

Abstract
Background
Proton pump inhibitors (PPIs) are frequently used to prevent or treat peptic ulcers. Recently, PPIs have been shown to increase the risk of myocardial infarction. The purpose of this study was to determine whether PPIs adversely affect ventricular remodeling in infarcted rats.
Methods
Male Wistar rats were randomly assigned to receive either vehicle, omeprazole, omeprazole + vitamin C, omeprazole + olmesartan, or famotidine treatment for 4 weeks starting 24 hours after inducing myocardial infarction by ligating coronary arteries.
Results
Compared with vehicle-treated infarcted rats, omeprazole-treated infarcted rats had significant changes with reduced myocardial vitamin C levels, increased oxidant production, and decreased dimethylarginine dimethylaminohydrolase 2 (DDAH2) activity, which in turn increased asymmetric dimethylarginine (ADMA) levels and impaired ventricular remodeling. With gastric protection similar to omeprazole, the H2 blocker famotidine had no effect on ventricular remodeling. In contrast to the in vivo results, the ex vivo study showed similar superoxide and DDAH2 protein levels between vehicle- and omeprazole-treated infarcted rats, suggesting involvement of gastric vitamin C uptake rather than myocardial vitamin C in mediating the impaired axis of vitamin C-superoxide-DDAH2 in the in vivo measurements. The administration of PPIs was associated with impaired DDAH2 expression and increased myocardial ADMA, which impaired ventricular remodeling after infarction. These effects were prevented by the coadministration of vitamin C or olmesartan.
Conclusions
Our results indicate that the administration of PPIs was associated with impaired DDAH2 expression and increased myocardial ADMA by reducing gastric vitamin C uptake, which impaired ventricular remodeling after infarction.
Keywords: Asymmetric dimethylarginine, Dimethylarginine dimethylaminohydrolase, Myocardial infarction, Superoxide anion, Ventricular remodeling
INTRODUCTION
Proton pump inhibitors (PPIs) are frequently used to prevent or treat peptic ulcers, especially in patients with acute coronary syndrome who need dual antiplatelet treatment.1 However, their safety has never been approved by regulatory authorities after myocardial infarction (MI). A few epidemiological studies have reported mixed results regarding the association between PPI use and cardiovascular events. Very recently, a data-mining study showed that PPIs might be associated with an elevated risk of MI in the general population.2 In contrast, clinical studies have not shown an association between PPI use and cardiovascular events.3 The reason for this discrepancy remains unclear, and it is important to understand the limitations of such observational studies. Much of the current evidence linking PPI use to serious long-term adverse consequences is weak and insubstantial.
PPIs inhibit gastric H+/K+-adenosine triphosphatase (H+/K+-ATPase). It is known that the effects of PPIs are not strictly limited to gastric parietal cells, and competitive reverse transcription-polymerase chain reactions (RT-PCRs) using probes derived from specific gastric H+/ K+-ATPase complementary DNA (cDNA) have shown functional H+/K+-ATPase in rat hearts.3 Furthermore, myocardial H+/K+-ATPase activity is upregulated after MI.4 PPIs have been shown to markedly reduce the proportion of vitamin C in its biologically active antioxidant form of ascorbic acid,5 because PPIs cause a more marked and sustained rise in intragastric pH compared with H2 blockers (H2B),6 and this contributes to the instability and degradation of vitamin C. Vitamin C acts as a potent water-soluble antioxidant in biological fluids by scavenging physiologically relevant reactive oxygen species (ROS) such as superoxide anions. The data on PPIs and ROS levels are inconsistent and conflicting, and some studies have demonstrated a positive correlation7 while others have not.8,9 It has been reported that PPIs inhibit the proliferation of tumor cells by increasing the production of ROS.7 On the other hand, Biswas and co-workers7 showed that omeprazole acts as a scavenger of hydroxyl radicals, through which it plays a significant role in gastroprotection. Differences in measurements of ROS and cell types could contribute to the discrepancies. However, the effect of PPIs on myocardial ROS remains unclear. Furthermore, to date, neither experimental nor clinical studies have shown the effect of PPIs on ventricular remodeling, a condition of increased ROS levels.10
Asymmetric dimethylarginine (ADMA) is an endogenous nitric oxide synthase (NOS) inhibitor that blocks nitric oxide (NO) production. ADMA is eliminated principally by dimethylarginine dimethylaminohydrolase (DDAH) with a small contribution from renal excretion. It has been reported that DDAH activity is affected by a posttranslational modification mediated by ROS.11 The sensitivity of DDAH to oxidative stress is conferred by a critical sulfhydryl in the active site of the enzyme that is required for the metabolism of ADMA. Two isoforms of DDAH have been identified: DDAH1 is predominantly expressed in tissues containing nNOS, and DDAH2 is mainly expressed in cardiovascular tissues expressing eNOS or iNOS.12 Therefore, DDAH2 is rich in cardiac tissue and mainly responsible for the degradation of ADMA. Plasma and tissue levels of ADMA have been shown to be higher following MI, and this may be a mechanism to regulate NOS activity.13 Increased plasma ADMA has ben reported to be a predictor of mortality in patients after MI.12
Cardiac remodeling is an unfavorable evolution associated with myocardial hypertrophy, fibrosis and ventricular dysfunction after MI.14 The postinfarction remodeling process involves increased intracellular oxidative stress through multiple mechanisms. We previously showed that pharmacological interventions to scavenge ROS can ameliorate cardiac remodeling after MI.10,15 However, there is currently no evidence indicating the impact of PPIs on ventricular remodeling after infarction. Recently, Ghebremariam et al.16 showed that PPIs were associated with the inhibition of DDAH activity and thus increased plasma ADMA levels. However, these experiments did not address the possible pathways by which DDAH activity was inhibited by PPIs. Given that PPI therapy can induce hypochlorhydria resulting in low levels of gastric juice ascorbate and subsequent increases in ROS levels, we tested the hypothesis that PPI-induced oxidative stress impairs DDAH activity through a vitamin C-dependent pathway. To further explain why there are no reports regarding the adverse effects of PPIs on cardiac remodeling in a clinical setting after MI for which angiotensin receptor blockers (ARBs) are suggested,17 we also assessed infarcted rats treated with ARBs.
METHODS
Animals
All experiments were performed according to the Animal Protection Law of the Council of Agriculture, Executive Yuan and the Animal Welfare Act. The Institutional Animal Care and Use Committee of China Medical University reviewed and approved the research protocols (approval number: 2016-316), and the protocols conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication 2011). In addition, all animal care and experimental procedures complied with the ARRIVE guidelines.18 Male Wistar rats aged 8 weeks (220-250 g) were maintained in a 12 h light/12 h dark cycle (lights on from 08 h to 20 h) with ad libitum access to food and water.
Part 1: in vivo
Male Wistar rats were subjected to ligation of the anterior descending artery as previously described,14 resulting in infarction of the left ventricular (LV) free wall. The rats were randomly assigned to receive either vehicle (saline), omeprazole (5 mg/kg body weight per day), omeprazole + vitamin C (200 mg/kg body weight per day), omeprazole + olmesartan (0.01 mg/kg body weight per day), or famotidine (a H2B, 3 mg/kg body weight per day). These doses of vitamin C19 and famotidine20 have been shown to effectively modulate biological activities. For clinical relevance, the dose of omeprazole was based on an oral human dose of 40 mg normalized by body surface area, and this dose was smaller compared with a previous study.20 We previously demonstrated that a low dose of olmesartan can improve ventricular remodeling without significantly changing blood pressure.21
The drugs were started 24 hours after infarction, at a time when they could produce maximum benefits.22 The study duration was designed to be 4 weeks because the majority of the myocardial remodeling process in rats (70-80%) is completed within 3 weeks.22 The drugs were administered by daily oral gavage. Sham-operated rats served as controls to exclude the possibility that the drugs themselves directly modulated ventricular remodeling. In each treated group, the drugs were withdrawn about 24 hours before the end of the experiments in order to eliminate their pharmacological actions.
Part 2: ex vivo
Although results of the above study showed that PPIs significantly inhibited DDAH activity (see Results), the involved mechanism remained unclear. To determine whether the effect of PPIs on DDAH was mediated directly by inhibiting myocardial DDAH activity or whether its effect indirectly depended on a vitamin C-mediated ROS pathway, we performed an ex vivo experiment in which the effect of gastric pH on vitamin C uptake could be excluded. Four weeks after the induction of MI by coronary ligation, infarcted rat hearts were isolated and subjected to no treatment (vehicle), omeprazole (2 mg/ml), famotidine (25 mg/ml), and omeprazole + vitamin C (5 μM). The hearts were perfused with noncirculating modified Tyrode’s solution containing (in mM): glucose 5.5, NaCl 117.0, NaHCO3 23.0, KCl 4.6, NaH2PO4 0.8, MgCl2 1.0, and CaCl2 2.0, equilibrated at 37 °C and oxygenated with a 95% O2 to 5% CO2 gas mixture. The drugs were infused for 60 minutes. At the end of the study, all hearts (n = 5 in each group) were used for Western analysis for DDAH2 and immunohistochemical staining for dihydroethidium (DHE) at the border zone (< 2 mm outside the infarct).
Echocardiography
At 28 days after the operation, the rats were lightly anesthetized with intraperitoneal injections of ketamine HCl (25 mg/kg). Echocardiographic measurements were done using a GE Healthcare Vivid 7 Ultra-sound System (Milwaukee, WI) equipped with a 14-MHz probe as previously described.23 M-mode tracing of the LV was obtained from the parasternal long-axis view to measure LV end-diastolic diameter dimension (LVEDD) and LV end-systolic diameter dimension (LVESD), and fractional shortening (FS) (%) was calculated according to the American Society for Echocardiology. Measurements represented the mean of at least five consecutive cardiac cycles. Analyses were performed by an experienced observer blinded to the treatment groups to which the animals were allocated. Subsequently, hemodynamic measurement were quickly performed after systemic heparinization.
Hemodynamics and infarct size measurements
Hemodynamic parameters were measured in anesthetized rats with an additional intraperitoneal dose of ketamine-xylazine (90 mg/kg-9 mg/kg) at the end of the echocardiogram. A polyethylene Millar catheter was inserted into the LV and connected to a transducer (Model SPR-407, Miller Instruments, Houston, TX) to measure LV systolic and diastolic pressure as the mean of measurements of five consecutive pressure cycles as previously described.23 The maximal rates of LV pressure increase (+dP/dt) and decrease (-dP/dt) were measured. After arterial pressure measurements, the atria and right ventricle were trimmed off, and the LV was rinsed in cold physiological saline, weighed, and immediately frozen in liquid nitrogen after obtaining a coronal section of the LV to estimate the infarct size. A section, taken from the equator of the LV, was fixed in 10% formalin and embedded in paraffin to determine the infarct size. Each section was stained with hematoxylin and eosin, and trichrome. The infarct size was determined as previously described.23 With respect to clinical importance, only rats with a large infarction (> 30%) were selected for analysis.
Morphometric determination of myocyte size
Because ventricular remodeling after infarction is a combination of reactive fibrosis and myocyte hypertrophy, we measured the size of cardiomyocytes in addition to myocardial weight to avoid the confounding influence of nonmyocytes on cardiac hypertrophy. LV sections from the border zone were stained with hematoxylin and eosin. To obtain consistent results, myocytes positioned perpendicularly to the plane of the section with a visible nucleus and cell membrane clearly outlined and unbroken were then selected for cross-sectional area measurements.21 This area was determined by manually tracing the cell contour on a digitized image acquired on an image-analysis system at a magnification of 400 using computerized planimetry (Image Pro Plus) as previously described.24 A total of 100 myocytes were selected in the LV of each heart and analyzed by an observer blinded to the experimental treatment.
In situ detection of superoxide and nitrotyrosine
To evaluate myocardial intracellular superoxide production using in situ dihydroethidium (DHE) (1 μM, Invitrogen Molecular Probes, Eugene, OR, USA) fluorescence, paraffin-embedded tissues (5 μm) were incubated with DHE in phosphate buffered saline (PBS) (10 mM) in a dark, humidified container at room temperature for 30 minutes. Generation of superoxide radicals by the tissue was demonstrated by a red fluorescent signal, and the density of the images was reported as arbitrary units per millimeter square field.10
To demonstrate nitrative stress, nitrotyrosine was detected in LV myocardial sections by immunohistochemistry from the remote zone. Nitrotyrosine has been identified as a biomarker for the formation of peroxynitrite, the by-product of •NO and O2•–. After antigen retrieval and quenching of endogenous peroxidase, slides were immunostained with a rabbit anti-nitrotyrosine antibody (1:200, Millipore, Bedford, MA, USA) overnight at 4 °C.
Western blot analysis of DDAH2, phosphorylated eNOS (Ser1177) and total eNOS
Samples obtained from the border zone underwent Western blot analysis as previously described.9 The primary antibodies were DDAH2 (Abcam Inc., Cambridge, USA), phosphorylated-eNOS (p-eNOS, ser1177; Cell Signaling Technology), eNOS (Cell Signaling Technology) and β-actin (Santa Cruz Biotechnology). Experiments were replicated three times, and the results were expressed as the mean value.
Real-time polymerase chain reaction (RT-PCR) of Na+-dependent L-ascorbic acid transporter (SVCT) and DDAH2
Real-time quantitative RT-PCR was performed from samples obtained from the border zone with a TaqMan system (Prism 7700 Sequence Detection System, PE Biosystems). Given that SVCT is necessary for the uptake of vitamin C in many tissues including myocardium,25 we measured the SVCT2 expression. For SVCT2, the primers were 5′-TTGACCATTACACCCACGGT-3′ and 5′-GTAATTCCCAAAACTCCAAT-3′. For DDAH2, the primers were 5′-CAGCTGCTGACTGCCTCTTTC-3′ and 5′-AGGTACCAGGGTGACATCAGAGA-3′. For cyclophilin, the primers were 5′-ATGGT CAACCCCACCGTGTTCTTCG-3′ and 5′-CGTGTGAAGTCACCACCCTGACACA-3′. Cyclophilin mRNA was chosen as the internal standard because it is expressed at a relatively constant level in virtually all tissues. For quantification, the expression was normalized to the expressed housekeeping gene cyclophilin. Reaction conditions were programmed on a computer linked to the detector for 40 cycles of the amplification step.
Laboratory measurements
Blood samples from the aorta were assayed at the end of the study. Blood samples were immediately centrifuged at 3,000 g for 10 min, and the serum was stored at -70 °C until further analysis. Myocardium at the border zone was processed according to the manufacturer’s instructions. The serum and myocardial levels of vitamin C and ADMA were measured according to the manufacturer’s instructions (Ferric Reducing Ascorbate Assay Kit, BioVision, CA, USA and Cayman Chemical, respectively).
Superoxide production by myocardium from the border zone was measured using lucigenin (5 μmol/L bis-N-methylacridinium nitrate (Sigma, St. Louis, MO) enhanced chemiluminescence as previously described.24 The specific chemiluminescence signal was calculated after subtracting background activity and expressed as counts per minute per milligram weight (cpm/mg). To estimate myocardial peroxynitrite formation, we measured free nitrotyrosine (as a marker for peroxynitrite formation) by enzyme-linked immunosorbent assay (ELISA) (Cayman Chemical, Ann Arbor, MI, USA) in myocardial homogenates. DDAH enzyme activity in homogenized myocardium was assayed by determining L-citrulline formation as previously described.26
Statistical analysis
Results were presented as mean ± standard deviation (SD). Statistical analysis was performed using the SPSS statistical package (SPSS, version 18.0, Chicago, Illinois). Differences among the groups of rats were tested by ANOVA. In cases of a significant effect, the measurements between the groups were compared with Bonferroni’s correction. The significance level was assumed at a p value of < 0.05.
RESULTS
Part 1. In vivo study
Differences in mortality among the infarcted groups were not found throughout the study. Four weeks after infarction, the infarcted area of the LV was very thin and had been totally replaced by fully differentiated scar tissue. The weight of the LV inclusive of the septum remained essentially constant for 4 weeks among the infarcted groups (Table 1). Compared to the vehicle group, the infarcted group treated with omeprazole alone had an increase in right ventricular weight/body weight ratio and lung weight/body weight ratio, and this was improved after adding vitamin C and olmesartan. Unlike omeprazole, famotidine did not affect remodeling of the right ventricle and lung after infarction. Significant reductions in +dP/dt and -dP/dt were evident in the infarcted group treated with omeprazole, and this was improved after the coadministration of either vitamin C or olmesartan. LV end-systolic pressure, LV end-diastolic pressure, and infarct size did not differ among the infarcted groups.
Table 1. Cardiac morphology and hemodynamics at the end of study.
| Parameters | Sham | Infarction treated with | ||||
| Vehicle | Vehicle | OM | OM + Vit C | OM + ARB | FM | |
| No. of rats | 10 | 10 | 11 | 11 | 12 | 10 |
| Body weight, g | 315 ± 12 | 311 ± 11 | 325 ± 13 | 314 ± 10 | 324 ± 12 | 326 ± 14 |
| Heart rate, bpm | 383 ± 14 | 405 ± 15 | 397 ± 12 | 407 ± 12 | 374 ± 16 | 387 ± 16 |
| LVESP, mmHg | 110 ± 5 | 97 ± 10 | 96 ± 8 | 105 ± 6 | 102 ± 10 | 97 ± 7 |
| LVEDP, mmHg | 7 ± 3 | 18 ± 2* | 19 ± 4* | 16 ± 5* | 15 ± 5* | 17 ± 5* |
| +dP/dt, mmHg/s | 8052 ± 418 | 2482 ± 264* | 2241 ± 249*# | 4142 ± 352*#† | 3798 ± 262*#† | 2624 ± 342*† |
| -dP/dt, mmHg/s | 6873 ± 305 | 2402 ± 234* | 2135 ± 242*# | 3222 ± 316*#† | 2982 ± 265*#† | 2592 ± 308*† |
| Infarct size, % | -- | 40 ± 4 | 41 ± 2 | 42 ± 4 | 40 ± 3 | 41 ± 3 |
| LVW/BW, mg/g | 2.13 ± 0.20 | 3.04 ± 0.25* | 2.95 ± 0.26* | 2.87 ± 0.35* | 2.83 ± 0.37* | 2.98 ± 0.38* |
| RVW/BW, mg/g | 0.51 ± 0.11 | 0.64 ± 0.10* | 0.75 ± 0.11*# | 0.55 ± 0.09#† | 0.58 ± 0.08† | 0.62 ± 0.13*† |
| LungW/BW, mg/g | 4.05 ± 0.37 | 5.27 ± 0.38* | 5.96 ± 0.33*# | 4.52 ± 0.32#† | 4.93 ± 0.35*#† | 4.95 ± 0.43*† |
Values are mean ± SD.
ADMA, asymmetric dimethylarginine; ARB, olmesartan; BW, body weight; LungW, lung weight; FM, famotidine; LVEDP, left ventricular end-diastolic pressure; LVESP, left ventricular end-systolic pressure; LVW, left ventricular weight; OM, omeprazole; RVW, right ventricular weight; Vit C, vitamin C.
* p < 0.05 compared with sham; # p < 0.05 compared with infarcted group treated with vehicle; † p < 0.05 compared with infarcted group treated with OM.
MI significantly decreased vitamin C levels and increased ADMA levels in serum and myocardium relative to the sham-operated rats (Figure 1). The infarcted groups treated with omeprazole or omeprazole + olmesartan had further decreases in vitamin C level compared with the vehicle group. The famotidine-treated infarcted group had similar values of vitamin C as the vehicle group. ADMA levels were significantly increased after infarction, and were further increased in the omeprazole-treated infarcted group. However, the ADMA levels were reduced after adding either vitamin C or olmesartan compared with omeprazole alone. Famotidine treatment did not affect the ADMA levels compared with vehicle treatment.
Figure 1.
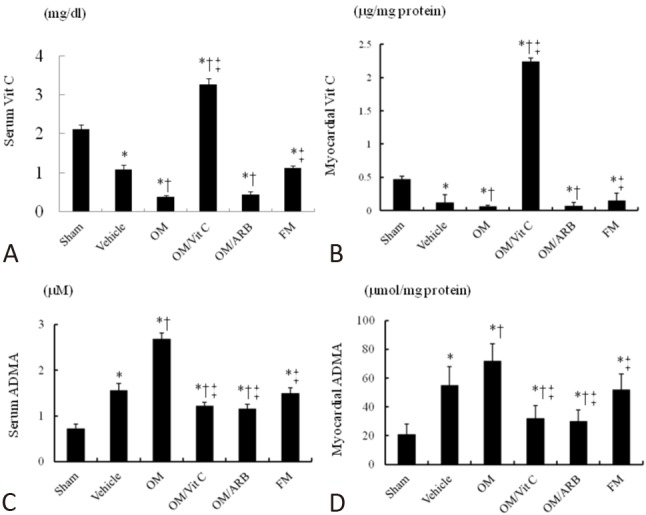
Vitamin C and ADMA levels in serum and myocardium. (A) Serum vitamin C, (B) myocardial vitamin C, (C) serum ADMA, and (D) myocardial ADMA levels from the border zone. Each column and bar represents mean ± SD. Sham (n = 10); Vehicle (n = 10); OM, omeprazole (n = 11); OM/Vit C, vitamin C (n = 11); OM/ARB, olmesartan (n = 12); FM, famotidine (n = 10). * p < 0.05, compared with sham; † p < 0.05, compared with vehicle-treated rats; ‡ p < 0.05, compared with OM-treated rats. Abbreviations as in Table 1. SD, standard deviation.
Cardiac hypertrophy
To characterize the cardiac hypertrophy on a cellular level, morphometric analyses of LV sections from the border zone were performed in different treatment groups (Figure 2). Myocytes were significantly hypertrophied in the vehicle-treated infarcted group compared with the sham-operated group. Furthermore, the omeprazole-treated infarcted rats had a significant increase in cardiomyocyte size compared with the vehicle-treated infarcted rats. Vitamin C and olmesartan reduced cell areas by 31% and 26%, respectively, compared with omeprazole alone (both p < 0.05). The famotidine-treated rats developed similar cardiomyocyte hypertrophy compared with the vehicle-treated rats.
Figure 2.
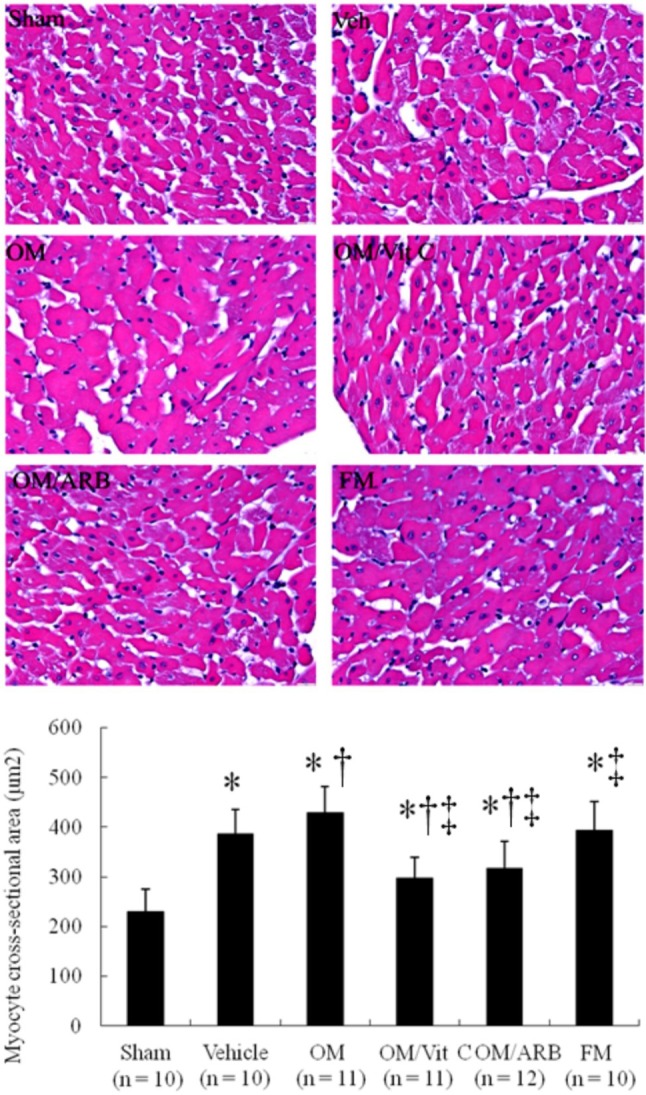
Cardiomyocyte sizes. Morphometric analyses of LV sections from the border zone at 4 wk after infarction. Bar = 50 μm. Each column and bar represents mean ± SD. The number of animals in each group is indicated in parentheses. ARB, olmesartan; FM, famotidine; OM, omeprazole; Vit C, vitamin C. * p < 0.05, compared with sham; † p < 0.05, compared with vehicle-treated rats; ‡ p < 0.05, compared with OM-treated rats.
Echocardiography
Compared with the sham-operated animals, MI hearts showed structural changes such as increased LV diastolic and systolic diameters (Table 2), consistent with LV remodeling. Echocardiography showed a significant increase in LVESD and LVEDD in the omeprazole-treated infarcted rats compared with the vehicle-treated infarcted rats (Figure 3). LV fractional shortening was significantly lower in the omeprazole-treated infarcted rats compared with the vehicle-treated infarcted rats (15 ± 3% vs. 20 ± 3% in vehicle, p < 0.05). Omeprazole significantly increased hypertrophy of the remote myocardium (i.e. LV posterior wall, p < 0.05) but did not affect the thickness of the infarcted portion (i.e. interventricular septum). Infarcted groups treated with vitamin C or olmesartan had improved LV remodeling and function. These data were corroborated by the results that +dP/dt and -dP/dt were significantly decreased in the omeprazole-treated group compared with the vehicle-treated group. There were no significant differences in echocardiographic parameters between the famotidine-treated infarcted rats and vehicle-treated rats.
Table 2. Echocardiographic findings.
| Parameters | Sham | Infarction treated with | ||||
| Vehicle | Vehicle | OM | OM + Vit C | OM + ARB | FM | |
| No. of rats | 10 | 10 | 11 | 11 | 12 | 10 |
| IVS (mm) | 1.4 ± 0.1 | 0.5 ± 0.2* | 0.6 ± 0.2* | 0.6 ± 0.2* | 0.6 ± 0.1* | 0.6 ± 0.1* |
| LVEDD (mm) | 5.7 ± 0.2 | 8.6 ± 0.2* | 9.1 ± 0.2*# | 7.7 ± 0.2*#† | 7.5 ± 0.1*#† | 8.5 ± 0.2*† |
| LVESD (mm) | 3.5 ± 0.2 | 6.9 ± 0.2* | 7.7 ± 0.2*# | 5.8 ± 0.2*#† | 5.5 ± 0.3*#† | 6.9 ± 0.1*† |
| LV posterior wall (mm) | 1.4 ± 0.1 | 1.7 ± 0.2* | 2.0 ± 0.1*# | 1.7 ± 0.1*† | 1.7 ± 0.2*† | 1.8 ± 0.2* |
| FS (%) | 39 ± 2 | 20 ± 3* | 15 ± 3*# | 24 ± 3*† | 26 ± 3*#† | 19 ± 2*† |
Values are mean ± SD.
Abbreviations as in Table 1. FS, fractional shortening; IVS, interventricular septum; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension.
* p < 0.05 compared with the sham group; # p < 0.05 compared with infarcted group treated with vehicle; † p < 0.05 compared with infarcted group treated with OM.
Figure 3.
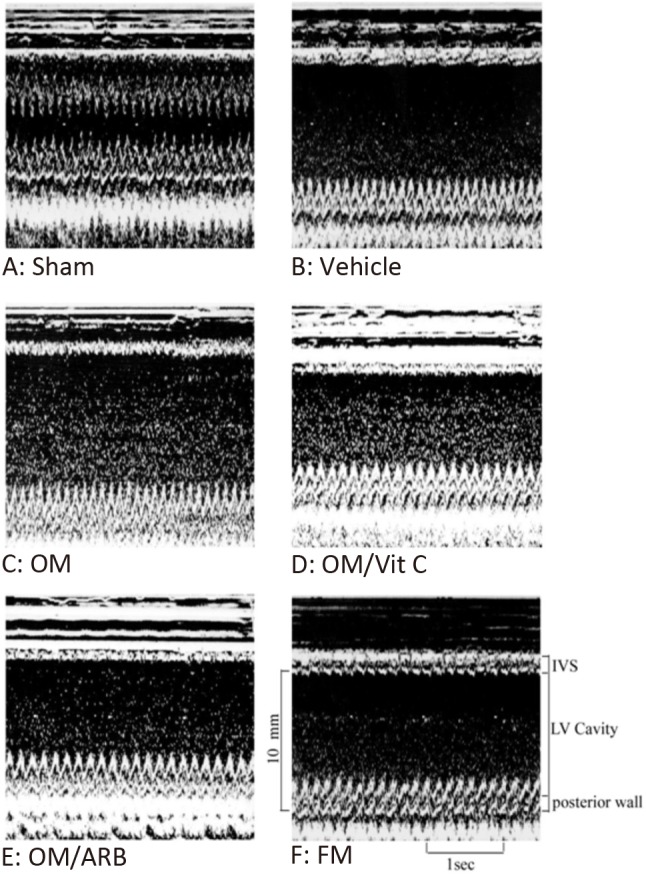
Representative M-mode image reveals a hypokinetic-to-akinetic anterior wall and LV dilation in the infarcted hearts (B to F) in contrast to normal anterior wall motion in sham-operated heart (A). There are markedly dilated LV end-diastolic diameter and LV end-systolic diameter in the OM-treated group (C) than those treated with vehicle (B) and FM (D). Besides, infarcted rats after adding either Vit C or ARB showed a significant decrease of LV end-diastolic diameter and LV end-systolic diameter compared with OM alone. ARB, olmesartan; FM, famotidine; IVS, interventricular septum thickness ; OM, omeprazole; Vit C, vitamin C.
Myocardial superoxide and nitrotyrosine levels
Superoxide production assessed by chemiluminescence was significantly increased at the border LV tissues from the vehicle-treated infarcted rats compared with the sham-operated rats, and was further increased in the omeprazole-treated infarcted rats (Figure 4A). Superoxide production was significantly decreased in the vitamin C- and olmesartan-treated rats compared to the sham-operated rats. Famotidine treatment did not affect myocardial superoxide production compared with vehicle treatment.
Figure 4.
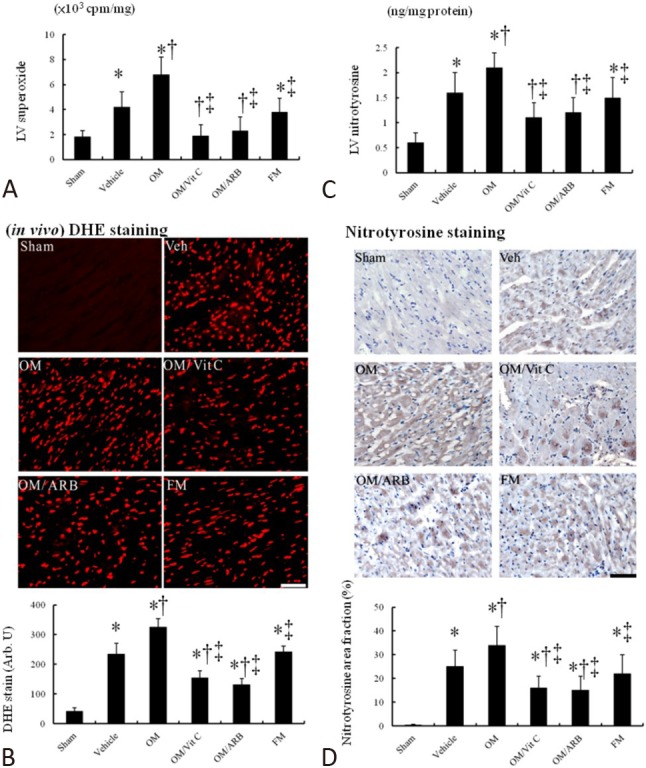
In vivo myocardial superoxide and nitrotyrosine levels. Myocardial (A) superoxide by chemiluminescence, (B) DHE staining and quantitative analysis, (C) nitrotyrosine by ELISA, and (D) nitrotyrosine immunoreactive staining and quantitative analysis from the border zone. Myocardial DHE (red fluorescent) and nitrotyrosine (brown) staining showed more intense signals (nuclear position for DHE and cytoplasm for nitrotyrosine) after MI. Compared with vehicle, myocardial superoxide and nitrotyrosine levels in the OM-treated infarcted group were significantly increased, which can be significantly reduced after adding either Vit C or ARB. Bar = 50 μm. Sham (n = 10); Vehicle (n = 10); OM, omeprazole (n = 11); OM/Vit C, vitamin C (n = 11); OM/ARB, olmesartan (n = 12); FM, famotidine (n =10). * p < 0.05, compared with sham; † p < 0.05, compared with vehicle-treated rats; ‡ p < 0.05, compared with OM-treated rats. Abbreviations as in Table 1
DHE reacts with superoxide radicals to form ethidium bromide, which in turn intercalates with DNA to provide nuclear fluorescence as a marker of superoxide radical generation. As shown in Figure 4B, postinfarction remodeling markedly enhanced the intensity of DHE staining at the border zone in the vehicle-treated rats compared with the sham-operated rats. Compared with the vehicle-treated infarcted rats, the omeprazole-treated infarcted rats had a significantly increased intensity of the fluorescent signal. The increased intensity of the fluorescent signal after infarction was significantly attenuated by administering vitamin C or olmesartan.
Myocardial nitrotyrosine in the vehicle-treated infarcted rats significantly increased compared to the sham-operated rats (p < 0.01, Figure 4C). Similarly, nitrotyrosine immunoreactivity, as evidenced by increased brown staining (Figure 4D), was significantly increased after MI, and this was further increased after adding omeprazole. The increased intensity of the fluorescent signal in the infarcted group treated with omeprazole was significantly attenuated by administering vitamin C or olmesartan.
DDAH2 activity, protein and mRNA expression
Myocardial DDAH enzyme activity was significantly higher in the rats after MI, compared with the LV of the sham-operated rats (Figure 5A). Compared with the infarcted rats treated with either vehicle or famotidine, the omeprazole-treated infarcted rats had significantly decreased DDAH enzyme activity.
Figure 5.
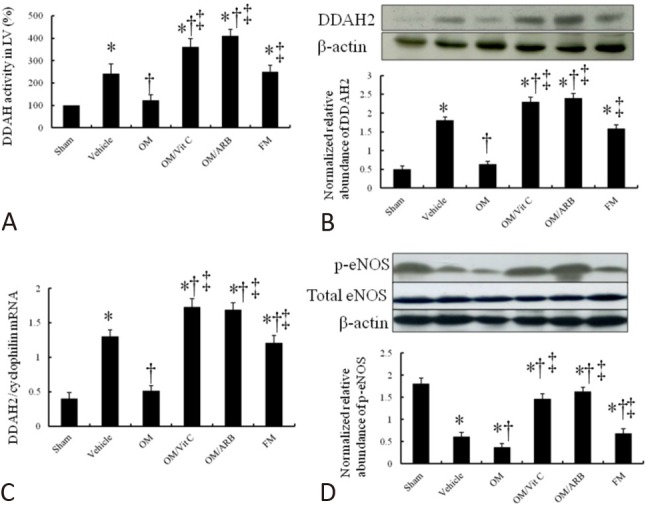
Myocardial DDAH and eNOS activity assessment. (A) Myocardial DDAH activity, (B) DDAH2 Western blot, (C) DDAH2 RT-PCR, and (D) eNOS activity from the border zone. Compared with vehicle in omeprazole-treated rats, DDAH activity, protein level and mRNA expression were significantly decreased, which can be reversed by adding vitamin C. When compared with vehicle-treated infarcted rats, omeprazole-treated infarcted rats had significantly lower p-eNOS levels. Relative abundance was obtained by normalizing the density of eNOS protein against that of β-actin. Sham (n = 10); Vehicle (n = 10); OM, omeprazole (n = 11); OM/Vit C, vitamin C (n = 11); OM/ARB, olmesartan (n = 12); FM, famotidine (n =10). * p < 0.05, compared with sham; † p < 0.05, compared with vehicle-treated rats; ‡ p < 0.05, compared with OM-treated rats. Abbreviations as in Table 1. DDAH, dimethylarginine dimethylaminohydrolase; RT-PCR, real-time polymerase chain reaction.
Western blot analysis showed that DDAH2 levels were significantly upregulated by 3.6-fold at the border zone in the vehicle-treated infarcted rats compared to the sham-operated rats (p < 0.0001, Figure 5B). Compared with the vehicle-treated infarcted rats, the omeprazole-treated infarcted rats had significantly lower DDAH2 levels at the border zone (1.81 ± 0.11 in vehicle vs. 0.63 ± 0.08, p < 0.05). The reduced PPI-induced DDAH2 protein levels were significantly reversed by administering either vitamin C or olmesartan. The famotidine-treated infarcted rats showed similar DDAH2 levels compared with the vehicle-treated infarcted rats.
Polymerase chain reaction (PCR) amplification of the cDNA revealed that the DDAH2 mRNA levels were upregulated by 3.2-fold at the border zone in the vehicle-treated infarcted rats compared with the sham-operated rats (p < 0.0001, Figure 5C). In the omeprazole-treated infarcted rats, the DDAH2 mRNA expression was significantly decreased compared with the famotidine-treated infarcted rats. PCR amplification of SVCT2 showed similar expressions among the infarcted groups (data not shown), implying that differences in vitamin C levels were not related to the gene expression.
eNOS activity
An increase in ADMA would be expected to reduce the activity of eNOS. Accordingly, we found that active eNOS (p-eNOS) levels were significantly decreased after infarction compared with the sham-operated rats (Figure 5D). After administering omeprazole, eNOS activity was further reduced, and this was increased after adding vitamin C. These findings are consistent with a reduction in eNOS activity resulting from an accumulation of ADMA.
Part 2. Ex vivo study
To exclude the effect of gastric pH on vitamin C uptake-related myocardial superoxide levels, we performed an ex vivo experiment (Figure 6A). Surprisingly, myocardial superoxide levels assessed by either chemiluminescence (data not shown) or DHE staining were not significantly increased in the omeprazole-treated group compared with the vehicle-treated group in the ex vivo study, in contrast to the in vivo study. These findings implied the importance of gastric pH on myocardial superoxide levels. Furthermore, we found that omeprazole-treated rat hearts did not have significant differences in DDAH2 levels by Western blot compared with vehicle-treated rat hearts (Figure 6B) in the ex vivo study, in contrast to the in vivo findings.
Figure 6.
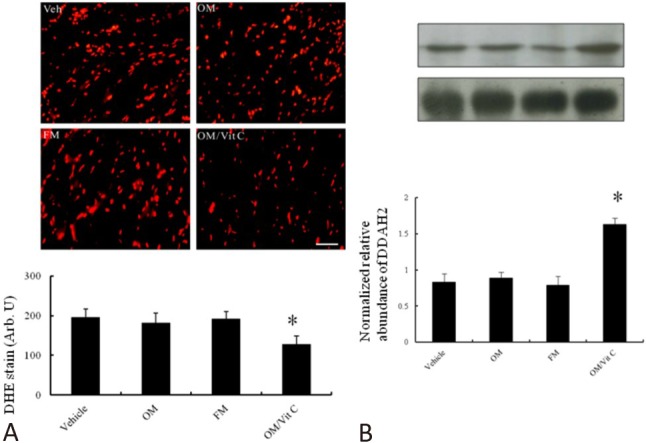
Ex vivo myocardial superoxide and DDAH2 levels. (A) Myocardial superoxide levels assessed by DHE staining and (B) DDAH2 protein levels from the border zone. Compared with vehicle, myocardial superoxide and DDAH2 levels in the omeprazole-treated infarcted group were not significantly changed, which contrasted with the in vivo study. Bar = 50 μm. Vehicle (n = 5); OM, omeprazole (n = 5); FM, famotidine (n = 5); OM/Vit C, vitamin C (n = 5). * p < 0.05, compared with vehicle-, omeprazole- and famotidine-treated rats. Abbreviations as in Table 1
DISCUSSION
This study is the first to show that chronic treatment for 4 weeks with PPIs led to negative effects on ventricular remodeling after MI in an in vivo study. The salient findings of the present investigation are as follows: 1) DDAH activity was inhibited by PPIs, not H2B, through attenuated vitamin C uptake- and increased ROS-dependent pathways. Ameliorating the deleterious effects of ADMA by exogenous antioxidant vitamin C drastically increased NO activity and subsequently improved ventricular remodeling. 2) ARB administration also improved ventricular remodeling by scavenging ROS. Given that ARBs are recommended for patients after MI,17 this may explain at least in part why few clinical studies have addressed the unexpected results in patients treated with PPIs. These insights may lead to new therapeutic reevaluations after MI, and may explain the clinical discrepancies. Our results were consistent with the effects of PPI administration on impaired ventricular remodeling in infarcted rats, as documented structurally by increases in cardiomyocyte size and DHE and nitrotyrosine staining, molecularly by myocardial DDAH2 protein and mRNA expression, biochemically by measurements of myocardial vitamin C, ADMA, superoxide, and nitrotyrosine levels, pharmacologically by antioxidation vitamin C and ARBs, and functionally by impaired echocardiographically-derived heart contractility. Our results are consistent with a recent study showing a graded association between the duration of PPI exposure and the risk of death.27 Our data may provide a unifying mechanism for the association between PPI use and impaired ventricular remodeling after MI.
The effect of PPIs on impaired ventricular remodeling was supported by 3 lines of evidence as shown in Figure 7.
Figure 7.
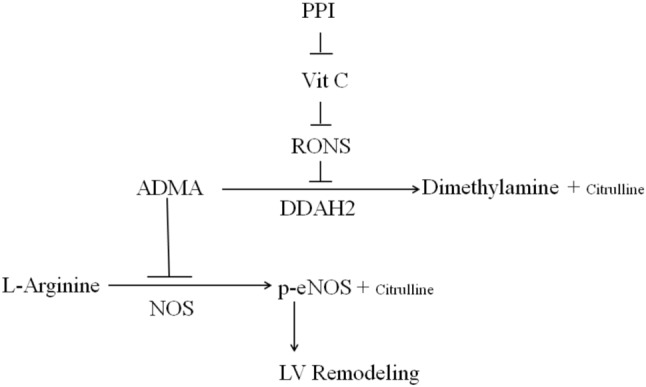
Central illustration. Schematic representation illustrates the involvements of PPI and its related components in DDAH-related ventricular remodeling in postinfarcted rats. PPIs increase superoxide production through vitamin C-related decreased uptake. Thereby, attenuated DDAH activity increases ADMA levels and subsequent decreases eNOS activity. Inhibition of these signaling pathways by their respective inhibitors is indicated by the vertical lines. RONS, reactive oxygen and nitrogen species. ADMA, asymmetric dimethylarginine; DDAH2, dimethylarginine dimethylaminohydrolase 2; LV, left ventricular; NOS, nitric oxide synthase; PPI, proton pump inhibitors; p-eNOS, phosphorylated-eNOS; RONs, reactive oxygen and nitrogen species; Vit C, vitamin C.
(1) Omeprazole administration was associated with a decrease in vitamin C and increase in free radicals after infarction. Our results showed that vitamin C levels in serum and myocardium were significantly decreased after infarction compared with the sham-operated rats, which is consistent with previous studies.28 Omeprazole was more effective than famotidine in keeping gastric pH high and lowering the total gastric acid output. Thus, a PPI-mediated sustained rise in intragastric pH may contribute to the instability and degradation of vitamin C. The reduced vitamin C levels in serum and myocardium subsequently increased ROS production. Our results are consistent with the very recent study by Yepuri et al.,29 who reported that esomeprazole was associated with increased generation of superoxide anions. To determine whether the increased superoxide anion production was related to vitamin C, vitamin C was coadministered, and this reversed the increased ROS generation.
Although emerging evidence suggests that PPIs have effects beyond potent blockade of gastric H+/K+-ATPase, this study did not support an association between the effect of PPIs on DDAH activity and the direct inhibition of myocardial H+/K+-ATPase, because our ex vivo study showed no significant changes in DDAH2 levels in the rats treated with PPIs. To assess the role of gastric H+/ K+-ATPase in modulating antioxidant vitamin C activity, we used a similar dose of omeprazole in the ex vivo study to that in the in vivo study. It is known that PPIs need an acidic environment to be fully activated,30 and a small change in pH from 7.4 to 6.1 has been shown to decrease the IC50 value of omeprazole by 9-fold.30 Given the lower expression of H+/K+-ATPase and relatively basic pH (7.4) in myocardial tissue than in the stomach,4 myocardial H+/K+-ATPase may be inhibited only at higher doses. Thus, it is expected that the similar dose in the ex vivo study did not inhibit myocardial H+/K+-ATPase, and that subsequently ROS and DDAH2 levels would not be affected, consistent with our findings.
(2) Omeprazole administration was associated with decreased DDAH activity and increased ADMA levels. In addition, we showed that regulation of DDAH2 was altered after administering PPIs in infarcted rat myocardium. PPI administration increased myocardial ADMA levels via inhibition of the gene expression, protein level, and activity of DDAH2. However, compared with the sham-operated rats, DDAH activity was enhanced after MI, implying high ROS generation conditions. This finding seems to be in contrast to previous reports in which high oxidative stress was associated with impaired DDAH activity and decreased DDAH protein levels. Many factors have been identified that regulate the transcription or translation of the DDAH gene. Besides redox modification, another explanation may be due to inflammatory cells activating DDAH2 expression, however this was not assessed in this study.
(3) Omeprazole administration was associated with impaired ventricular remodeling after MI. The influence of omeprazole administration on ventricular remodeling has never been examined before. Reduced NO availability signals important pathophysiological changes leading to impaired ventricular remodeling. ADMA concentrations ranging from 1 to 10 μM have been reported to inhibit NO in a dose-dependent manner.31 The increase in ADMA in this study may have pathophysiological significance, because its concentration falls into the range shown to inhibit the activity of NOS. Thus, the impaired ventricular remodeling in the infarcted rats treated with PPIs could be a consequence of decreased NOS activity.
Other mechanisms
Although the present study suggests that the mechanisms of PPI-induced impairment of ventricular remodeling may be related to increased vitamin C-dependent superoxide production and reduced DDAH-dependent eNOS levels, other potential mechanisms need to be studied. First, PPIs can influence circulating ADMA through interference with the absorption of vitamin B12.32 This vitamin is required for the conversion of homocysteine to cysteine. Elevated plasma homocysteine levels have been reported to increase plasma levels of ADMA, and may increase susceptibility to coronary artery disease.33 Previous studies have observed that acute increases in homocysteine level acutely increase plasma ADMA and impair endothelium dependent vasodilation.34 Second, DDAH might be able to regulate remodeling signaling independently of the NOS pathway. DDAH2 has been reported to enhance vascular endothelial growth factor mRNA expression in endothelial cells,35 an important player in ventricular remodeling. DDAH2 protein directly binds to protein kinase A and subsequently phosphorylates the transcription factor Sp1 without increasing NO production.34 Therefore DDAH protein seems to be involved in regulation of intracellular pathways by protein-protein interactions in addition to its known function in ADMA metabolism. Third, other sources for increasing free radicals after PPI administration should be considered. A very recent study showed that PPI administration was associated with the increased production of free radicals by impairing the lysosomal proton pump of the endothelium.29 However, vitamin C administration can reverse the impairment of ventricular remodeling, implying the role of vitamin C in the PPI-mediated negative impact on ventricular remodeling.
Clinical implication
Safety issues associated with PPIs have recently attracted widespread attention. The present study provides strong evidence that PPIs had adverse effects on ventricular remodeling in an animal model of homogenous baseline. The oral dose of omeprazole of 5 mg/kg/day used in the rats in this study is equal to an oral human dose of 40 mg on a body surface area basis.36 Larsso et al.37 reported that omeprazole inhibited basal and stimulated acid secretion in rats at an oral dose of 5 mg/kg per day. Given that our studies were conducted in a clinical dose range, the conclusions of this study are clinically relevant. The metabolism of omeprazole is dependent on the isoenzyme CYP2C19, which exhibits polymorphisms. Approximately 3% of Caucasians and 15 to 20% of Asians are poor metabolizers.38 In repeated single 20 mg omeprazole doses, an increase in AUC of approximately four-fold was noted in Asian subjects compared with Caucasians.37 Although the FDA-approved drug label for omeprazole does not comment on dose adjustments based on CYP2C19 status, this reduced enzyme activity may have a negative effect on clinical cardiovascular outcomes, especially in Asian people with poor metabolism. Thus, we suggest that regulatory agencies should reevaluate the safety of these agents.
Whether or not the negative impact of PPIs on cardiovascular outcomes translates to clinically meaningful adverse events has been the subject of great debate. Currently, no clinical research regarding the impaired remodeling of PPI has been reported. Given that PPIs are recommended for patients who require antiplatelet therapy and have a history of upper gastrointestinal bleeding, the sequences of PPI use after MI need to be defined. Clinical research may be confounded by unexpected errors due to the influence of other drugs being used by patients after infarction. ARBs have long been established as a useful agent for ventricular remodeling after infarction.17 Therefore, it is important to prove that ARBs may affect the effect of PPI on remodeling. In the present study, we demonstrated that the combination of a PPI and an ARB effectively prevented the inhibition of DDAH activity compared with PPI monotherapy. Thus, ARB use is an important confounding factor when analyzing the effect of PPIs on ventricular remodeling. However, our results indicate that PPIs can be taken with confidence when combined with ARBs with respect to a myocardial risk. Our results are consistent with a previous study, in which the renal DDAH2 expression was reduced by telmisartan administration to diabetic rats.39 Our study provides a potential explanation for the seemingly paradoxical findings in impaired PPI-induced DDAH2 activity and patients treated with ARBs who do not have impaired ventricular remodeling. Furthermore, our results also suggest that the FDA do not need to warn of an increased risk of heart problems for patients treated with omeprazole.40
Previous studies have shown that PPIs are associated with increased arrhythmias in patients with focal tachyarrhythmias after adjusting for potential confounders,41 however the mechanism remains unclear. Our previous study showed that favorable ventricular remodeling has benefits not only in anatomical structure but also in arrhythmia susceptibility.42 PPI-induced impaired ventricular remodeling may explain, at least in part, the increased incidence of arrhythmias, however further studies are needed to clarify these findings under clinical conditions. Given the potential complications of PPIs, we suggest that patients with a proven indication for a PPI should continue to receive it in the lowest effective dose, and that continued chronic PPI therapy in those unresponsive to initial empiric therapy should be discouraged.
Study limitations
There are some limitations to the present study. First, gene expression analysis could only be performed on tissues from surviving animals, and this may have biased the results toward a probably less severe situation, since only animals that coped well with postinfarction remodeling were investigated. Furthermore it is currently not clear whether increased tissue ADMA is derived from inflammatory cells, from the endothelium or from myocytes. Thus, further experiments are planned to elucidate the exact mechanism of DDAH inhibition and to clarify the source of ADMA. Second, ADMA levels were measured only once at the end of the study, and not after the acute phase of MI. The analysis may have been more valuable if the change in ADMA over time had been shown with several measures.
CONCLUSIONS
Our findings show that infarcted hearts exposed to PPIs, but not H2B, had decreased DDAH2 gene expression, reduced DDAH activity, and consequently increased ADMA accumulation and decreased eNOS activity, probably through reduced vitamin C uptake- and increased ROS production-dependent pathways. These effects were prevented by the coadministration of vitamin C or ARBs. Further studies are necessary to determine the relevance of these findings in the setting of PPI-associated impaired ventricular remodeling in humans.
Acknowledgments
This work was supported by the grant of An-Nan Hospital (ANHRF 104-02), China Medical University (CMU 103-S-07).
DECLARATION OF CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Li YH, Fang CY, Hsieh IC, et al. 2018 expert consensus on the management of adverse effects of antiplatelet therapy for acute coronary syndrome in Taiwan. Acta Cardiol Sin. 2018;34:201–210. doi: 10.6515/ACS.201805_34(3).20180302A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah NH, LePendu P, Bauer-Mehren A, et al. Proton pump inhibitor usage and the risk of myocardial infarction in the general population. PLoS One. 2015;10:e0124653. doi: 10.1371/journal.pone.0124653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landi SN, Sandler RS, Pate V, Lund JL. No increase in risk of acute myocardial infarction in privately insured adults prescribed proton pump inhibitors vs histamine-2 receptor antagonists (2002-2014). Gastroenterology. 2018;154:861–873. doi: 10.1053/j.gastro.2017.10.042. [DOI] [PubMed] [Google Scholar]
- 4.Beisvag V, Falck G, Loennechen JP, et al. Identification and regulation of the gastric H+/K+-ATPase in the rat heart. Acta Physiol Scand. 2003;179:251–262. doi: 10.1046/j.0001-6772.2003.01191.x. [DOI] [PubMed] [Google Scholar]
- 5.van der Loo B, Bachschmid M, Spitzer V, et al. Decreased plasma and tissue levels of vitamin C in a rat model of aging: implications for antioxidative defense. Biochem Biophys Res Commun. 2003;303:483–487. doi: 10.1016/s0006-291x(03)00360-7. [DOI] [PubMed] [Google Scholar]
- 6.Netzer P, Gaia C, Sandoz M, et al. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 hours. Am J Gastroenterol. 1999;94:351–357. doi: 10.1111/j.1572-0241.1999.857_y.x. [DOI] [PubMed] [Google Scholar]
- 7.Reilly J, Fennerty MB. Stress ulcer prophylaxis: the prevention of gastrointestinal bleeding and the development of nosocomial infections in critically ill patients. J Pharm Prac. 1998;11:418–432. [Google Scholar]
- 8.Biswas K, Bandyopadhyay U, Chattopadhyay I, et al. A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J Biol Chem. 2003;278:10993–11001. doi: 10.1074/jbc.M210328200. [DOI] [PubMed] [Google Scholar]
- 9.Goodman SG, Clare R, Pieper KS, et al. Platelet Inhibition and Patient Outcomes Trial Investigators. Association of proton pump inhibitor use on cardiovascular outcomes with clopidogrel and ticagrelor: insights from the platelet inhibition and patient outcomes trial. Circulation. 2012;125:978–986. doi: 10.1161/CIRCULATIONAHA.111.032912. [DOI] [PubMed] [Google Scholar]
- 10.Lee TM, Lin SZ, Chang NC. Antiarrhythmic effect of lithium in rats after myocardial infarction by activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014;77:71–81. doi: 10.1016/j.freeradbiomed.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Jia SJ, Jiang DJ, Hu CP, et al. Lysophosphatidylcholine-induced elevation of asymmetric dimethylarginine level by the NADPH oxidase pathway in endothelial cells. Vascul Pharmacol. 2006; 44:143–148. doi: 10.1016/j.vph.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol. 2007;293:H3227–H3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 13.Zeller M, Korandji C, Guilland JC, et al. Impact of asymmetric dimethylarginine on mortality after acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2008;28:954–960. doi: 10.1161/ATVBAHA.108.162768. [DOI] [PubMed] [Google Scholar]
- 14.Shyu KG. The role of endoglin in myocardial fibrosis. Acta Cardiol Sin. 2017;33:461–467. doi: 10.6515/ACS20170221B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TM, Lai PY, Chang NC. Effect of N-acetylcysteine on sympathetic reinnervation in post-infarcted rat hearts. Cardiovasc Res. 2010;85:137–146. doi: 10.1093/cvr/cvp286. [DOI] [PubMed] [Google Scholar]
- 16.Ghebremariam YT, LePendu P, Lee JC, et al. Unexpected effect of proton pump inhibitors: elevation of the cardiovascular risk factor asymmetric dimethylarginine. Circulation. 2013;128:845–853. doi: 10.1161/CIRCULATIONAHA.113.003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Gara PT, Kushner FG, Ascheim DD, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127:e362–e425. doi: 10.1161/CIR.0b013e3182742cf6. [DOI] [PubMed] [Google Scholar]
- 18.Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sai K, Umemura T, Takagi A, et al. The protective role of glutathione, cysteine and vitamin C against oxidative DNA damage induced in rat kidney by potassium bromate. Jpn J Cancer Res. 1992;83:45–51. doi: 10.1111/j.1349-7006.1992.tb02350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sener-Muratoğlu G, Paskaloğlu K, Arbak S, et al. Protective effect of famotidine, omeprazole, and melatonin against acetylsalicylic acid-induced gastric damage in rats. Dig Dis Sci. 2001;46:318–330. doi: 10.1023/a:1005652815921. [DOI] [PubMed] [Google Scholar]
- 21.Lee TM, Lin MS, Chou TF, Chang NC. Additive effects of combined blockade of AT1 receptor and HMG-CoA reductase on left ventricular remodeling in infarcted rats. Am J Physiol. 2006;291:H1281–H1289. doi: 10.1152/ajpheart.00792.2005. [DOI] [PubMed] [Google Scholar]
- 22.Xia QG, Chung O, Spitznagel H, et al. Significance of timing of angiotensin AT1 receptor blockade in rats with myocardial infarction-induced heart failure. Cardiovasc Res. 2001;49:110–117. doi: 10.1016/s0008-6363(00)00227-3. [DOI] [PubMed] [Google Scholar]
- 23.Lee TM, Lin MS, Chang NC. Effect of ATP-sensitive potassium channel agonists on ventricular remodeling in healed rat infarcts. J Am Coll Cardiol. 2008;51:1309–1318. doi: 10.1016/j.jacc.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 24.Lee TM, Lin MS, Chou TF, et al. Adjunctive 17β-estradiol administration reduces infarct size by altered expression of canine myocardial connexin43 protein. Cardiovasc Res. 2004;63:109–117. doi: 10.1016/j.cardiores.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 25.May JM. The SLC23 family of ascorbate transporters: ensuring that you get and keep your daily dose of vitamin C. Br J Pharmacol. 2011;164:1793–1801. doi: 10.1111/j.1476-5381.2011.01350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stühlinger MC, Tsao PS, Her JH, et al. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 27.Xie Y, Bowe B, Li T, et al. Risk of death among users of proton pump inhibitors: a longitudinal observational cohort study of United States veterans. BMJ Open. 2017;7:e015735. doi: 10.1136/bmjopen-2016-015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Numair KS, Chandramohan G, Alsaif MA, et al. Morin, a flavonoid, on lipid peroxidation and antioxidant status in experimental myocardial ischemic rats. Afr J Tradit Complement Altern Med. 2014;11:14–20. doi: 10.4314/ajtcam.v11i3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yepuri G, Sukhovershin R, Nazari-Shafti TZ, et al. Proton pump inhibitors accelerate endothelial senescence. Circ Res. 2016;118:e36–e42. doi: 10.1161/CIRCRESAHA.116.308807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keeling DJ, Fallowfield C, Milliner KJ, et al. Studies on the mechanism of action of omeprazole. Biochem Pharmacol. 1985;34:2967–2973. doi: 10.1016/0006-2952(85)90023-1. [DOI] [PubMed] [Google Scholar]
- 31.Mowat C, Carswell A, Wirz A, McColl KE. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology. 1999;116:813–822. doi: 10.1016/s0016-5085(99)70064-8. [DOI] [PubMed] [Google Scholar]
- 32.Howden CW. Vitamin B12 levels during prolonged treatment with proton pump inhibitors. J Clin Gastroenterol. 2000;30:29–33. doi: 10.1097/00004836-200001000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Stühlinger MC, Oka RK, Graf EE, et al. Endothelial dysfunction induced by hyperhomocyst(e)inemia: role of asymmetric dimethylarginine. Circulation. 2003;108:933–938. doi: 10.1161/01.CIR.0000085067.55901.89. [DOI] [PubMed] [Google Scholar]
- 34.Hasegawa K, Wakino S, Tanaka T, et al. Dimethylarginine dimethylaminohydrolase 2 increases vascular endothelial growth factor expression through Sp1 transcription factor in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1488–1494. doi: 10.1161/01.ATV.0000219615.88323.b4. [DOI] [PubMed] [Google Scholar]
- 35.Smith CL, Birdsey GM, Anthony S, et al. Dimethylarginine dimethylaminohydrolase activity modulates ADMA levels, VEGF expression, and cell phenotype. Biochem Biophys Res Commun. 2003;308:984–989. doi: 10.1016/s0006-291x(03)01507-9. [DOI] [PubMed] [Google Scholar]
- 36.Segawa K, Nakazawa S, Tsukamoto Y, et al. Effect of omeprazole on gastric acid secretion in rat: evaluation of dose, duration of effect, and route of administration. Gastroenterol Jpn. 1987;22:413–418. doi: 10.1007/BF02773807. [DOI] [PubMed] [Google Scholar]
- 37.Larsson H, Carlsson E, Junggren U, et al. Inhibition of astric acid secretion by omeprazole in the dog and rat. Gastroenterology. 1983;85:900–907. [PubMed] [Google Scholar]
- 38.Roman M, Ochoa D, Sanchez-Rojas SD, et al. Evaluation of the relationship between polymorphisms in CYP2C19 and the pharmacokinetics of omeprazole, pantoprazole and rabeprazole. Pharmacogenomics. 2014;15:1893–1901. doi: 10.2217/pgs.14.141. [DOI] [PubMed] [Google Scholar]
- 39.Onozato ML, Tojo A, Leiper J, et al. Expression of NG,NG-dimethylarginine dimethylaminohydrolase and protein arginine N-methyltransferase isoforms in diabetic rat kidney: effects of angiotensin II receptor blockers. Diabetes. 2008;57:172–180. doi: 10.2337/db06-1772. [DOI] [PubMed] [Google Scholar]
- 40.FDA Statement. FDA’s safety reviews of Prilosec and Nexium find no evidence of increased rates of cardiac events. Cited 10 December 2007;Available at http://www.fda.gov/bbs/topics/NEWS/2007/NEW01754.html [Google Scholar]
- 41.El-Charabaty E, Saifan C, Abdallah M, et al. Effects of proton pump inhibitors and electrolyte disturbances on arrhythmias. Int J Gen Med. 2013;6:515–518. doi: 10.2147/IJGM.S46932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TM, Chou TF, Tsai CH. Effects of pravastatin on cardiomyocyte hypertrophy and ventricular vulnerability in normolipidemic rats after myocardial infarction. J Mol Cell Cardiol. 2003;35:1449–1459. doi: 10.1016/j.yjmcc.2003.09.009. [DOI] [PubMed] [Google Scholar]