

Abstract
MicroRNA-32 (miR-32) is upregulated in colorectal cancer (CRC) tissues; its overexpression leads to increased cell proliferation, migration and invasion, as well as reduced apoptosis of CRC cells, at least partly by inhibiting the target gene phosphatase and tensin homolog. However, the mechanisms of its upregulation have remained elusive. In the present study, the effects of methylation and acetylation on the expression of miR-32 were investigated. The promoter methylation status of miR-32 in the CRC cell lines HT-29 and HCT-116 and the normal colonic epithelial cell line NCM460 was investigated by bisulfate sequencing polymerase chain reaction (BSP). The potential role of methylation and histone acetylation in the regulation of miR-32 expression in CRC cells was investigated using the demethylation reagent 5-aza-2′-deoxycytidine (5-Aza-dC), the histone deacetylase inhibitor trichostatin A (TSA) and transfection of DNA methyltransferase 1 (DNMT1) overexpression plasmid. BSP revealed that CpG sites in the miR-32 promoter region of CRC and normal colonic epithelial cells were all hypomethylated, with methylation rates of 0.12, 1.14 and 0.64% in HCT-116, HT-29 and NCM460 cells, respectively. Treatment with 5-Aza-dC and/or TSA and transfection with DNMT1 plasmid did not significantly alter the expression of miR-32. Therefore, the present results suggest that methylation and histone acetylation do not affect miR-32 expression in CRC cells.
Keywords: microRNA-32, colorectal cancer, promoter, methylation, histone acetylation
Introduction
Colorectal cancer (CRC) is a leading cause of cancer and associated mortalities worldwide. With the improvement in living standards and change in lifestyle and dietary structure, the incidence of CRC has demonstrated annual increases. CRC arises from the accumulation of genetic alterations in colonic epithelial cell and in the majority of cases, the cancer develops via the normal adenoma-carcinoma sequence (1). MicroRNAs (miRNAs/miRs) are a class of small, endogenous, non-coding, regulatory RNA molecules that inhibit gene translation after forming a complex referred to as the RNA-interference-induced silencing complex or after induction by mRNA cleavage (2). miRNAs may function as oncogenes by targeting tumor suppressor genes or as tumor suppressors by either inhibiting cellular oncogene expression or regulating cell apoptosis, as well as tumor occurrence and development (3,4).
DNA methylation is a type of epigenetic modification that refers to changes in gene expression without alteration of the gene sequence. Under DNA methyltransferase (DNMT) catalysis, a methyl group is covalently bound to the 5′carbon of cytosine in the genomic CpG dinucleotide, which is unstable and may spontaneously undergo deamination to form thymine, thereby affecting gene expression (5). Aberrant methylation, including DNA hypomethylation and/or promoter gene CpG hypermethylation, is implicated in the development of a variety of tumor types, including CRC (6). The expression of miR-145 in CRC cells has been reported to be either activated or inhibited by the methylation of histones at the core promoter regions in the upper regulatory sequence of pre-miR-145 (7). miR-663 expression was downregulated and the promoter was consistently and significantly methylated in human myeloid leukemia cell lines; following treatment with the demethylation reagent 5-aza-2′-deoxycytidine (5-Aza-dC), miR-663 expression was significantly upregulated (8). Another epigenetic phenomenon is acetylation of histones, which is regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC). HDAC is able to remove the acetyl group from the histone lysine residue, increase the DNA-binding ability of histones and make the promoter less accessible to transcriptional regulatory elements, finally causing transcriptional repression. The effect of HAT is, however, just the opposite. Together, they synergistically maintain the normal acetylation level of histone (9). HDAC inhibition with suberoylanilide hydroxamic acid increased acetylated histone enrichment on the Cdc20b/miR-449a promoter, consequently upregulating miR-449a levels during diabetes (10). Together, these studies suggested the susceptibility of miRNAs to epigenetic regulation.
miR-32 is an intronic miRNA, located within intron 14 of C9orf5, the gene encoding transmembrane protein 245 (TMEM245). In previous studies by our group, miR-32 was identified to be upregulated in CRC tissues (11,12). Overexpression of miR-32 led to increased cell proliferation, migration and invasion, and reduced apoptosis of CRC cells, by inhibiting the target gene phosphatase and tensin homolog. However, the mechanism underlying the increase in miR-32 levels remains elusive. In the present study, the methylation status of miR-32 promoter in CRC and normal colonic epithelial cells was investigated using bisulfate sequencing polymerase chain reaction (PCR) (BSP), and the role of methylation and histone acetylation in the regulation of miR-32 expression in CRC cells was examined.
Materials and methods
Cell culture
The CRC cell lines HT-29 and HCT-116 (Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI-1640 medium (GE Healthcare Life Sciences, Little Chalfont, UK) and the normal colonic epithelial cell line NCM460 (BeNa Culture Collection, Jiangsu, China) was cultured in Dulbecco's modified Eagle's medium/F12 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), each supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 IU/ml penicillin and 100 µg/ml streptomycin, at 37°C in a humidified atmosphere with 5% CO2.
BSP of CpG islands in the miR-32 promoter
Genomic DNA was extracted from the HT-29, HCT-116 and NCM460 cells using the TIANamp Genomic DNA kit (cat. no. DP304; Tiangen Biotech, Beijing, China). It was subjected to bisulfite modification using the EpiTect Bisulfite kit (cat. no. 59826; Qiagen GmBH, Hilden, Germany), according to the manufacturer's protocol. The target region of the promoter of TMEM245, the host gene of miR-32, containing 94 CpG sites, located between −195 and +521 bp numbered from the start codon ATG, was amplified by PCR using DreamTaq Hot Start DNA Polymerase (Thermo Fisher Scientific, Inc.) and specific primers (Fig. 1; Table I). The primers were designed by MethPrimer (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi) and synthesized by YINGBIO technology Co., Ltd. (Shanghai, China). The PCR products were purified and sub-cloned to T vector (T-Vector pMD 19; cat. no. 3271, Takara Bio Inc., Otsu, Japan). Colony PCR was performed using M13 primers (forward, 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′ and reverse, 5′-AGCGGATAACAATTTCACACAGGA-3′). A total of 10 independent colonies for each tested region were selected and sequenced, and the extent of methylation was assessed by identifying the number and position of methylated cytosine residues.
Figure 1.

The sequence in bold letters is the CpG island in the promoter region of the TMEM245 gene, as predicted by MethPrimer. A total of 94 CpG sites (underlined) were identified.
Table I.
Primer sequences for bisulfite sequencing.
| Name | Sequence |
|---|---|
| F1 | 5′-TTTTGTAAATTTGTTAGTAGGATTTTG-3′ |
| R1 | 5′-TCCAAAATAAAATAAACCAACACC-3′ |
| F2 | 5′-TGTTGGTTTATTTTATTTTGGAGG-3′ |
| R2 | 5′-TCACCCACAAACTACTAAAATAATCC-3′ |
The target region of miR-32 promoter, containing 94 CpGs, was divided into two parts, and each part was amplified by polymerase chain reaction using the abovementioned primers. F, forward; R, reverse.
5-Aza-dC and trichostatin A (TSA) treatment
HT-29 cells were incubated for 72 h in media containing 10 µM of the demethylating agent 5-Aza-dC (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The culture medium was replaced with new medium containing 5-Aza-dC every 24 h. For inhibition of histone deacetylation, 1 µM TSA (Sigma-Aldrich; Merck KGaA), which specifically inhibits HDAC, was added to the medium for 24 h. For combined inhibition, the cells were incubated with 10 µM 5-Aza-dC for 48 h, followed by an additional 24 h with 1 µM TSA, as described previously (13,14). Untreated HT-29 cells were used as a control. Total RNA was purified and subjected to reverse transcription-quantitative (RT-q)PCR for evaluating miR-32 expression.
Cell transfection
DNMT1 overexpression plasmid pRP-hDNMT1 was synthesized by VectorBuilder Inc. (Guangzhou, China). HT-29 cells were seeded onto 6-well plates, at a density of 3×105 cells per well. The cells were cultured at 37°C in a 5% CO2 incubator in antibiotic-free medium (RPMI-1640 medium with 10% fetal bovine serum) 24 h prior to transfection. HT-29 cells were transfected with 2 µg of pRP-hDNMT1 or pRP vector as a negative control. Cells were transfected using Lipofectamine 2000™ reagent (Thermo Fisher Scientific, Inc.) in Opti-MEM I (Gibco; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. The relative levels of miR-32 in transfected cells were examined by RT-qPCR.
RT-qPCR
The expression of miR-32 in various treated cells (as described above) was evaluated using RT-qPCR. Total RNA was extracted with RNAiso Plus (Takara Bio Inc.), and reverse transcribed using the Mir-X miRNA First Strand Synthesis kit (cat. no. 638315; Clontech Laboratories, Inc., Mountainview, CA, USA). U6 small nuclear RNA was used as an internal control for miR-32. PCR was performed using TB Green Premix Ex Taq II (Tli RNaseH Plus; cat. no. RR820A; Takara Bio, Inc.) in a LightCycler 480 II system (Roche Diagnostics, Basel, Switzerland). Sequence-specific forward primers for mature miR-32 and U6 internal control were 5′-CGGTATTGCACATTACTAAGTTGCA-3′ and 5′-CTCGCTTCGGCAGCACA-3′, respectively. The primers were synthesized by Sangon Biotech (Shanghai, China); mRQ 3′ Primer was included in the Mir-X miRNA First Strand Synthesis kit. The PCR conditions were 30 sec at 95°C, followed by 40 cycles of 5 sec at 95°C and 20 sec at 60°C. The relative levels of miR-32 were determined using the 2−∆∆Cq method (15).
Statistical analysis
5-Aza-dC and TSA treatments as well as cell transfections were repeated in triplicate. Values are expressed as the mean ± standard deviation. Student's t-test was used for comparison between two groups, while multiple-group comparisons were performed using analysis of variance. All analyses were performed with SPSS 19.0 (IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate statistical significance.
Results
Methylation analysis of the miR-32 promoter in CRC cells and normal colonic epithelial cells
To identify promoters harboring CpG islands, a Bioinformatics search of the promoter of TMEM245, the host gene of miR-32, was performed. Analysis of the TMEM245 promoter sequence, predicted by MethPrimer, identified one CpG island (Fig. 2A). The methylation status of the CpG sites was examined by BSP. As presented in Fig. 2B, CpG sites in the promoter region were rarely methylated (solid circles) in HCT-116 cells in all of the randomly selected clones, with a methylation rate of 0.12%. The CpG sites were also observed to be rarely methylated in clones derived from HT-29 and NCM460 cells, with methylation rates of 1.14 and 0.64 %, respectively (Fig. 3A and B).
Figure 2.

Bisulfate sequencing PCR of the promoter region of microRNA-32. (A) CpG island regions identified in the promoter of the host gene transmembrane protein 245. (B) The methylation status of 94 CpG sites in the promoter was investigated using genomic DNA from HCT-116. A total of 10 individual clones were randomly picked for sequencing. The figures below each column of the circle represents the position of the CG site in the sequence. Solid and hollow circles represent methylated and unmethylated sites, respectively. ‘X’ denotes unmethylated sites, as continuous repeated bases appeared in some regions following bisulfite modifications, so success rate was reduced when detected by this instrument. These regions were thought as ‘base loss’ by the instrument. PCR, polymerase chain reaction.
Figure 3.

Bisulfate sequencing polymerase chain reaction analysis of the promoter region of microRNA-32. The methylation status of 94 CpG sites in the promoter was assessed using genomic DNA from the (A) HT-29 and (B) NCM460 cell lines. A total of 10 individual clones were randomly picked for sequencing. The figures below each column of the circle represents the position of the CG site in the sequence. Solid and hollow circles represent methylated and unmethylated sites, respectively.
miR-32 expression following 5-Aza-dC and/or TSA treatment
To determine the effect of methylation and histone deacetylation on the expression levels of miR-32, HT-29 cells were treated with 5-Aza-dC and/or TSA. Compared with that in untreated controls, treatment with 5-Aza-dC or TSA alone or in combination exerted no marked effect on the expression of miR-32 (P>0.05; Fig. 4).
Figure 4.
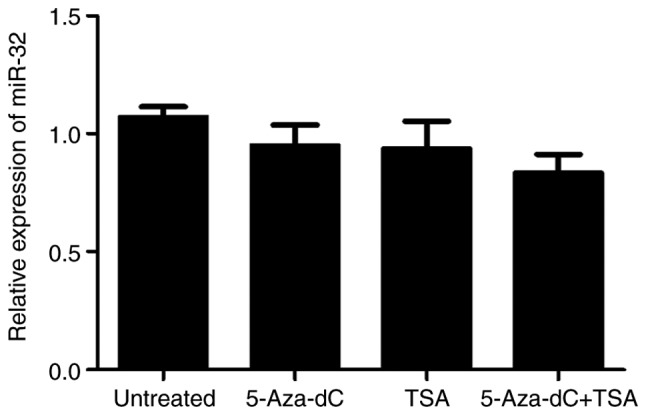
miR-32 expression levels in HT-29 cells treated with 5-Aza-dC and/or TSA. Values are expressed as the mean ± standard deviation from three independent experiments. P>0.05. TSA, trichostatin A; miR, microRNA; 5-Aza-dC, 5-aza-2′-deoxycytidine.
miR-32 expression following transfection with DNMT1 plasmid
HT-29 cells were transfected with DNMT1 plasmid to enhance methylation. The RT-qPCR results indicated that DNMT1 expression was significantly higher in HT-29 cells transfected with pRP-DNMT1 compared with that in cells transfected with pRP vector (P<0.05; Fig. 5A). However, transfection with pRP-DNMT1 did not change the expression level of miR-32. Together, the results suggested that miR-32 expression is not markedly affected by DNA methylation or histone deacetylation in HT-29 cells (P>0.05; Fig. 5B).
Figure 5.

Effect of DNMT1 plasmid on miR-32 expression. (A) DNMT1 expression and (B) miR-32 expression in HT-29 cells after transfection with pRP-DNMT1 or pRP vector (negative control); Values are expressed as the mean ± standard deviation from three independent experiments. *P<0.05. miR, microRNA; DNMT1, DNA methyltransferase 1.
Discussion
DNA methylation and histone acetylation represent two types of epigenetic modification that regulate chromatin structure and gene transcription. The CpG dinucleotides tend to cluster in regions called CpG islands. DNA methylation mostly occurs on CpG islands, with 60% of genes in mammalian genomes containing promoter-proximal CpG islands (16). CpG island methylation in those regions is an important mechanism of gene expression regulation (16), and may be involved in the regulation of miRNA expression.
Dysregulation of miRNA expression in different cancer types may be attributed to the improper binding of transcription factors to response elements of the promoter regions, and to epigenetic changes, including aberrant DNA methylation and histone modification (17). Numerous miRNAs are epigenetically regulated by DNA methylation and/or histone modification (18). Xia et al (19) reported that decreased miR-98 levels in glioma tissues and cell lines are associated with DNA methylation. Treatment with 5-Aza-dC significantly increased the expression of miR-98 in glioma cells. Furthermore, downregulation of miR-200b was reported to be associated with CpG island hypermethylation in bladder cancer cells and pharmacological de-methylation using 5-Aza-dC was able to restore miR-200b expression (20). miR-21 upregulation, induced by histone acetylation, was even significantly elevated after TSA treatment (21). Another study indicated that miR-214 was upregulated in HeLa cells upon treatment with 5-Aza-dC and/or TSA; in fact, the most significant increase of miR-214 was seen in HeLa cells treated with 5-Aza-dC and TSA simultaneously (22).
miR-32 is an intronic miRNA encoded by TMEM245 (23). Intragenic miRNAs are reported to be embedded within the host gene and thought to be regulated by the host gene promoter (24). They are transcribed in parallel with their host transcripts (25). Human papillomavirus type16 protein E6 may indirectly regulate miR-23b, an intronic miRNA, through the methylation of its host gene C9orf3 (26). Chen et al (27) indicated that TSA caused an upregulation of the expression of miR-15a/16-1, residing in the host tumor suppressor Dleu2 gene, by increasing the histone acetylation in the promoter region of Dleu2/miR-15a/16-1 in lung cancer cells. Yang et al (28) indicated that SAHA, an HDAC inhibitor, repressed the transcription of miR-20a, miR-93 and miR-106b by repressing their host genes (miR-17–92 cluster and minichromosome maintenance complex component 7) in hepatocellular carcinoma cells. The intronic miRNA miR-449a and the host gene cell division cycle (Cdc)20b were highly upregulated in response to HDAC inhibition. Bioinformatics analyses identified a common promoter for Cdc20b and miR-449a, featuring significant histone acetylation, which was further verified by augmented occupancy of acetylated histones on the Cdc20b/miR-449a promoter in SAHA-treated C2C12 cells (10). It was also reported that the transcript levels of C9orf5 (TMEM245) and miR-32 are positively and significantly correlated (23).
The present study examined whether miR-32 expression is affected by epigenetic mechanisms, namely DNA methylation and histone acetylation. The level of miR-32 promoter methylation was identified to be low, with a methylation rate of 0.12, 1.14 and 0.64% in HCT-116, HT-29 and NCM460 cells, respectively. Regardless of whether normal colonic mucosal cells or CRC cells were observed, the promoter of miR-32 was hypomethylated, and the present study aimed to further clarify whether methylation has a significant effect on the expression of miR-32. For this, the levels of miR-32 expression after treatment with 5-Aza-dC and/or TSA were determined by RT-qPCR, and the results demonstrated no significant effect. Furthermore, DNMT1 plasmid transfection had no effect on the expression levels of miR-32. It therefore appears unlikely that these epigenetic modifiers are involved in the regulation of miR-32 expression, but these results require to be verified in other CRC cell lines and in CRC tissues in future studies. Since this type of epigenetic modification appears to have no effect on the expression of miR-32, it is further required to clarify whether other regulatory pathways, including transcription factors or interaction with non-coding RNAs, are able to regulate miR-32 expression.
Acknowledgements
Not applicable.
Funding
This study was supported by the Guangdong Natural Science Foundation of China (grant no. 2017A030313546).
Availability of data and materials
Available from the corresponding authors on reasonable request.
Authors' contributions
WW and ZY designed the experiments. WW, YS and TW performed the experiments. WW and QJ performed statistical analysis of the data obtained and drafted the manuscript. ZY revised the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 2.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Liu J, Wang G. The role of microRNAs in human breast cancer progression. Tumour Biol. 2014;35:6235–6344. doi: 10.1007/s13277-014-2202-8. [DOI] [PubMed] [Google Scholar]
- 4.Zheng Q, Chen C, Guan H, Kang W, Yu C. Prognostic role of microRNAs in human gastrointestinal cancer: A systematic review and meta-analysis. Oncotarget. 2017;8:46611–46623. doi: 10.18632/oncotarget.16679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tse JWT, Jenkins LJ, Chionh F, Mariadason JM. Aberrant DNA Methylation in colorectal cancer: What should we target? Trends Cancer. 2017;3:698–712. doi: 10.1016/j.trecan.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Ji G, Xiao X, Chen X, Qin WW, Yang F, Li YF, Fan LN, Xi WJ, Huo Y, et al. Epigenetically regulated miR-145 suppresses colon cancer invasion and metastasis by targeting LASP1. Oncotarget. 2016;7:68674–68687. doi: 10.18632/oncotarget.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan-Fang T, Jian N, Jun L, Na W, Pei-Fang X, Wen-Li Z, Dong W, Li P, Jian W, Xing F, Jian P. The promoter of miR-663 is hypermethylated in Chinese pediatric acute myeloid leukemia (AML) BMC Med Genet. 2013;14:74. doi: 10.1186/1471-2350-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Poddar S, Kesharwani D, Datta M. Histone deacetylase inhibition regulates miR-449a levels in skeletal muscle cells. Epigenetics. 2016;11:579–587. doi: 10.1080/15592294.2016.1188247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu W, Yang J, Feng X, Wang H, Ye S, Yang P, Tan W, Wei G, Zhou Y. MicroRNA-32 (miR-32) regulates phosphatase and tensin homologue (PTEN) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol Cancer. 2013;12:30. doi: 10.1186/1476-4598-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu W, Yang P, Feng X, Wang H, Qiu Y, Tian T, He Y, Yu C, Yang J, Ye S, Zhou Y. The relationship between and clinical significance of MicroRNA-32 and phosphatase and tensin homologue expression in colorectal cancer. Genes Chromosomes Cancer. 2013;52:1130–1140. doi: 10.1002/gcc.22108. [DOI] [PubMed] [Google Scholar]
- 13.Mizuno Y, Maemura K, Tanaka Y, Hirata A, Futaki S, Hamamoto H, Taniguchi K, Hayashi M, Uchiyama K, Shibata MA, et al. Expression of delta-like 3 is downregulated by aberrant DNA methylation and histone modification in hepatocellular carcinoma. Oncol Rep. 2018;39:2209–2216. doi: 10.3892/or.2018.6293. [DOI] [PubMed] [Google Scholar]
- 14.Guo LH, Li H, Wang F, Yu J, He JS. The tumor suppressor roles of miR-433 and miR-127 in gastric cancer. Int J Mol Sci. 2013;14:14171–14184. doi: 10.3390/ijms140714171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Weber M, Schubeler D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–280. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31:1609–1622. doi: 10.1038/onc.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Yao H, Lin S, Zhu X, Shen Z, Lu G, Poon WS, Xie D, Lin MC, Kung HF. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett. 2013;331:1–10. doi: 10.1016/j.canlet.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Xia Z, Qiu D, Deng J, Jiao X, Yang R, Sun Z, Wan X, Li J. Methylation-induced downregulation and tumor-suppressive role of microRNA-98 in glioma through targeting Sal-like protein 4. Int J Mol Med. 2018;41:2651–2659. doi: 10.3892/ijmm.2018.3464. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Shindo T, Niinuma T, Nishiyama N, Shinkai N, Kitajima H, Kai M, Maruyama R, Tokino T, Masumori N, Suzuki H. Epigenetic silencing of miR-200b is associated with cisplatin resistance in bladder cancer. Oncotarget. 2018;9:24457–24469. doi: 10.18632/oncotarget.25326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song WF, Wang L, Huang WY, Cai X, Cui JJ, Wang LW. MiR-21 upregulation induced by promoter zone histone acetylation is associated with chemoresistance to gemcitabine and enhanced malignancy of pancreatic cancer cells. Asian Pac J Cancer Prev. 2013;14:7529–7536. doi: 10.7314/APJCP.2013.14.12.7529. [DOI] [PubMed] [Google Scholar]
- 22.Wang F, Liu M, Li X, Tang H. MiR-214 reduces cell survival and enhances cisplatin-induced cytotoxicity via down-regulation of Bcl2l2 in cervical cancer cells. FEBS Lett. 2013;587:488–495. doi: 10.1016/j.febslet.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdogan B, Bosompem A, Peng D, Han L, Smith E, Kennedy ME, Alford CE, Wu H, Zhao Z, Mosse CA, et al. Methylation of promoters of microRNAs and their host genes in myelodysplastic syndromes. Leuk Lymphoma. 2013;54:2720–2727. doi: 10.3109/10428194.2013.790542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeung CL, Tsang TY, Yau PL, Kwok TT. Human papillomavirus type 16 E6 suppresses microRNA-23b expression in human cervical cancer cells through DNA methylation of the host gene C9orf3. Oncotarget. 2017;8:12158–12173. doi: 10.18632/oncotarget.14555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CQ, Chen CS, Chen JJ, Zhou LP, Xu HL, Jin WW, Wu JB, Gao SM. Histone deacetylases inhibitor trichostatin A increases the expression of Dleu2/miR-15a/16-1 via HDAC3 in non-small cell lung cancer. Mol Cell Biochem. 2013;383:137–148. doi: 10.1007/s11010-013-1762-z. [DOI] [PubMed] [Google Scholar]
- 28.Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, Tian Z, Zhang C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17–92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer. 2015;112:112–121. doi: 10.1038/bjc.2014.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Available from the corresponding authors on reasonable request.