attenuated vaccine candidates (Hondalus et al, 2000; Smith et al., 2001; Pavelka et al., 2003b). Additionally, multiple groups have shown the requirement of several tryptophan biosynthesis enzymes for survival in mice and macrophages (Smith et al., 2001; Parish, 2003). Salmonella auxotrophs for aromatic amino acids, histidine and methionine have also been shown to be attenuated in vivo (Hoiseth and Stocker, 1981; Fields et al., 1986; O’Callaghan et al., 1988).
Measuring the pathogen’s transcriptional response to the intracellular niche paints the same picture of amino acid starvation. Since pathogens often regulate gene expression in response to environmental signals, expression profiles can serve as bioprobes of the conditions they encounter. Many intracellular pathogens upregulate amino acid biosynthesis genes during infection (Chatterji and Ojha, 2001). This is true for bacterial pathogens as well as fungal pathogens like Candida albicans (Rubin-Bejerano et al., 2003). Many fungal species encode a kinase, Gcn4, that senses exogenous amino acid availability. Upon infection, Gcn4 is activated and coordinates Candida amino acid biosynthesis (Tripathi et al., 2002). Additionally, the stringent response, a conserved bacterial signalling pathway that senses amino acid starvation, among other stresses, is often required for intracellular growth (Magnusson et al., 2005; Potrykus and Cashel, 2008). RelA senses amino acid starvation and synthesizes ppGpp, which induces necessary adaptive changes, including transcriptional upregulation of amino acid biosynthesis genes (Traxler et al., 2008). In many intracellular pathogens, including M. tuberculosis and Salmonella, RelA is upregulated and specifically required for intracellular growth (Primm et al., 2000; Pizarro-Cerdá and Tedin, 2004; Song et al., 2004; Thompson et al., 2006).
The mechanisms
While there is much evidence that the host can create an amino acid depleted niche, the mechanisms for producing this starved niche are not well characterized. One major clue, however, comes from the autophagic response to intracellular pathogens. Autophagy is an important cellular process through which eukaryotic cells respond to a variety of stresses, including metabolite starvation and damaged organelles (Levine, 2005; Deretic, 2009; Deretic and Levine, 2009). Bacterial invasion also induces an autophagic response, called xenophagy, wherein invading pathogens are delivered to autophagosomes to be broken down (Levine, 2005; Deretic, 2009; Deretic and Levine, 2009). This process of xenophagy is required for optimal suppression of the growth of multiple intracellular pathogens, including Listeria, Shigella and Mycobacterium species (Gutierrez et al., 2004; Singh et al., 2006; 2010; Suzuki et al., 2007; Zhao et al., 2008; Yuk et al., 2009).
Nutritional signals play a key regulatory role in autophagy. In particular, autophagy is inhibited when the cytosol contains high concentrations of amino acids. Tattoli and colleagues found that intracellular amino acid levels must be low in order for xenophagy to be fully induced (Sancak et al., 2008; 2010). They found that the activity of mTOR, a checkpoint kinase that links nutrient sensing to metabolic activity, decreased upon infection with the intracellular pathogens Salmonella and Shigella (Tattoli et al., 2012b). This indicated a possible amino acid starvation, as mTOR activity decreases when amino acid levels are low. Moreover, cytosolic levels of isoleucine and leucine fell within an hour after infection (Tattoli et al., 2012b). Interestingly, Shigella-induced amino acid starvation lasted at least 4 h, while Salmonella induced a more transient 1 h starvation. The authors hypothesized that membrane damage might be responsible for amino acid starvation as the temporal pattern of such damage (transient for Salmonella; lasting for Shigella) matched the loss of amino acids. In fact, even aseptic membrane damage, induced by digitonin, could induce amino acid starvation. While this does not rule out alternative mechanisms, this study strongly supports the possibility that bacteria induce amino acid starvation and presents compelling evidence that membrane damage could play a major role (Tattoli et al., 2012a,b). Interestingly, Salmonella could reverse this amino acid starvation, which activated mTOR and inhibited autophagy, ultimately restoring bacterial growth. When the authors overrode the starvation-induced autophagy block, Salmonella growth was restored (Tattoli et al., 2012b).
In addition to general amino acid depletion, host cells can also specifically deplete intracellular tryptophan during pathogen invasion (Hayashi et al., 2001; Silva et al., 2002). Tryptophan depletion as an anti-bacterial mechanism was first described in Chlamydia-infected cells (Byrne et al., 1986; Murray et al., 1989; Beatty et al., 1994). The addition of interferon-gamma (IFN-γ) to infected cells almost completely abolishes intracellular chlamydial growth. Stunningly, simply adding tryptophan reverses the anti-chlamydial effect of IFN-γ (Byrne et al., 1986; Beatty et al., 1994; Leonhardt et al., 2007). IFN-γ activates indoleamine-2,3-dioxygenase (IDO), the enzyme responsible for the first step in kynurenine synthesis from tryptophan (Leonhardt et al., 2007; Zelante et al., 2009). When strongly induced – as in the case of IFN-γ activation – IDO can deplete about 95% of intracellular tryptophan (Fujigaki, 2002). Thus, IDO is required for limiting the growth of tryptophan-requiring intracellular pathogens, such as some Chlamydia species, Leishmania donovani, Toxoplasma gondii, as well as lab-generated strains tryptophan-auxotrophic bacteria (Daubener et al., 2001; Fujigaki, 2002; Leonhardt et al., 2007; Ibana et al., 2011a,b).
It is worth noting, however, that the effect of IDO on pathogen growth is multi-faceted (Medzhitov et al., 2011). The first appreciated role for IDO in pathogen defence was intracellular tryptophan depletion. Recently, the role of its enzymatic products, kynurenines, has also been characterized. Kynurenines are potent negative regulators of inflammation and T cell activity (Munn et al., 2005; Zelante et al., 2009; Favre et al., 2010; Medzhitov et al., 2011). So while the role of IDO on intracellular tryptophan is clear, its pleiotropic effects in pathogen defence have been more difficult to pin down (Blumenthal et al., 2012). Nevertheless, IDO represents a mechanism by which an infected host can drastically reduce the amounts of an often-essential amino acid available to pathogen.
Pathogen responses to host AA starvation
Many pathogens are able to synthesize the full set of amino acids. It would be reasonable to think, given how common amino acid depleted environments are, that amino acid biosynthetic pathways would be a requirement for being an intracellular human pathogen. However, many known successful human pathogens are auxotrophic for some amino acids. How can this be? Some bacteria are simply able to escape by altering their metabolic requirements. For example, they can differentiate into non-replicating, less metabolically demanding cells. Other organisms are able to avoid amino acid starvation altogether by manipulating host amino acid uptake and production systems to increase available amino acids in the pathogen’s intracellular niche. We will review three model pathogens that fit these three moulds: (i) Chlamydia, which employs growth arrest and differentiation, (ii) M. tuberculosis, which exemplifies amino acid self-sufficiency, and (iii) L. pneumophila, which exploits host machinery to extract amino acids from the host cell.
Chlamydia: hiding out and holding out for a better day – growth arrest and differentiation
Chlamydia trachomatis causes a variety of human diseases, including the leading cause of infectious blindness and genital tract infections that can have long-term severe consequences for infected individuals. All members of the Chlamydia family are intracellular pathogens that share two distinct developmental stages. Elementary bodies (EBs) initiate the intracellular infection through endocytic uptake. In optimal growth conditions, the metabolically inert EBs then differentiate into metabolically active reticulate bodies (RBs), which then replicate and grow within the bacterial vacuole. Eventually RBs develop back into EBs, which upon release, go on to infect other cells.
When stimulated with IFN-γ, the host cell imposes a much greater stress upon the bacteria. Chlamydia responds to this stress by morphing into a third, aberrant form (Beatty et al., 1994; Leonhardt et al., 2007). The aberrant form cells are similar to reticulate bodies in morphology, but are unable to divide or differentiate back to EBs. In some studies, adding tryptophan restores growth of these aberrant RBs, demonstrating that persistence is driven by tryptophan starvation (Byrne et al., 1986; Beatty et al., 1994; Leonhardt et al., 2007; Ibana et al., 2011a). Differentiation into this aberrant RB form allows the pathogen to hide from its tryptophan-depleted environment until tryptophan availability resumes (Fig. 1). Isoleucine also seems to be limited during infection, and Chlamydia growth in one intracellular infection model could be restored upon the addition of isoleucine (Hatch, 1975). Interestingly, Chlamydia does not encode the classical bacterial stringent response, which enables many bacteria to sense amino acid starvation and enter into persistent states (Ouellette et al., 2006). Thus, Chlamydia must utilize a unique mechanism to sense tryptophan depletion and differentiate into these aberrant forms.
Fig. 1.
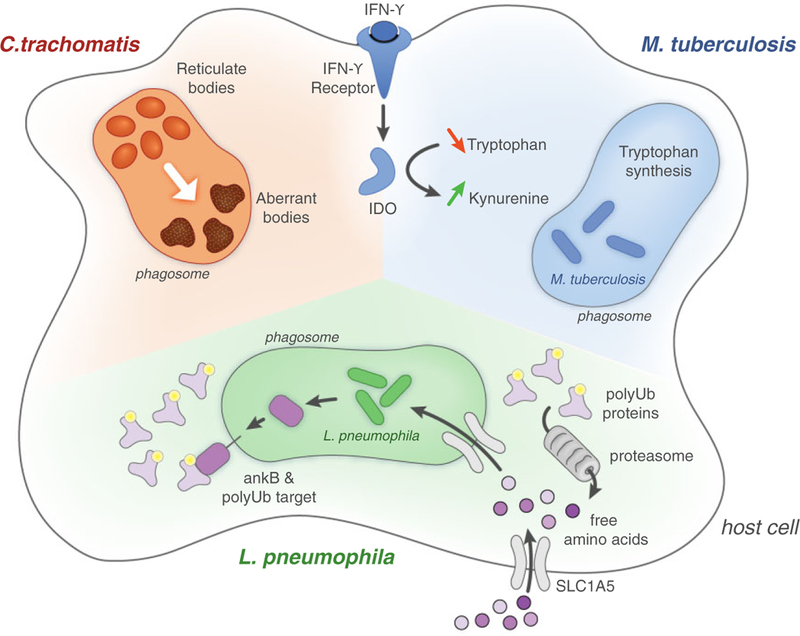
Three bacterial strategies for evading amino acid starvation. Chlamydia trachomatis responds to amino acid starvation by differentiating to aberrant reticulate bodies, viable but metabolically inactive forms that can differentiate back to active forms when amino acid become available again. While Mycobacterium tuberculosis faces amino acid starvation during infection, its constitutive expression of amino acid synthesis makes it generally resistant to host-driven starvation. Legionella pneumophila exploits host proteins to override starvation and deliver free amino acids into its intracellular niche. AnkB, a secreted virulence factor, drives the polyubiquitination of LCV membrane proteins. The host proteasome is then recruited to proteolyze ubiquinated targets, producing free amino acids that are transported into the LCV by the upregulated host transporter, SLC1A5.
Early investigations on the role of tryptophan and IDO in driving aberrant form differentiation yielded mixed results. While some groups found tryptophan supplementation alone could reverse the effects of IFN-γ, others found no effect (Murray et al., 1989). When the C. trachomatis genome was sequenced – and especially when multiple clinical strains were sequenced – a major clue emerged. As expected, Chlamydia lacks the full suite of tryptophan biosynthetic enzymes, and thus cannot synthesize tryptophan from chorismate or most other biosynthetic precursors (Stephens, 1998). It does, however, encode tryptophan synthase (TrpAB), a more limited enzyme that allows the organism to synthesize tryptophan from indole. In addition Chlamydia encodes TrpR, a tryptophan- dependent aporepressor, which enables the pathogen to react specifically to environmental tryptophan availability (Stephens, 1998; Belland et al., 2003; Wood et al., 2003). Interestingly, C. trachomatis strains isolated from ocular infections and synovial tissue contain frameshift mutations in trpAB, while strains from genital infections do not (Fehlner-Gardiner et al, 2002; Caldwell et al, 2003; Gerard et al., 2010). Within strains isolated from sexually transmitted infections, there are three trpAB variants; while the effect of each trpAB variant on tryptophan synthesis is unknown, identity of the trpAB allele correlates perfectly with IFN-γ susceptibility in vitro (Morrison, 2000; Fehlner-Gardiner et al, 2002; Caldwell et al., 2003; McClarty et al., 2007). So while all C. trachomatis contain the trpAB gene, not all strains are able to make tryptophan, and the ability to make tryptophan appears to track with STI virulence.
Is the C. trachomatis response to tryptophan starvation differentiation to a non-growing cell type or regulated tryptophan biosynthesis? It appears that tryptophan biosynthesis is possible, but under a limited number of circumstances. First, the strain has to encode a functional enzyme. Second, it has to have a source of indole, which the host does not make. Some investigators have noticed that many functional TrpAB-containing strains were isolated from patients with bacterial vaginosis, and thus hypothesize that the vaginal flora might be a source of indole (McClarty et al., 2007). So some C. trachomatis strains can synthesize tryptophan in certain environments, increasing their resistance to IFN-γ mediated immunity (Belland et al., 2003). Where tryptophan biosynthesis is not available, Chlamydia use their ability to enter into a persistent form that enables it to survive when all sources of tryptophan are lost.
M. tuberculosis: energy independence – making its own amino acids in an unreliable environment
Whereas Chlamydia tryptophan biosynthesis is limited in scope, M. tuberculosis seems to leave nothing up to chance. It contains the entire biosynthetic toolset for all 20 amino acids (Cole et al., 1998). During infection of both mice and macrophages, it appears that M. tuberculosis constitutively expresses amino acid biosynthesis genes (Schnappinger et al., 2003; Talaat et al., 2004; 2007; Rohde et al., 2012). Even when grown in vitro, where tryptophan is freely available, the tryptophan biosynthetic locus continues to be expressed (Y. Zhang and E. Rubin, unpubl. data). Thus, M. tuberculosis seems to choose to independently synthesize amino acids regardless of environmental availability (Fig. 1). M. tuberculosis does the same with energetic sources as well – unlike Escherichia coli, M. tuberculosis will continue to metabolize energy- poor carbon sources even in the presence of more energy efficient carbon sources (de Carvalho et al., 2010).
The M. tuberculosis intracellular niche is similar to Chlamydia’s. M. tuberculosis is taken up by phagocytosis and enters into a vacuole that escapes lysosome fusion. As with Chlamydia, there is strong evidence that IFN-γ activation of infected macrophages plays a major role in controlling M. tuberculosis growth (Flynn et al., 1993; Fabri et al., 2011). Unlike Chlamydia, M. tuberculosis is always able to make tryptophan, which is essential for its survival of IDO activation by IFN-γ. Its ability to make other amino acids is also crucial for virulence, as lysine, proline and leucine auxotrophs also fail to grow normally in macrophages and are severely attenuated in mice (Hondalus et al., 2000; Smith et al., 2001; Parish, 2003; Pavelka et al., 2003a). Perhaps because amino acid biosynthesis is constitutive in M. tuberculosis, amino acid biosynthesis has not been seen as a virulence factor. However, these processes are clearly essential and, in fact, could serve as the basis for drug design.
If amino acid production is constitutive, is there any role for amino acid sensing during infection? M. tuberculosis does have a stringent response, and it encodes a RelA homologue, which in E. coli and other organisms responds to amino acid starvation (Primm et al., 2000). ReIA knockouts are attenuated for growth in mice and guinea pigs (Primm et al., 2000; Dahl et al., 2003; Klinkenberg et al., 2010). It is likely that RelA can respond to amino acid levels – this is true in a related species, Mycobacterium smegmatis – but neither RelA sensing of amino acid starvation nor RelA-dependent co-ordination of amino acid synthesis is well documented in M. tuberculosis (Dahl et al., 2005). The stringent response signals through ppGpp, which in E. coli requires DksA to be fully active (Paul et al., 2004; Magnusson et al., 2005). M. tuberculosis encodes CarD, an alternative DksA, which is also required for the stringent response to starvation, and mutants are slightly attenuated in mice (Connolly and Cox, 2009). However, CarD is essential in rich media, suggesting a role outside of the stringent response that might explain its requirement for growth in vivo (Connolly and Cox, 2009; Stallings et al, 2009). Notably, RelA mutants are far less attenuated than amino acid auxo-trophs (Primm et al., 2000). It is most likely that the stringent response in M. tuberculosis is primarily responsible for sensing and responding to other forms of starvation or immune-mediated stress. Thus, M. tuberculosis is an example of a pathogen that has decoupled amino acid metabolism from host availability. It persists in making its own amino acids, making it resistant to host mechanisms of amino acid starvation.
Legionella: host exploitation – extracting amino acids from a stingy host
Not all bacteria are as energy independent as M. tuberculosis. The host, after all, is a tremendous resource for essential nutrients if the pathogen can manage access (Rohmer et al., 2011). In this respect, L. pneumophila has been quite successful. Like M. tuberculosis and Chlamydia, L. pneumophila replicates in an intracellular vacuole, the Legionella-containing vacuole (LCV). The LCV avoids lysosomal fusion and further develops by recruiting mitochondria and rough ER-derived vesicles. L. pneumophila translocates ~ 300 effectors into the host cell cytoplasm via its type IV secretion system to specifically refine its intracellular niche (Al-Quadan et al., 2012).
Unlike its other intracellular pathogen cousins, the evidence seems to suggest that L. pneumophila does not face an amino acid starved milieu. L. pneumophila does employ the stringent response, which is necessary for survival during infection. In fact, the stringent response in L. pneumophila drives the expression of most known virulence factors (Hammer and Swanson, 1999; Molofsky and Swanson, 2004). However, in macrophages, its stringent response machinery does not rely on RelA, suggesting that sensing amino acid starvation is unimportant during infection (Dalebroux et al., 2009; 2010). Instead L. pneumophila responds to fatty acid starvation through another stringent response sensor, SpoT, and uses this as a signal to turn on its intracellular pathogenesis programme (Dalebroux et al., 2009). Amino acid catabolism is necessary for growth in vivo and some evidence points to serine as a key intracellular carbon source for L. pneumophila (Eylert et al., 2010). Finally, a bacterial transporter, PhtA, is used by L. pneumophila as a high-affinity threonine transporter and is necessary for growth in cells, showing that at least one amino acid is acquired from the host (Sauer et al., 2005).
The LCV, then, seems to be an amino acid rich environment designed for a hungry bacterium. How then, does L. pneumophila manipulate host cells, normally stingy with their amino acids, to serve up amino acids into the LCV? After all, it is likely that amino acid starvation programmes are triggered in host cells infected with L. pneumophila. The bacterium has at least two characterized mechanisms of reversing this starvation.
First, L. pneumophila upregulates a host neutral amino acid transporter, SLC1A5, during macrophage infection (Fig. 1) Knock-down of the transporter does not affect host cell viability, but limits Legionella growth (Wieland et al., 2005). Even when cells were grown in amino acid- rich media, an 80% decrease in SLC1A5 expression resulted in ~ 1000-fold decreased L. pneumophila growth (Wieland et al., 2005). The same transporter is specifically upregulated during infection with another intracellular pathogen, Francisella, and is also required for bacterial replication in THP-1 cells (Barel et al., 2012). It is unclear whether SLC1A5 exists on the LCV membrane, the plasma membrane, or both, but it is essential in providing amino acids to L. pneumophila.
Second, a recent study of host–pathogen interactions at the LCV membrane revealed that L. pneumophila utilizes the host proteasome to generate free amino acids for bacterial growth (Price et al., 2011). The authors noted that the LCV is decorated with Lys48-linked polyubiquitinated proteins anchored to the bacterial virulence factor AnkB (Price et al., 2011). These polyubiquitinated proteins recruit the host proteasome, generating small peptides at the LCV membrane. Inhibition of both the proteasome and amino acid-releases peptidases results arrests L. pneumophila growth, a phenomenon that is reversed by the addition of excess free amino acids (Price et al., 2011). Using a standard transcriptional reporter of amino acid starvation (green fluorescent protein fused to the flaA promoter), the authors found that while wild-type L. pneumophila did not experience amino acid starvation during infection – confirming previous stringent response studies – AnkB null mutants did. AnkB growth was not further suppressed by proteasome inhibition, and was rescued by amino acid supplementation. Thus, by recruiting proteins and targeting them for proteasomal degradation, AnkB provides a ready source of amino acids (Price et al., 2011). L. pneumophila actively moulds the LCV into a growth-supporting niche, subverting host amino acid starvation machinery and exploiting host AA-acquisition mechanisms (Al-Quadan et al., 2012).
Concluding remarks
Many of us take a very pathogen-centric view of host defence. However, the vast majority of bacteria encountered by humans are incapable of causing infection. ‘Non-specific’ defences, such as nutrient deprivation, play a large role in eliminating organisms that are not specifically adapted to a pathogenic niche. Pathogens, therefore, have evolved specific mechanisms to subvert these defences and to take advantage of the nutrient-rich human host. Amino acids, necessary for protein synthesis and, sometimes, as a carbon source, are often depleted in the intracellular environment as a means of starving the pathogen. Pathogen responses range from growth suppression to manipulating host pathways to reverse amino acid starvation.
Understanding the interplay between host and pathogen could prove to be useful in designing new therapies (Escaich, 2008; Rasko and Sperandio, 2010). The three bacteria discussed in this review, C. trachomatis, L. pneumophila and M. tuberculosis are major human pathogens that, for various reasons, have proven to be difficult to control. All three represent organisms for which new anti-bacterial therapies are needed. By knowing how our immune system tries to kill these bacteria as well as the bacterial evasion strategies, we could target these processes to synergize with the immune system to kill invading pathogens. For example, blocking tryptophan biosynthesis, could increase IFN-γ-mediated killing of M. tuberculosis. Amino acid biosynthesis genes could be reasonable drug targets. It is already targeted by many herbicides, and a small molecule used to treat mouse Pseudomonas aureginosa infection was shown to have activity against bacterial tryptophan biosynthesis (Epelbaum et al., 1996; Lesic et al., 2007). Furthermore, host tryptophan needs are supplied through the diet, suggesting that, barring off-target effects, small molecules targeting tryptophan synthesis will not be toxic to the host. Alternatively, the susceptibility of Legionella to the inhibition of the proteasome suggests that host-directed therapies could alter that balance in virulence (Price et al., 2011; Al-Quadan et al., 2012). Thus, altering both bacterial and host responses critical for amino acid starvation could provide new avenues for the development of therapeutics.
References
- Al-Quadan T, Price CT, and Abu Kwaik Y. (2012) Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends Microbiol 20: 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barel M, Meibom K, Dubail I, Botella J, and Charbit A (2012) Francisella tularensis regulates the expression of the amino acid transporter SLC1A5 in infected THP-1 human monocytes. Cell Microbiol 14: 1769–1783. [DOI] [PubMed] [Google Scholar]
- Beatty WL, Belanger TA, Desai AA, Morrison RP, and Byrne GI (1994) Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun 62: 3705–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, et al. (2003) Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA 100: 8478–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal A, Nagalingam G, Huch JH, Walker L, Guillemin GJ, Smythe GA, et al. (2012) M. tuberculosis induces potent activation of IDO-1, but this is not essential for the immunological control of infection. PLoS ONE 7: e37314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne GI, Lehmann LK, and Landry GJ (1986) Induction of tryptophan catabolism is the mechanism for gamma- interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun 53: 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HD, Wood H, Crane D, Bailey R, Jones RB, Mabey D, et al. (2003) Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest 111: 1757–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho LP, Zhao H, Dickinson CE, Arango NM, Lima CD, Fischer SM, et al. (2010) Activity-based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase. Chem Biol 17: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji D, and Ojha A (2001) Revisiting the stringent response, ppGpp and starvation signaling. Curr Opin Microbiol 4: 160–165. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. [DOI] [PubMed] [Google Scholar]
- Connolly LE, and Cox JS (2009) CarD tricks and magic spots: mechanisms of stringent control in mycobacteria. Cell Host Microbe 6: 1–2. [DOI] [PubMed] [Google Scholar]
- Dahl JL, Kraus CN, Boshoff HI, Doan B, Foley K, Avarbock D, et al. (2003) The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc Natl Acad Sci USA 100: 10026–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JL, Arora K, Boshoff HI, Whiteford DC, Pacheco SA, Walsh OJ, et al. (2005) The relA homolog of Mycobacterium smegmatis affects cell appearance, viability, and gene expression. J Bacteriol 187: 2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalebroux ZD, Edwards RL, and Swanson MS (2009) SpoT governs Legionella pneumophila differentiation in host macrophages. Mol Microbiol 71: 640–658. [DOI] [PubMed] [Google Scholar]
- Dalebroux ZD, Yagi BF, Sahr T, Buchrieser C, and Swanson MS (2010) Distinct roles of ppGpp and DksAin Legionella pneumophila differentiation. Mol Microbiol 76: 200–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubener W, Spors B, Hucke C, Adam R, Stins M, Kim KS, and Schroten H (2001) Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3- dioxygenase. Infect Immun 69: 6527–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V (2009) Strange bedfellows expose ancient secrets of autophagy in immunity. Immunity 30: 479–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, and Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5: 527–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich W, Dandekar T, Heesemann J, and Goebel W (2010) Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 8: 401–412. [DOI] [PubMed] [Google Scholar]
- Epelbaum S, Chipman D, and Barak Z (1996) Metabolic effects of inhibitors of two enzymes of the branched-chain amino acid pathway in Salmonella typhimurium. J Bacteriol 178: 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escaich S (2008) Antivirulence as a new antibacterial approach for chemotherapy. Curr Opin Chem Biol 12: 400–408. [DOI] [PubMed] [Google Scholar]
- Eylert E, Herrmann V, Jules M, Gillmaier N, Lautner M, Buchrieser C, et al. (2010) Isotopologue profiling of Legionella pneumophila: role of serine and glucose as carbon substrates. J Biol Chem 285: 22232–22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, et al. (2011) Vitamin D is required for IFN- gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 3: 104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre D, Mold J, Hunt PW., Kanwar B, Loke P, Seu L, et al. (2010) Tryptophan catabolism by indoleamine 2,3- dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med 2: 32ra36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlner-Gardiner C, Roshick C, Carlson JH, Hughes S, Belland RJ, Caldwell HD, and McClarty G (2002) Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J Biol Chem 277: 26893–26903. [DOI] [PubMed] [Google Scholar]
- Fields PI, Swanson RV, Haidaris CG, and Heffron F (1986) Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci USA 83: 5189–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, and Bloom BR (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 178: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujigaki S (2002) L-tryptophan-L-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: cross regulation between inducible nitric oxide synthase and indoleamine-2,3-dioxygenase. Infect Immun 70: 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard HC, Stanich JA, Whittum-Hudson JA, Schumacher HR, Carter JD, and Hudson AP (2010) Patients with Chlamydia-associated arthritis have ocular (trachoma), not genital, serovars of C. trachomatis in synovial tissue. Microb Pathog 48: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766. [DOI] [PubMed] [Google Scholar]
- Hammer BK, and Swanson MS (1999) Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol Microbiol 33: 721–731. [DOI] [PubMed] [Google Scholar]
- Hatch TP (1975) Competition between Chlamydia psitacci and L cells for host isoleucine pools: a limiting factor in chlamydial multiplication. Infect Immun 12: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rao SP, Takabayashi K, Van Uden JH, Kornbluth RS, Baird SM, et al. (2001) Enhancement of innate immunity against Mycobacterium avium infection by immunostimulatory DNA is mediated by indoleamine 2,3- dioxygenase. Infect Immun 69: 6156–6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiseth SK, and Stocker BA (1981) Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291: 238–239. [DOI] [PubMed] [Google Scholar]
- Hondalus MK, Bardarov S, Russell R, Chan J, Jacobs WR, and Bloom BR (2000) Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect Immun 68: 2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood MI, and Skaar EP (2012) Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10: 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibana JA, Belland RJ, Zea AH, Schust DJ, Naga-matsu T, AbdelRahman YM, et al. (2011a) Inhibition of indoleamine 2,3-dioxygenase activity by levo-1-methyl tryptophan blocks gamma interferon-induced Chlamydia trachomatis persistence in human epithelial cells. Infect Immun 79: 4425–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibana JA, Belland RJ, Zea AH, Schust DJ, Naga-matsu T, AbdelRahman YM, et al. (2011b) Inhibition of indoleamine 2,3-dioxygenase activity by levo-1-methyl tryptophan blocks gamma interferon-induced Chlamydia trachomatis persistence in human epithelial cells. Infect Immun 79: 4425–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkenberg LG, Lee JH, Bishai WR, and Karakousis PC (2010) The stringent response is required for full virulence of Mycobacterium tuberculosis in guinea pigs. J Infect Dis 202: 1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt RM, Lee S-J, Kavathas PB, and Cresswell P (2007) Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infect Immun 75: 5105–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesic B, Lepine F, Deziel E, Zhang J, Zhang Q, Pad-field K, et al. (2007) Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog 3: 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B (2005) Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120: 159–162. [DOI] [PubMed] [Google Scholar]
- McClarty G, Caldwell HD, and Nelson DE (2007) Chlamydial interferon gamma immune evasion influences infection tropism. Curr Opin Microbiol 10: 47–51. [DOI] [PubMed] [Google Scholar]
- Magnusson LU, Farewell A, and Nyström T (2005) ppGpp: a global regulator in Escherichia coli. Trends Microbiol 13: 236–242. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Shevach EM, Trinchieri G, Mellor AL, Munn DH, Gordon S, et al. (2011) Highlights of 10 years of immunology in Nature Reviews Immunology. Nat Rev Immunol 11: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, and Swanson MS (2004) Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol Microbiol 53: 29–40. [DOI] [PubMed] [Google Scholar]
- Morrison RP (2000) Differential sensitivities of Chlamydia trachomatis strains to inhibitory effects of gamma interferon. Infect Immun 68: 6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, and Mellor AL (2005) GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22: 633–642. [DOI] [PubMed] [Google Scholar]
- Murray HW, Szuro-Sudol A, Wellner D, Oca MJ, Granger AM, Libby DM, et al. (1989) Role of tryptophan degradation in respiratory burst-independent antimicrobial activity of gamma interferon-stimulated human macrophages. Infect Immun 57: 845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan D, Maskell D, Liew FY, Easmon CS, and Dougan G (1988) Characterization of aromatic- and purine-dependent Salmonella typhimurium: attention, persistence, and ability to induce protective immunity in BALB/c mice. Infect Immun 56: 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette SP, Hatch TP, AbdelRahman YM, Rose LA, Belland RJ, and Byrne GI (2006) Global transcriptional upregulation in the absence of increased translation in Chlamydia during IFNgamma-mediated host cell tryptophan starvation. Mol Microbiol 62: 1387–1401. [DOI] [PubMed] [Google Scholar]
- Parish T (2003) Starvation survival response of Mycobacterium tuberculosis. J Bacteriol 185: 6702–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, and Gourse RL (2004) DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118: 311–322. [DOI] [PubMed] [Google Scholar]
- Pavelka MS, Chen B, Kelley CL, Collins FM, and Jacobs WR Jr, (2003a) Vaccine efficacy of a lysine auxotroph of Mycobacterium tuberculosis. Infect Immun 71: 4190–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka MS, Chen B, Kelley CL, Collins FM, and Jacobs WR (2003b) Vaccine efficacy of a lysine auxotroph of Mycobacterium tuberculosis. Infect Immun 71: 4190–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerdá J, and Tedin K (2004) The bacterial signal molecule, ppGpp, regulates Salmonella virulence gene expression. Mol Microbiol 52: 1827–1844. [DOI] [PubMed] [Google Scholar]
- Potrykus K, and Cashel M (2008) (p)ppGpp: still magical? Annu Rev Microbiol 62: 35–51. [DOI] [PubMed] [Google Scholar]
- Price CTD, Al-Quadan T, Santic M, Rosenshine I, and Abu Kwaik Y. (2011) Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science 334: 1553–1557. [DOI] [PubMed] [Google Scholar]
- Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, and Barry CE (2000) The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol 182: 4889–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, and Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 9: 117–128. [DOI] [PubMed] [Google Scholar]
- Rohde KH, Veiga DFT, Caldwell S, Balazsi G, and Russell DG (2012) Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog 8: e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer L, Hocquet D, and Miller SI (2011) Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19: 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin-Bejerano I, Fraser I, Grisafi P, and Fink GR (2003)Phagocytosis by neutrophils induces an amino acid deprivation response in Saccharomyces cerevisiae and Candida albicans. Proc Natl Acad Sci USA 100: 11007–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, and Sabatini D (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010) Regulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J-D, Bachman MA, and Swanson MS (2005) The phagosomal transporter A couples threonine acquisition to differentiation and replication of Legionella pneumophila in macrophages. Proc Natl Acad Sci USA 102: 9924–9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva NM, Rodrigues CV, Santoro MM, Reis LFL, Alvarez-Leite JI, and Gazzinelli RT (2002) Expression of indoleamine 2,3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect Immun 70: 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Davis AS, Taylor GA, and Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313: 1438–1441. [DOI] [PubMed] [Google Scholar]
- Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. (2010) Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12: 1154–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar EP (2010) The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog 6: e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DA, Parish T, Stoker NG, and Bancroft GJ (2001) Characterization of auxotrophic mutants of Mycobacterium tuberculosis and their potential as vaccine candidates. Infect Immun 69: 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Kim H-J, Kim EY, Shin M, Lee HC, Hong Y, et al. (2004) ppGpp-dependent stationary phase induction of genes on Salmonella pathogenicity island 1. J Biol Chem 279: 34183–34190. [DOI] [PubMed] [Google Scholar]
- Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, and Glickman MS (2009) CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138: 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RS (1998) Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282: 754–759. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaat AM, Lyons R, Howard ST, and Johnston SA (2004) The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci USA 101: 4602–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaat AM, Ward SK, Wu C-W, Rondon E, Tavano C, Bannantine JP, et al. (2007) Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J Bacteriol 189: 4265–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I, Sorbara MT, Philpott DJ, and Girardin SE (2012a) Bacterial autophagy: the trigger, the target and the timing. Autophagy 8: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, et al. (2012b) Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11: 563–575. [DOI] [PubMed] [Google Scholar]
- Thompson A, Rolfe MD, Lucchini S, Schwerk P, Hinton JCD, and Tedin K (2006) The bacterial signal molecule, ppGpp, mediates the environmental regulation of both the invasion and intracellular virulence gene programs of Salmonella. J Biol Chem 281: 30112–30121. [DOI] [PubMed] [Google Scholar]
- Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, and Conway T (2008) The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68: 1128–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi G, Wiltshire C, Macaskill S, Tournu H, Budge S, and Brown AJP (2002) Gcn4 co-ordinates morphogenetic and metabolic responses to amino acid starvation in Candida albicans. EMBO J 21: 5448–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal A, Bryk R, Shi S, Rhee K, Rath P, Schnap-pinger D, et al. (2011) Virulence of Mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe 9: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland H, Ullrich S, Lang F, and Neumeister B (2005) Intracellular multiplication of Legionella pneumophila depends on host cell amino acid transporter SLC1A5. Mol Microbiol 55: 1528–1537. [DOI] [PubMed] [Google Scholar]
- Wood H, Fehlner-Gardner C, Berry J, Fischer E, Graham B, Hackstadt T, et al. (2003) Regulation of tryptophan synthase gene expression in Chlamydia trachomatis. Mol Microbiol 49: 1347–1359. [DOI] [PubMed] [Google Scholar]
- Yuk J-M, Shin D-M, Lee H-M, Yang C-S, Jin HS, Kim K-K, et al. (2009) Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6: 231–243. [DOI] [PubMed] [Google Scholar]
- Zelante T, Fallarino F, Bistoni F, Puccetti P, and Romani L (2009) Indoleamine 2,3-dioxygenase in infection: the paradox of an evasive strategy that benefits the host. Microbes Infect 11: 133–141. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. (2008) Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]