

Abstract
Vascularization of engineered bone tissue is critical for ensuring its survival after implantation. In vitro pre-vascularization of bone grafts with endothelial cells is a promising strategy to improve implant survival. In this study, we pre-cultured human smooth muscle cells (hSMCs) on bone scaffolds for 3 weeks followed by seeding of human umbilical vein endothelial cells (HUVECs), which produced a desirable environment for microvasculature formation. The sequential cell-seeding protocol was successfully applied to both natural (decellularized native bone, or DB) and synthetic (3D-printed Hyperelastic “Bone” scaffolds, or HB) scaffolds, demonstrating a comprehensive platform for developing natural and synthetic-based in vitro vascularized bone grafts. Using this sequential cell-seeding process, the HUVECs formed lumen structures throughout the DB scaffolds as well as vascular tissue bridging 3D-printed fibers within the HB. The pre-cultured hSMCs were essential for endothelial cell (EC) lumen formation within DB scaffolds, as well as for upregulating EC-specific gene expression of HUVECs grown on HB scaffolds. We further applied this co-culture protocol to DB scaffolds using a perfusion bioreactor, to overcome the limitations of diffusive mass transport into the interiors of the scaffolds. Compared with static culture, panoramic histological sections of DB scaffolds cultured in bioreactors showed improved cellular density, as well as a nominal increase in the number of lumen structures formed by ECs in the interior regions of the scaffolds. In conclusion, we have demonstrated that the sequential seeding of hSMCs and HUVECs can serve to generate early microvascular networks that could further support the in vitro tissue engineering of naturally or synthetically derived bone grafts and in both random (DB) and ordered (HB) pore networks. Combined with the preliminary bioreactor study, this process also shows potential to generate clinically sized, vascularized bone scaffolds for tissue and regenerative engineering.
Keywords: bone regeneration, vascularized bone graft, decellularized bone scaffold, 3D printing bone scaffold, endothelial cell
Introduction
In tissue engineering, one of the major limitations is the inability to provide sufficient blood supply to maintain tissue viability in the initial phases after implantation. Before proper vasculature has been established, the implant must rely on diffusion for the supply of nutrients and the removal of waste. This can lead to nutrient limitations, which can result in improper integration or even death of the implant. Rapid vascularization of implants can help restore the oxygenation of the surgical site, which is important for cell metabolism. Oxygen also prevents infection and induces angiogenesis, which in turn support the survival of implants1,2.
Osteogenesis is a vascular-dependent process, and thus depends on the angiogenic capacity of the implanted bone graft. Angiogenesis plays a vital role in both embryonic skeletal development and in osteogenic repair of critical bone defects3,4. The lack of vascularization within engineered bone constructs has been reported to be one of the major causes for poor implant integration and survival. Recent studies have focused on tissue engineering strategies for improving vascularization in bone scaffolds prior to and post-implantation, which include incorporating pro-angiogenic factors into bone scaffolding materials, as well as introducing cell-derived “bioactive” coatings to the materials’ surfaces.
Incorporation and sustained release of exogenous vascular endothelial growth factor (VEGF) from bone scaffolds (e.g. 35S5 bioactive glass scaffolds) has previously been shown to promote both neovascularization and bone regeneration5–7. However, growth factor delivery in turn depends upon a host response for rapid formation of microvasculature, which may vary depending upon the health of the implant recipient as well as the location of the implant. Thus, the cooperation of endothelial cells (ECs) has become a point of focus in generating pre-vascularized engineered bone constructs. Numerous efforts have been made in this direction. Co-culture of human umbilical vein endothelial cells (HUVECs) and human smooth muscle cells (hSMCs) has been shown effective in facilitating the formation of osteogenic tissue containing an EC network8,9. Similarly, endothelial progenitor cell (EPC)/mesenchymal stem cell (MSC) co-cultures on Matrigel-coated tissue culture plates have been used to investigate the effects of cell ratio on osteogenic and angiogenic marker expression10. This group further cultured the EPC/MSC on calcium polyphosphate bone scaffolds. With increasing time of EPC/MSC culture on scaffolds, the levels of the early osteogenic marker ALP and of the angiogenic factor VEGF increased in culture supernatants. However, EC lumen formation was only observed by co-culture of EPC/MSC on tissue culture plate, and little was learned about vascular lumen formation within bone scaffolds, which would be required for any type of clinical use. Correia and colleagues have cultured HUVECs and hMSCs within a decellularized bone scaffold, which led to only limited formation of capillary-like structures, probably due to the inefficient cell coating on the scaffold11. Here, our study was directed at developing a method to produce robust and numerous vascular luminal structures within both naturally derived, decellularized bone matrix (DB), and synthetically derived 3D-printed Hyperelastic Bone (HB) engineered scaffolds12 with improved cell coating, in order to support bone growth uniformly across a scaffold area.
In this study, we utilized a hSMC pre-culture step, in order to provide a desirable EC growth environment within both synthetic hydroxyapatite and natural extracellular matrix-based bone scaffolds. This is in contrast to many previous approaches, which have utilized MSCs. The conflicting reports regarding the ability of MSCs to support network formation of ECs raise concerns in the clinical efficacy of using MSCs for angiogenic purpose13–15. Overall, the cultivation system comprises three steps: (1) hSMCs are seeded onto, and adhere to, a porous, three-dimensional (3D) bone scaffold surface (naturally or synthetically derived); (2) After 3 weeks of culture, the hSMCs/scaffold construct is seeded with HUVECs and cultured in an endothelial-specific culture medium; (3) fibrin is added to fill the pore spaces after 6 days, to provide a hydrogel environment into which HUVECs can sprout and form microvascular structures. We hypothesized that pre-seeded hSMCs would provide a suitable substrate to support HUVECs survival and angiogenic function in both natural and synthetic scaffold systems. We demonstrated that the co-culture protocol is successful in generating large numbers of lumenized structures within naturally derived bone scaffolds, and also promotes vascular tissue bridging between 3D-printed fibers comprising synthetic scaffolds that are separated by up to 1,000 μm. Robust lumen formations, or tube-like structures, were formed predominantly in specimens that were cultured with all three constituents: hSMCs, HUVECs, and fibrin hydrogel within the pore spaces. In addition, EC-specific gene expression was upregulated when HUVECs were cultured in the presence of hSMCs on the HB scaffolds. As we applied this successfully to both natural and synthetic bone scaffolds, this general approach may inform the microvascularization of other types of connective tissue scaffolds.
Materials and Methods
Fabrication of Bone Scaffolds
Decellularized bone scaffold
Fresh cancellous bone samples were harvested from the spongy bone of porcine rib immediately after slaughter from a local slaughterhouse. Bone blocks (6×6×10 mm) were stored at –20°C until decellularization. Decellularization of porcine bone blocks started with washing with phosphate buffered saline (PBS) to remove the bone marrow. Then, a cycle of solution washes was performed, comprising a step with 2% sodium dodecyl sulfate/10 mM in Tris (American Bioanalytical, MA, USA) for 12 h at room temperature (RT), followed by sequential washes with methanol (RT, 10 min), chloroform (RT, 10 min), ethanol (RT, 10 min), and 0.25% trypsin (37°C for 20 min). Afterwards, bone blocks were rinsed with sonication in ddH2O for 5 min, followed by treatment with an enzyme solution (50U/mL DNAse, 1U/mL RNAse, 10 mM Tris, Roche Applied Science) for 3 h at 37°C. To remove residual detergent, DB samples were washed two times in ddH2O for 24 h, and lyophilized before use. At this point, decellularized bone blocks were cut to thin blocks for further characterization (Fig. 1A–B) and cell culture.
Figure 1.

Imaging of decelluarized bone scaffolds (A: SEM and B: photograph) and HB scaffolds (C: SEM and D: photograph). Scale bars in panels A and C are 500 μm.
Hyperelastic “bone” inks and 3D-printing hyperelastic “bone”
HB inks were synthesized according to previously described methods12,16; in brief, by thorough mixing polylactic-co-glycolic acid (82:18; PLGA; Evonik; 10% solids by weight) micron-scale hydroxyapatite powder (HA; Sigma; 90% by solids weight), and three solvents: dichloromethane (DCM; Sigma), 2-butoxyathanol (2-Bu; Sigma), and dibutyl phthalate (DBP; Sigma). The resulting mixtures were sonicated in a chemical hood, while open, and occasionally stirred, permitting excess DCM to evaporate and the ink to thicken to 30 to 35 Pa·s. All 3D-printed HB structures were fabricated in Ramille Shah’s laboratory at Northwestern University using a 3D-Bioplotter (EnvisionTEC GmbH, Manufacturing Series Model, Germany)12,16. The HB ink was 3D-printed via direct RT extrusion into three, separate 5 cm-diameter cylindrical sheets, using a 250 µm-diameter nozzle at 60–80 mm/s linear deposition speeds. Each sheet comprised 14 layers, with 125 µm inter-layer (Z-direction) spacing (resulting in structures approximately 2 mm thick), and an alternating 0–90° pattern, with every other layer offset in X and Y at a distance of half the designated in-plane strut spacing, similar to a previous report by Jakus et al12. Each of the three sheets was defined by varying in-plane strut (pore) spacing of approximately 300, 700, and 1000 µm (Fig. 1C–D). The resulting 5 cm cylinders were shipped to the Niklason Lab at Yale University, where they were cut/punched to the appropriate size, and washed in 70% ethanol and sterile, deionized water to remove residual solvents as well as sterilize the material prior to cell seeding.
Cell Isolation and Culture
hSMCs were isolated from discarded human aorta tissue as previously described17 and cultured in SMC medium (Dulbecco’s Modified Eagles Medium with 20% fetal bovine serum, and 1% penicillin/streptomycin solution). Primary HUVECs were purchased from the tissue culture core laboratory at the Yale University Vascular Biology Core, and maintained in VascuLife® VEGF endothelial cell medium (Lifeline Cell Technologies, MD, USA). Both cell types were routinely passaged at 80% confluence and used at less than passage #4 for all experiments.
Vascular Cell Culture on Bone Scaffolds
DB scaffolds and HB scaffolds were cut to sizes of 6 mm × 6 mm × 2 mm, and were sterilized in 70% ethanol for 3 h, and then rinsed several times in sterile PBS. Next, 14 µl of DMEM solution containing 1×105 SMCs was added over each bone scaffold. Scaffold/SMC constructs were maintained in culture plates containing SMC medium for 3 weeks, with medium changes twice per week. The constructs were then seeded with HUVECs at a cell density of 5×105 per scaffold, and the scaffold/SMC/EC constructs were then incubated in VascuLife® medium for 6 days. Finally, to further support the formation of vascular structures, the remaining voids in the scaffolds were filled with human fibrin (EVICEL Fibrin Sealant, Ethicon, NJ, USA, fibrinogen 2.75–4.25 mg/ml, thrombin 2–3 IU/ml) for 1, 3, or 5 days (Fig. 2A), before tissue harvesting and analysis.
Figure 2.

Cell culture protocol timelines. (A) Human SMCs were seeded onto the bone scaffold—either DB or HB—and maintained in SMC medium for 3 weeks. Afterwards, HUVECs were seeded, and the constructs cultured in EC medium for 6 days. Fibrin hydrogel was finally added, with ongoing culture in EC medium, for 1–5 days. (B) HUVEC-only culture timeline. DB scaffold was pre-treated with fibronectin for 1 h, and then only HUVECs were seeded and cultured for 6 days, followed by application of fibrin sealant for 1–5 days. Red arrows indicate the work flow direction.
In some DB scaffolds, seeding with SMCs was omitted and only HUVECs were seeded, followed by application of fibrin sealant. For ECs-only studies, the DB scaffold was pre-treated with fibronectin 50 μg/mL to enhance cell attachment (Fig. 2B). Furthermore, the impact of bioreactor culture incorporating medium flow through the seeded scaffolds was studied in sub-sets of DB scaffolds (Fig. 3, see methods below). At the conclusion of culture, histological stains and gene expression analysis were used to characterize SMC and EC viability, distribution, and phenotype.
Figure 3.
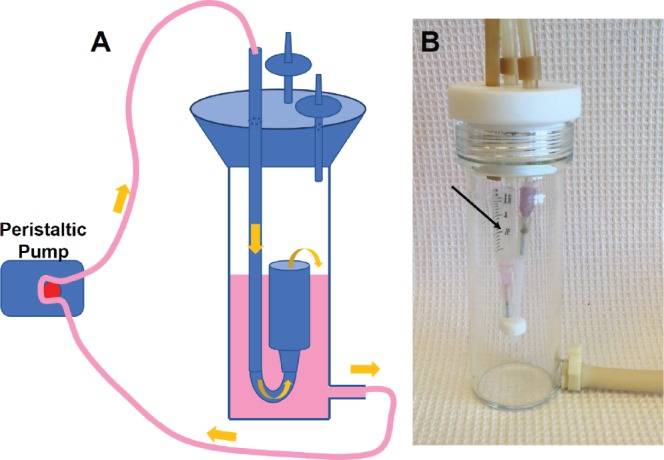
A. Schematic showing the assembled perfusion bioreactor with syringe chamber inside, connected to flow pump and medium flow through tubing in direct of yellow arrows. B. Assembled glass bioreactor, black arrow indicates the syringe chamber.
Bioreactor Culture of Vascular Cells on DB Scaffolds
The bioreactor used in this study was custom-made (Yale University glass-blowing facility) and consisted of a borosilicate glass chamber (51 mm OD, 43 mm ID) capped with a threaded white PTFE cap, as previously described18 (Fig. 3A). Three holes, each 7 mm in diameter, were bored through the cap, one for silicon tubing and two for sterile air filter attachment ports. A medium outlet port, 5 mm, was incorporated at the base. For closed-loop flow, Master-Flex L/S 16 tubing (Cole-ParmerZW-06508-16, IL, USA) was used to attach to this base via a sealing Luer lock (Cole-Parmer ZW-06464-90, IL, USA) to male connectors on the cap. The lumens were 3 cc Luer lock syringes (BD 209657, NJ, USA) cut at the 1.5 cc mark, and the cell/scaffold constructs were inserted into the syringe chamber to undergo medium perfusion. A peristaltic pump was used to provide medium flow (Cole-Parmer, IL, USA).
Prior to flow bioreactor experiments, the hSMC-seeded DB scaffolds were cultured at 37°C for 1 day to allow firm cell attachment. The constructs were then transferred into the vessel chamber (Fig. 3B, black arrow), and 50 mL of SMC medium was added to the bioreactor reservoir. The construct was exposed to a medium flowed at a rate of 1 mL/min for 3 weeks, pumped with a peristaltic pump, and then seeded with HUVECs (5×105 per scaffold). The scaffold/SMC/EC constructs were incubated in VascuLife medium for 1 day under static conditions, and then cultured in the bioreactor with a flow rate of 1 mL/min for 5 days. Finally, the scaffolds were filled with human fibrin for 5 days and cultured under static conditions. The medium was changed twice per week during the entire process.
Histological Evaluations
Histochemical analysis of DB constructs
The samples (DB scaffolds, and cell/DB-scaffold constructs) were formalin-fixed, paraffin-embedded and sectioned at 5 μm thickness. Analysis was performed with standard Hematoxylin and Eosin (H&E) staining and fluorescent immunohistochemistry (IHC) staining for CD31 and von Willebrand factor (vWF) as markers of endothelium (see specifics, below). H&E images were taken using a Zeiss microscope (Axioskop 2 plus, Germany) and EVOS Cell Imaging Systems (Thermo Fisher Scientific, USA), and IHC images were taken using Leica 6000 microscope.
Rhodamine phalloidin staining
The cell/DB-scaffold and cell/HB-scaffold constructs were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 for 5 min at RT. Fluorescent rhodamine phalloidin and DAPI (Molecular Probes, Invitrogen, CA, USA) were used for labeling F-actin and nuclei, respectively. Multiple images were obtained with confocal laser scanning microscopy (Leica TCS SP5 Spectral Confocal Microscope, Germany) using a z-stack scanning mode with a step size of 3 µm.
Immunofluorescence staining of HB constructs
The HB scaffold is not suitable for routine paraffin embedding and thin sectioning, so immunofluorescence stains were evaluated on thick sections using confocal microscope imaging. The cell/HB-scaffold constructs were washed with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at RT. Cells were then permeabilized and blocked for non-specific antigen binding with PBS + 10% FBS + 0.1% Triton X-100 for 1 h at RT. The constructs were then incubated with primary antibodies against PECAM-1 (CD31, Dako JC70A, CA, USA), VE-cadherin (Santa Cruz, sc9989, TX, USA), and Von Willebrand factor (vWF, Abcam ab6994, MA, USA), overnight at 4°C. The next day, constructs were washed three times with 1× PBS and incubated with the corresponding secondary antibody (1:500) for 2 h at RT. DAPI (1 μg/mL) was used to label nuclei before confocal microscope imaging.
Real-Time Quantitative RT-PCR Analysis
qRT-PCR was used to determine the expression of endothelial markers in HB scaffolds containing SMC/EC/Fibrin. HB/EC/Fibrin constructs were used as controls, and served as the normalization standard for PCR data. Briefly, total cellular RNA was prepared using Trizol Reagent (Life Technologies, 15596026, CA, USA). Single-stranded cDNA was synthesized using the reverse transcription-PCR protocol of the First Strand cDNA Synthesis Kit from Invitrogen. Quantitative real-time PCR was performed using SYBR Green PCR Supermix (Bio-Rad, CA, USA). Concentrations of all primers were optimized before use. PCR conditions included an initial denaturation step of 4 min at 95°C, followed by 40 cycles of PCR consisting of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Each sample was run in triplicate. The comparative Ct value method using GAPDH as a housekeeping gene for an internal standard was employed to determine relative levels of gene expression. Ct values from the triplicate PCR reactions for a gene of interest (GOI) were normalized against average GAPDH Ct values from the same cDNA sample. Fold change of GOI transcript levels between sample A and sample B = 2-ΔΔCt, where ΔCt = Ct(GOI) – Ct(GAPDH) and ΔΔCt = ΔCt(A) – ΔCt(B). Primers were used in this study are listed in Table 1.
Table 1.
Sequences of Primers Used in qRT-PCR.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | GACAACAGCCTCAAGATCATCAG | ATGGCATGGACTGTGGTCATGAG |
| vWF | CCGATGCAGCCTTTTCGGA | TCCCCAAGATACACGGAGAGG |
| CD31 | TGCAGTGGTTATCATCGGAGTG | CGTTGTTGGAGTTCAGAAGTG |
| VE-Cadherin | GTTCACGCATCGGTTGTTCAA | CGCTTCCACCACGATCTCATA |
Statistics
The qPCR experiment was repeated three times and each sample was analyzed in triplicate. Data are presented as the mean ± SD for quantitative variables. One-way variance (ANOVA) analysis was used to determine whether there was an effect of HB scaffold spacing on gene expression. A post-hoc Tukey test was performed to evaluate whether the two groups were significantly different from each other. A p value of p < 0.05 (two-tailed) was considered statistically significant.
Results
Mono-Culture of SMCs and ECs Within the DB Scaffold
To evaluate the suitability of the DB scaffold for SMC and EC culture, we first performed mono-culture of these two cell types on the scaffold. hSMCs that were cultured under static conditions for 3 weeks demonstrated extensive coverage of the previously acellular matrix, that was visible by both H&E staining and by scanning electron microscopy (SEM, Fig. 4A). In some instances, hSMC growth was vigorous enough to produce circular structures on histology as seen by phalloidin staining (Fig. 4A), though these structures did not stain with endothelial markers (data not shown).
Figure 4.

Vascular cell culture on DB scaffolds. (A) Human SMCs cultured for 3 weeks on DB scaffold show extensive coverage of the matrix by H&E, SEM, and phalloidin staining. (B) Human EC (HUVECs) cultured on DB scaffolds for 6 days, with or without fibrin, showed overall little cell growth and a slight tendency for lumen formation (arrows).
Human ECs cultured on the DB scaffold also proliferated in the presence of EC medium after pre-treatment with fibronectin 50 μg/mL, but merely coated the scaffold surface and did not spontaneously participate in lumen formation after 6 days of culture (Fig. 4B). However, if fibrin sealant was added after 6 days of EC culture, then in some instances a small number of lumen-like structures were visible (Fig. 4B last panel, arrows).
Co-Culture of SMC and EC Within the DB Scaffold
We next assessed the ability of hSMCs to assist HUVECs in forming vascular lumen-like structures throughout the scaffold (Fig. 5). When ECs were cultured for 6 days after pre-culture of hSMCs for 3 weeks, H&E staining was notable for more frequent formation of lumen-like structures within the pores of the DB scaffold (Fig. 5A). When fibrin sealant was added and histology evaluated sequentially, we noted an apparent increase in lumen-like formation after 5 days of fibrin culture (Fig. 5B–D).
Figure 5.
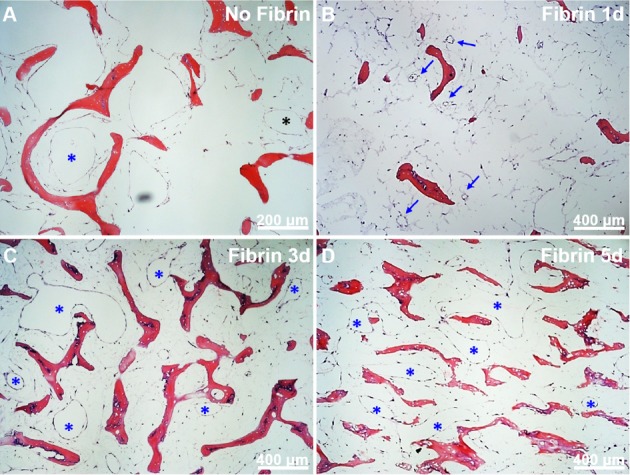
Vascular cell co-culture on DB scaffolds. A: SMC and EC show few lumen-like structures (asterisks); B: One day after fibrin addition shows multiple small lumens appearing (arrows); C, D: Three and 5 days after fibrin addition shows increasing numbers and sizes of lumen-like structures (asterisks).
To determine whether the lumen-like structures forming within the DB scaffold were lined with ECs, we performed IHC for two EC markers: vWF and CD31. Immunofluorescence of SMC-EC-fibrin cultures showed clear luminal structures in the interstices of the DB scaffold, that were lined by cells staining positively for vWF and for CD31 (Fig. 6A–C). In no case did we observe luminal structures staining for SMC markers such as alpha smooth muscle actin or calponin (data not shown).
Figure 6.

Immunostaining for EC markers in SMC-EC-fibrin cultures. A: vWF (green) and DAPI for nuclei (blue) shows green-lined lumens (asterisks) in scaffold interstices after 3 days of fibrin culture. Slight non-specific staining of DB matrix by DAPI is visible. B, C: CD31 (red), vWF (green) and DAPI staining show EC-lined luminal structures.
Bioreactor Culture of DB with Vascular Cells Study
In the static co-culture studies above, it is interesting to note that most cell survival and lumen formation tended to occur on the outer surfaces of the 6 mm × 6 mm × 2 mm DB scaffold (Fig. 7A). This is due, presumably, to limitations in diffusive mass transport to the interior of the scaffold area. We and others have shown previously that enhancement of mass transport and the generation of hydrodynamic shear are critically important for EC viability, as well as tissue viability within DB scaffolds19,20. Therefore, we hypothesized that a bioreactor providing dynamic medium flow (1 mL/min) through the bulk of the scaffold would support more vascular lumen formation throughout the DB scaffold. Histological analysis clearly demonstrates that, in contrast to static culture, cell survival and lumen formation is visible throughout the entire scaffold after 5 days of dynamic perfusion culture of HUVECs that are seeded onto SMC-containing DB scaffolds (Fig. 7B). As with static culture, luminal structures also stain positively for EC markers under bioreactor flow (lower insets, Fig. 7A, B).
Figure 7.

Histological staining of hSMCs/ECs/fibrin/DB constructs cultured under static (A) and bioreactor flow conditions (B). Upper right insets in both panels show enlarged image of central DB scaffold area. Yellow arrows show lumen formation, which is absent in the center of the static culture (A). Lower left insets show immunostaining of representative luminal structures, with vWF (green) and CD31 (red).
Formation of Vascular Structures Within the HB Scaffold
We next evaluated the applicability of this vascular cell co-culture strategy to the synthetic HB scaffolds16. Three distinct HB scaffold groups, defined by 3D-printed fiber spacings of 300 μm, 700 µm, and 1,000 μm (1.0 mm) were used to evaluate the impact of pore size on expression of genes of vascular differentiation.
We first performed hSMCs culture to assess the ability of HB scaffolds to support adhesion and proliferation of hSMCs (Fig. 8). hSMCs seeded on the HB scaffolds proliferated over the course of 3 weeks to cover the entirety of the HB scaffold fiber surfaces. No bridging fibers or lumenized structures were observed with hSMC mono-culture for any of the three pore sizes (Fig. 8).
Figure 8.

hSMCs cultured on HB scaffolds of varying pore sizes for 1–3 weeks, evaluated by confocal imaging. A: hSMC cultured on HB scaffold with 300 µm pore size for 1 week. B: hSMC cultured on 700 µm pore size scaffold for 1 week; and C: hSMC cultured on 1000 µm scaffold for 1 week. D: Cells on HB with 1,000 µm pore size at 3 weeks, 3D reconstruction of several fibers using confocal microscopy. Blue is DAPI stain (nuclei); red is phalloidin (cytoskeleton).
Thereafter, HUVECs were added to hSMCs that had been cultured for 3 weeks and followed by the application of fibrin sealant for 5 days, according to the regimen in Fig. 2. The HB scaffolds remained robust and no degradation was observed during the entire cell culture time. At the end time point, hSMC-HUVEC confocal imaging revealed robust expression of vWF and VE-cadherin expressed by the HUVECs cultured on HB scaffolds (Fig. 9). Furthermore, and in contrast to SMC mono-culture, we also observed formation of a bridging network between fibers of the HB scaffolds, for all three pore sizes examined (Fig. 9B–D). Bridging fibers co-stained with EC markers (VE-cadherin or vWF) and with phalloidin, indicating the presence of ECs on the fiber bridging structure. But while hSMC/EC co-culture clearly led to more cross-fiber tissue formation as compared with hSMC culture alone, we did not observe true lumen formation at any of the scaffold pore sizes. This observation may be due to the relatively large pore sizes of the HB scaffolds as compared with typical capillary diameters, or perhaps because of the short time period of hSMC/HUVEC co-culture (only 6 days).
Figure 9.

In vitro EC cellular network formation on HB scaffolds. A1, B1, C1, D1: SEM images of HB scaffolds, yellow arrows indicating the corresponding confocal scanning area in subsequent panels. Pore sizes for A1–D1 are 700 μm, 300 μm, 700 μm, and 1,000 μm, respectively. A2–A5: Confocal scanning of an individual HB scaffold fiber with hSMC and HUVEC co-cultured. B2–B5: Confocal scanning of HB scaffold 300 µm pore spacing with hSMC and HUVEC co-culture. C2–C5: Confocal scanning of HB scaffold with 700 µm pore spacing. D2–D5: Confocal scanning of HB scaffold 1000 µm pore spacing. Blue is DAPI staining for nuclei, green is VE-cadherin (EC marker), red is phalloidin (cytoskeletal marker) and yellow is vWF (EC marker).
Because most solvents used in histology processing dissolve or embrittle the polymer component of the HB scaffold, the HB scaffolds are not amenable to standard formalin fixation and paraffin embedding. Therefore, qPCR was performed to evaluate expression level differences for EC markers (vWF, CD31, and VE-cadherin) between hSMC/EC co-culture and HUVECs monoculture on HB scaffolds (Fig. 10A). Gene expression analysis further supported the notion that cell co-culture strategy promoted vascular formation within porous bone scaffolds. HUVECs that were cultured with hSMCs showed clear up-regulation of EC-related genes: vWF (1.5–4.6-fold increase); CD31 (2.5–7.1-fold increase); and VE-cadherin (1.7–5.1-fold increase). We also observed a pore size effect of the HB scaffolds on EC gene expression. vWF and CD31 gene expression significantly increased with the increase of HB scaffold pore size going from 300 to 1,000 μm, while VE-cadherin gene expression showed a decreasing trend (ANOVA, p < 0.01). The decreasing trend for VE-cadherin only may have been due to the decreased EC–EC contact formation that occurred on the HB scaffold having very large (1,000 µm) pores, since VE-cadherin is an intercellular junctional protein.
Figure 10.

A: EC marker expression level differences between co-culture and mono-culture in HB scaffolds with various pore sizes, exhibiting gene up-regulation during co-culture, the degree of which depends on the pore size. Values of three independent experiments from the triplicate PCR reactions for genes of interest were normalized against average GAPDH Ct values from the same cDNA sample. Fold change of GOI transcript levels between co-culture and monoculture equals to 2-ΔΔCt, where ΔCt = Ct(GOI) – Ct(GAPDH) and ΔΔCt = ΔCt(co-culture) – ΔCt(monoculture). (n = 3, error bars refer to SD, *p < 0.05). B: EC marker expression level of HUVECs monoculture on HB scaffolds with various pore sizes, showing pore size independent expression levels.
Fig. 10B displays EC gene expression level in HB/HUVECs/Fibrin constructs with various pore sizes, showing similar gene expression levels for cells cultured on different pore size scaffolds. The fact that pore size only affected the gene expression levels in the case of co-culture suggested the pore size could have played a role during the culture of SMCs (the only difference between co-culture and monoculture).
Discussion
A timely and coordinated angiogenic response is important for successful bone repair. When a bone fracture extends beyond a size where passive diffusion for the exchange of nutrients is sufficient, the development of a vascular system is required for final healing. Because the considerable amount of time required for angiogenesis, the use of large, non-autologous bone grafts and flaps is not often feasible, since necrosis develops in the core with only a shell of bone remaining at the surface21,22. Thus, the ability to produce viable, pre-vascularized bone grafts could vastly expand the number of surgical scenarios wherein non-autologous bone grafts have therapeutic value. We have demonstrated that a sequential SMCs and HUVECs co-culture strategy that can be used to engineer bone graft scaffolds containing live vascular tissues, using both native and synthetically derived scaffolds.
The major goal of this study was to test the hypothetical approach that pre-vascularized bone constructs can be formed in vitro from either random porosity, naturally derived materials as well as ordered porosity, synthetically derived materials in conjunction with sequential seeding and culture of SMCs and ECs. To this end, we seeded the HUVECs on both naturally derived bone scaffolds (DB) defined by a random pore network, as well as synthetically derived bone scaffolds (HB) defined by ordered, 3D-printed pore networks, with and without the pre-cultured SMCs layer on the bone scaffolds. By filling bone scaffold pore space with fibrin afterwards, we expected that HUVECs would migrate into the gel and form micro-vessel tube structures in the bone scaffold’s spacing areas.
In the DB scaffold study, the presence of SMCs prior to the introduction of HUVECs was an important factor to improve HUVECs attachment on DB scaffold wall and to stimulate the angiogenic tube formation in DB pore space. A similar result was observed in the synthetic, HB scaffolding system, where substantial vessel-forming activity only occurred in sample groups that had been pre-cultured with SMCs. In addition, although the degree of EC activity varied across the HB pore size groups (300, 700, and 1000 µm), it was present in all groups if they had been previously seeded with SMCs. In the HB scaffold study, we further observed HUVECs creating a bridging network between the 3D-printed scaffold fibers, even when those fibers were 1000 µm apart, and increased EC marker gene expressions within groups with SMCs as opposed to groups without SMCs. These results illustrate that, despite the pore structure (random or ordered, small or very large pores) or composition (natural or synthetic) of the underlying scaffold, that SMCs, or the matrix that SMCs produce, are necessary for the ECs to create and support a viable microvascular environment in vitro.
SMCs and ECs are the two primary cell types in blood vessels, and their communication is critical during blood vessel formation. The individual functions of ECs, such as migration, proliferation, and tube formation, are dependent on proper communication between them and SMCs23,24. In our approach, we provide a direct physical contact between ECs and SMCs on the bone scaffolds. After applying fibrin to induce tubulogenesis in ECs, the direct contact enhanced EC lumen formation (DB study) and EC gene expression (HB study). Mechanistically, the cell contact-dependent signaling between ECs and SMCs facilitates cell differentiation and vessel maturation25–29. Many studies have demonstrated that formation of stable and functional vascular networks necessitates the co-implantation of ECs with SMCs30–32. Our study supports these previous observations, as the lumen structures formed by HUVECs only increased with culture time in the presence of SMCs.
In the histology study, we also observed the limitation of static cell culture: that EC lumen structures are mainly formed in the outer region of bone constructs. After applying medium flow through the interstices of the scaffolds for efficient transport of nutrients and waste materials between the cells and culture medium, spatially uniform EC lumen structures were formed in DB scaffolds. The results demonstrated our co-culture strategy was effective in generating vascularized bone grafts in dynamic culture conditions. Understanding the dynamic culture flow rate and flow pattern, which is important for determining the upper size limit of vascularized bone construct that can be grown in vitro, will need further investigation. In addition, evaluating the effects of in vivo vasculogenesis and osteogenesis, particularly with respect to anastomosis with host vessels, will be a necessary follow-on step to evaluate the functional impact of the bone scaffold vascularization procedure.
Conclusion
We have developed an EC and smooth muscle cell co-culture method to engineer vascularized bone constructs using both natural decellularized bone with random porosity, and synthetic 3D-printed Hyperelastic Bone with ordered porosity. The in vitro pre-vascularization procedure presents an effective way to generate micro-vessel lumen structures in the naturally derived demineralized bone scaffold and EC network in the synthetically derived hydroxyapatite bone scaffold. In addition, improved cellular density and EC lumen formation in the scaffold interior was observed after applying the procedure in a dynamic bioreactor culture. Overall, this study shows potential to generate pre-vascularized bone constructs of clinical relevant size.
Acknowledgements
This work was supported by Yale University, and by NIH R01 HL127386 and by an unrestricted research gift from Humacyte Inc (both to LEN), and by Synthes Research Grant (DMS). Hyperelastic Bone samples were produced at the Simpson Querrey Institute for BioNanotechnology (SQI) at Northwestern University. The U.S. Army Research Office, the U.S. Army Medical Research and Material Command, and Northwestern University provided funding to develop SQI. AEJ was partially supported through a postdoctoral fellowship by The Hartwell Foundation. Additional funding support was provided by an unrestricted research gift from Google (RNS).
Authors' Note: Dr. Derek M. Steinbacher at Yale University and Dr. Ramille Shah at Northwestern University accept direct responsibility for the manuscript.
Ethical Approval: Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights: Statement of Human and Animal Rights is not applicable for this article.
Statement of Informed Consent: Statement of Informed Consent is not applicable for this article.
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: LEN is a founder and shareholder in Humacyte, Inc, which is a regenerative medicine company. Humacyte produces engineered blood vessels from allogeneic smooth muscle cells for vascular surgery. LEN’s spouse has equity in Humacyte, and LEN serves on Humacyte’s Board of Directors. LEN is an inventor on patents that are licensed to Humacyte and that produce royalties for LEN. LEN has received an unrestricted research gift to support research in her laboratory at Yale. Humacyte did not influence the conduct, description, or interpretation of the findings in this report. AEJ and RNS are co-founders and shareholders in Dimension Inx, LLC, which designs, develops, manufactures, and sells new 3D-printable materials and end-use products for medical and non-medical applications. As of August 2017, AEJ is currently full time Chief Technology Officer of Dimension Inx, LLC, and RNS serves part time as Chief Science Officer of Dimension Inx LLC.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Yale University, NIHR01 HL127386, unrestricted research gift from Humacyte Inc, Synthes Research Grant, The U.S. Army Research Office, the U.S. ArmyMedical Research and Material Command, Northwestern University Postdoctoral fellowship from The Hartwell Foundation, an unrestricted research gift from Google.
References
- 1. Bishop A. Role of oxygen in wound healing. J Wound Care. 2008;17(9):399–402. [DOI] [PubMed] [Google Scholar]
- 2. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010;89(3):219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Portal-Núñez S, Lozano D, Esbrit P. Role of angiogenesis on bone formation. Histology Histopathol. 2012;27(4):559–566. [DOI] [PubMed] [Google Scholar]
- 4. Saran U, Piperni SG, Chatterjee S. Role of angiogenesis in bone repair. Arch Biochem Biophys. 2014;561:109–117. [DOI] [PubMed] [Google Scholar]
- 5. Leach JK, Kaigler D, Wang Z, Krebsbach PH, Mooney DJ. Coating of VEGF-releasing scaffolds with bioactive glass for angiogenesis and bone regeneration. Biomaterials 2006;27(17):3249–3255. [DOI] [PubMed] [Google Scholar]
- 6. Leu A, Leach JK. Proangiogenic potential of a collagen/bioactive glass substrate. Pharm Res. 2008;25(5):1222–1229. [DOI] [PubMed] [Google Scholar]
- 7. Leu A, Stieger SM, Dayton P, Ferrara KW, Leach JK. Angiogenic response to bioactive glass promotes bone healing in an irradiated calvarial defect. Tissue Eng Part A. 2008;15(4):877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rouwkema J, Boer JD, Blitterswijk CAV. Endothelial cells assemble into a 3-dimensional prevascular network in a bone tissue engineering construct. Tissue Eng. 2006;12(9):2685–2693. [DOI] [PubMed] [Google Scholar]
- 9. Rouwkema J, Westerweel PE, de Boer J, Verhaar MC, van Blitterswijk CA. The use of endothelial progenitor cells for prevascularized bone tissue engineering. Tissue Eng Part A. 2009;15(8):2015–2027. [DOI] [PubMed] [Google Scholar]
- 10. Fu W-L, Xiang Z, Huang F-G, Gu Z-P, Yu X-X, Cen S-Q, Zhong G, Duan X, Liu M. Coculture of peripheral blood-derived mesenchymal stem cells and endothelial progenitor cells on strontium-doped calcium polyphosphate scaffolds to generate vascularized engineered bone. Tissue Eng Part A. 2014;21(5-6):948–959. [DOI] [PubMed] [Google Scholar]
- 11. Correia C, Grayson WL, Park M, Hutton D, Zhou B, Guo XE, Niklason L, Sousa RA, Reis RL, Vunjak-Novakovic G. In vitro model of vascularized bone: synergizing vascular development and osteogenesis. PloS one. 2011;6(12):e28352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jakus AE, Rutz AL, Jordan SW, Kannan A, Mitchell SM, Yun C, Koube KD, Yoo SC, Whiteley HE, Richter C-P. Hyperelastic “bone”: A highly versatile, growth factor–free, osteoregenerative, scalable, and surgically friendly biomaterial. Sci Transl Med. 2016;8(358):358ra127. [DOI] [PubMed] [Google Scholar]
- 13. Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009;113(18):4197–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho IA, Toh HC, Ng WH, Teo YL, Guo CM, Hui KM, Lam PY. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells. 2013;31(1):146–155. [DOI] [PubMed] [Google Scholar]
- 15. Menge T, Gerber M, Wataha K, Reid W, Guha S, Cox CS, Jr, Dash P, Reitz MS, Jr, Khakoo AY, Pati S. Human mesenchymal stem cells inhibit endothelial proliferation and angiogenesis via cell–cell contact through modulation of the VE-cadherin/β-catenin signaling pathway. Stem Cells Dev. 2012;22(1):148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jakus AE, Shah R. Multi and mixed 3D-printing of graphene-hydroxyapatite hybrid materials for complex tissue engineering. J Biomed Mater Res B. 2017;105(1):274–283. [DOI] [PubMed] [Google Scholar]
- 17. Dahl SL, Kypson AP, Lawson JH, Blum JL, Strader JT, Li Y, Manson RJ, Tente WE, DeBernardo L, Hensley MT, Carter R, Williams TP, Prichard HL, Dey MS, Begelman KG, Niklason LE. Readily available tissue-engineering vascular grafts. Sci Transl Med. 2011;3(68):68ra9. [DOI] [PubMed] [Google Scholar]
- 18. Sivarapatna A, Ghaedi M, Le AV, Mendez JJ, Qyang Y, Niklason LE. Arterial specification of endothelial cells derived from human induced pluripotent stem cells in a biomimetic flow bioreactor. Biomaterials. 2015;53:621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu W, Le AV, Mendez JJ, Chang J, Niklason LE, Steinbacher DM. Osteogenic performance of donor-matched human adipose and bone marrow mesenchymal cells under dynamic culture. Tissue Eng Part A. 2015;21(9–10):1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grayson WL, Fröhlich M, Yeager K, Bhumiratana S, Chan ME, Cannizzaro C, Wan LQ, Liu XS, Guo XE, Vunjak-Novakovic G. Engineering anatomically shaped human bone grafts. Proc Natl Acad Sci USA. 2010;107(8):3299–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med 1973;138(4):745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsigkou O, Pomerantseva I, Spencer JA, Redondo PA, Hart AR, O’Doherty E, Lin Y, Friedrich CC, Daheron L, Lin CP. Engineered vascularized bone grafts. Proc Natl Acad Sci USA. 2010;107(8):3311–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marcelo KL, Goldie LC, Hirschi KK. Regulation of endothelial cell differentiation and specification. Circ Res. 2013;112(9):1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol. 2011;3(8):a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, Weis SM, Byzova TV, Ruggeri ZM, Iruela-Arispe ML. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. 2012;119(9):2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124(1):161–173. [DOI] [PubMed] [Google Scholar]
- 27. Korff T, Braun J, Pfaff D, Augustin HG, Hecker M. Role of ephrinB2 expression in endothelial cells during arteriogenesis: impact on smooth muscle cell migration and monocyte recruitment. Blood. 2008;112(1):73–81. [DOI] [PubMed] [Google Scholar]
- 28. Fang JS, Dai C, Kurjiaka DT, Burt JM, Hirschi KK. Connexin45 regulates endothelial-induced mesenchymal cell differentiation toward a mural cell phenotype. Arterioscler Thromb Vasc Biol. 2013;33(2):362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693. [DOI] [PubMed] [Google Scholar]
- 30. Williams C, Wick TM. Endothelial cell–smooth muscle cell co-culture in a perfusion bioreactor system. Ann Biomed Eng. 2005;33(7):920–928. [DOI] [PubMed] [Google Scholar]
- 31. Ziegler T, Alexander R, Nerem R. An endothelial cell-smooth muscle cell co-culture model for use in the investigation of flow effects on vascular biology. Ann Biomed Eng. 1995;23(3):216–225. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y, Rayatpisheh S, Chew SY, Chan-Park MB. Impact of endothelial cells on 3D cultured smooth muscle cells in a biomimetic hydrogel. ACS Appl Mater Interfaces. 2012;4(3):1378–1387. [DOI] [PubMed] [Google Scholar]