

Abstract
Patient: Male, 1
Final Diagnosis: PDAC
Symptoms: Respiratory failure
Medication: —
Clinical Procedure: —
Specialty: Pediatrics and Neonatology
Objective:
Congenital defects/diseases
Background:
The pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect (PDAC) syndrome is a rare medical condition presumably of autosomal recessive way of inheritance with only a few reported cases. Recessive mutations in the STRA6 and both recessive and dominant mutations in RARB gene have been identified as the cause of anophthalmia/microphthalmia and other abnormalities included in the PDAC spectrum. However, those mutations have not been found in all PDAC syndrome cases reviewed from the literature.
Case Report:
We report a case of a full-term living male infant with pulmonary hypoplasia, left diaphragmatic eventration, bilateral microphthalmia, congenital cardiac defects, and severe pulmonary hypertension.
Conclusions:
The main feature in the reported cases was anophthalmia/microphthalmia. Therefore, screening for the other associated congenital anomalies is highly suggested. Mutations in STRA6 and RARB genes are commonly encountered in this spectrum. However, whole exome sequencing of suspected cases and their parents is recommended to detect possible de novo mutations. Further reports are needed to identify risk factors and prognosis of this rare syndrome.
MeSH Keywords: Anophthalmos; Diaphragmatic Eventration; Heart Defects, Congenital; Lung Diseases
Background
The PDAC (pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect) syndrome or as it has been previously known as the Spear or Mathew-Wood syndrome [1], is currently called microphthalmic syndrome 9 (MCOPS 9, OMIM601186) and is a rare medical condition presumably of autosomal recessive way of inheritance [2]. Ostor et al. described a premature stillborn infant with bilateral pulmonary agenesis and anophthalmia [3]. Spear et al. reported a stillborn boy with bilateral pulmonary agenesis and bilateral microphthalmia associated with a complex cardiac defect, and eventration of the left hemidiaphragm [4]. Seller et al. reported 2 siblings of nonconsanguineous parents, who had bilateral anophthalmia and pulmonary hypoplasia and called this disorder Mathew-Wood syndrome as requested by the parents after the name of a first-born sib [2]. The combination of pulmonary, diaphragmatic, ocular and cardiac congenital malformations has been reported occasionally in the past 4 decades. Some of the reported cases were prenatally diagnosed [2,5,6] while others were observed in the postnatal periods [4,7–9]. Engellenner et al., reported the only case associated with hydrops fetalis [5]. The first reported living patient was presented by Chitayat et al. [10]. Mutations in the STRA6 (stimulated by retinoic acid 6) gene have been identified as the cause of anophthalmia/microphthalmia and other abnormalities included in the PDAC spectrum [11]. However, those mutations were not found in all PDAC syndrome cases in several reports [10,12,13]. Both recessive and dominant mutations in RARB (retinoic acid receptor beta) gene were found to cause anophthalmia and/or microphthalmia and diaphragmatic hernia, providing further evidence of the role of retinoic acid pathway in eye development and organogenesis [14].
We report a full term living male infant with pulmonary hypoplasia, left diaphragmatic eventration, bilateral microphthalmia, congenital cardiac defects, and severe primary pulmonary hypertension.
Case Report
Our patient case was a male born at 38-week gestation via caesarian section delivery for a 33-year old mother with insignificant prenatal history other than gestational diabetes managed with insulin. Her 3 previous pregnancies resulted in 3 healthy children.
The couple is nonconsanguineous, both of Saudi Arabian origin with no history of a similar condition, previous abortions, or congenital anomalies in the family. The mother had an ultrasound study at the 33rd week of pregnancy, which showed absent nasal bone, absent gastric shadow, unilateral clubfoot, and polyhydramnios. Head and spine were normal. Heart was not seen well. The scan was limited for anomaly detection due to fetal position and maternal size.
At birth, the baby was flat and required positive pressure ventilation for 4 minutes. Apgar score was 3, 7, and 9 at 1, 5, and 10 minutes of age, respectively. Birth weight was 2960 g (25%), length was 49 cm (50%), and head circumference was 35.5 cm (95%). The newborn was admitted to the neonatal intensive care unit (NICU) directly and required continuous positive airway pressure (CPAP). Physical examination revealed a male born with dysmorphic features including bilateral microphthalmia, wide nasal bridge, low set ears, and talipes equinovarus of the right foot. He was having respiratory distress and stridor. However, both nostrils were patent. Chest radiograph showed elevated left hemidiaphragm with the mediastinum shifted to the right side. Bowel loops were noted higher up in the distal part of the left hemithorax. The left lung was small in size, while the right lung appeared hyperinflated.
At 12 hours of age, the patient was intubated and mechanically ventilated due to respiratory failure.
Echocardiogram (Figures 1, 2A, 2B) on the first day of life showed mesocardia, normal systemic and pulmonary veins, dilated right atrium and right ventricle, small to moderate atrial septal defect (ASD) with bidirectional shunt, moderate tricuspid regurgitation, a large patent ductus arteriosus (PDA) with bidirectional shunt, and severe pulmonary hypertension; he was started on inhaled nitric oxide.
Figure 1.

Echocardiography showing right atrium (RA), left atrium (LA) and atrial septal defect (ASD).
Figure 2.

(A, B) Echocardiography showing the main pulmonary artery (PA), right pulmonary artery (RPA), aortic valve (AOV), and patent ductus arteriosus (PDA).
At the age of 12 days, echocardiogram showed a closed PDA, severe right ventricular hypertrophy and dilatation, and severe pulmonary hypertension, most likely due to lung hypoplasia and a small left pulmonary artery, hence he was commenced on sildenafil.
When he was 26 days old, PDA re-opened with a bidirectional shunt as evidenced by echocardiogram.
Chest computed tomography (CT) scan with IV contrast showed eventration of the posterior part of the left hemidiaphragm with no defect/gap (Figure 3A, 3B). The mediastinum was shifted to the right side, and spleen and stomach were highly positioned. The left pulmonary artery was not clearly visualized 1 cm beyond its bifurcation. Both lung fields showed evidence of pulmonary infiltrations.
Figure 3.

(A, B) Coronal section of chest computed tomography scan showing dextrocardia, shifted mediastinum to the right side, eventration of the left hemidiaphragm, and bilateral pulmonary infiltrations.
Plain brain CT scan (Figure 4) showed immature brain with no structural abnormalities. Abdominal ultrasound was unremarkable except for mild left renal pelviectasis. Lung perfusion scan (Figure 5A, 5B) revealed that the left lung perfusion was decreased as compared to the right lung contributing to only 20% of total lung perfusion. Chest ultrasound and fluoroscopy proved that the left hemidiaphragm was elevated with absent direct movement.
Figure 4.
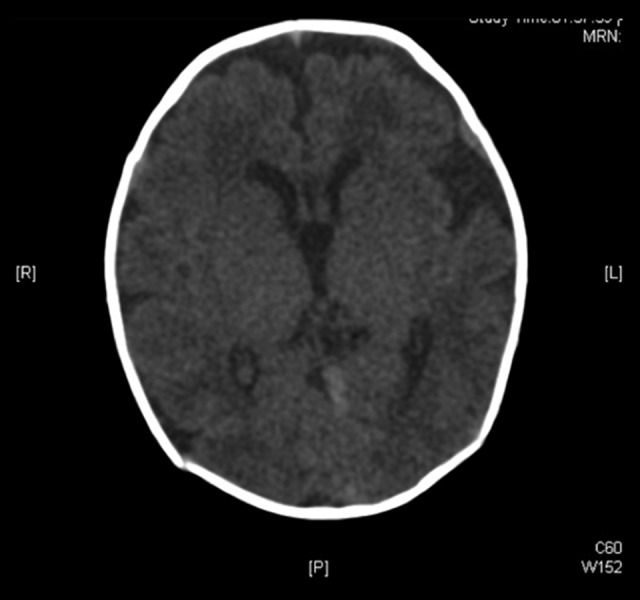
Axial section of plain brain computed tomography scan showing immature brain with no structural abnormalities.
Figure 5.

(A, B) Lung perfusion scan showing decreased left lung perfusion compared to the right lung, contributing to only 20% of total lung perfusion.
Due to the presence of stridor, flexible bronchoscopy was done and showed laryngomalacia and narrowing of the right-sided choana. At 3 weeks of age, we were able to extubate the patient to CPAP. Eight days later, he went into respiratory failure and was re-intubated. Due to his respiratory insufficiency and mechanical ventilation dependency, his family was counseled, and an informed consent was obtained regarding tracheostomy, which was done on the 35th day of age.
Chromosomal analysis showed a normal male karyotype (46, XY). Whole exome sequencing showed no causative pathogenic mutations, but de novo events were not detected because parental exome sequencing was not done.
Magnetic resonance imaging (MRI) of the brain at 4 months of age (Figure 6A–6F) showed thin corpus callosum and bilateral widened cerebrospinal fluid spaces anterior to temporal and frontal lobes but no hydrocephalus. Myelination was age appropriate. Posterior fossa structures appeared normal with no other gross structural morphological abnormalities.
Figure 6.

(A–F) Magnetic resonance imaging of the brain showing thin corpus callosum, bilateral widened cerebrospinal fluid spaces without hydrocephalus and age appropriate myelination. Posterior fossa structures appear normal with no gross structural abnormality.
Physical examination of the patient at 6 months of age revealed a conscious, global developmentally delayed, ventilator dependent tracheostomized male infant who was able to move his limbs minimally. He had broad forehead, small palpebral fissures, scant hair growth at the lateral third of eyebrows, deep-seated small eyes, broad nasal root, low-set ears, short neck, a Mongolian spot over the sacral area, and right sided talipes equinovarus. He had bilateral clear corneas and lenses, normal retinae and optic discs, and equal round reactive pupils but he did not fix and follow. Ophthalmology examination revealed blindness. He also failed newborn hearing test. His cardiac apex was on the right side with a continuous murmur heard on auscultation. The limbs, apart from right clubfoot, the abdomen and the external genitalia were normal.
Discussion
Only a few cases of PDAC syndrome have been reported in the last 4 decades (Table 1). However, not all of the cases had all the congenital anomalies occurring together. Yet, all the cases had overlapping features with anophthalmia/microphthalmia being the most remarkable sign. We report here a full-term male living infant who has dysmorphic features, bilateral microphthalmia, left-sided diaphragmatic eventration, left lung hypoplasia, ASD, PDA, severe primary pulmonary hypertension, laryngomalacia, narrow right-sided choana, global developmental delay, and normal brain structure apart from thin corpus callosum and bilateral widened cerebrospinal fluid (CSF) spaces. Despite the presence of all the main features of PDAC syndrome in our reported case, whole exome sequence did not detect any causative pathogenic gene mutations. Unfortunately, exome sequencing of his parents was not done which could have helped us in detecting possible de novo events. Ostor et al. reported 2 cases of bilateral pulmonary agenesis and anophthalmia [3], while Spear et al. [4] were the first to describe a case with the 4 major features of PDAC syndrome: anophthalmia, bilateral pulmonary agenesis, eventration of the left hemidiaphragm, and ventricular septal defect (VSD) with overriding aorta. Seller et al. reported 2 siblings, a male and a female, of a nonconsanguineous couple, with bilateral anophthalmia and pulmonary hypoplasia associated with other congenital anomalies in the female fetus [2]. Those 2 cases raised the possibility of an autosomal recessive mode of inheritance. While most of the reported cases were diagnosed based on prenatal scans or postnatal autopsies due to the fatal outcome of the syndrome, in a few reported cases, the infant lived beyond the neonatal period. Pasutto et al. reported 2 cases that lived for 6 months and 14 years of age, respectively [13]. Both cases had anophthalmia, cardiac defects, dysmorphic features, developmental delay, and normal brain structure except for absent optic nerves in the second child. In the same year, Chitayat et al., reported a male full-term living child who had 2 out of the 4 major features of PDAC syndrome [10]. That patient was 9.5 years old, and was born to nonconsanguineous healthy parents. He had severe bilateral microphthalmia, deep-set orbits, flat nasal bridge, left diaphragmatic hernia, and bilateral inguinal hernia. Cardiac and abdominal sonograms and brain CT were normal. The left diaphragmatic hernia was corrected surgically at 2 months of age. A lung CT scan did not show lung hypoplasia, and lung function tests at 19 months revealed normal lung capacity. He also had delayed development and truncal hypotonia. Many gene mutations have been associated with the development of anophthalmia/microphthalmia either alone or associated with other congenital malformations. Some genes implicated in the retinoic acid pathway have been identified to cause a broad spectrum of congenital anomalies including anophthalmia/microphthalmia, congenital heart, lung and diaphragmatic defects and others [12–15]. Segel et al. reported the first living child with bilateral microphthalmia, PDA, and congenital lobar emphysema in whom compound heterogeneous STRA6 mutations were found. This female child had a mild clinical phenotype and normal development expected for blindness at 30 months of age. The authors concluded that STRA6 analysis should be considered in patients with anophthalmia and parental counseling regarding the long-term outcome should be cautious [15].
Table 1.
Some of the reported cases of PDAC syndrome and their features.
| Findings | Our case | Spear et al. | Engellenner et al. | Seller et al. | Berkenstadt et al. | |
|---|---|---|---|---|---|---|
| Number of cases | 1 | 1 | 1 | 2 | 1 | |
| Gender | M | M | F | M | F | M |
| Anophthalmia/microphthalmia | Bil | Bil | Right eye | Bil | Bil | Bil |
| Pulmonary agenesis/dysgenesis/hypoplasia | Left hypoplasia | Bil | Bil | Dysgenesis | Bil Hypoplasia | Right agenesis |
| Diaphragmatic eventration/hernia | Left eventration | Left Event. | – | – | – | Right hernia |
| Cardiac anomalies | PDA + ASD Dilated RA + RV | VSD, absent ductus arteriosus, Overriding aorta | Dilated RA | – | Single ventricle, Hypoplastic LA | – |
| Pulmonary vascular abnormalities | N | Absent pul vessles | Absent pul vessels | –- | Enlarged Pul trunk | Absent pul vessels |
| Karyotype | 46, XY | 46, XY | 46, XX | 46, XY | 46, XX | 46, XY |
| Familial/sporadic | Sporadic | Sporadic | Sporadic | Familial | Sporadic | |
| Findings | Pierson et al. | Steiner et al. | Priolo et al. | Lee et al. | Li and Wei | |
|---|---|---|---|---|---|---|
| Number of cases | 1 | 2 | 1 | 1 | 1 | |
| Gender | F | F | M | M | F | F |
| Anophthalmia/microphthalmia | Bil | Cornneal cloudign | Bil | Bil | Bil | Bil |
| Pulmonary agenesis/dysgenesis/hypoplasia | Bil hypoplasia | Normal | Bil hypoplasia | Bil hypoplasia | Bil Hypoplasia | Bil hypoplasia |
| Diaphragmatic eventration/hernia | Left hernia | Bilateral eventration | Left hernia | Left hernia | – | |
| Cardiac anomalies | Bil Ventricular Hypertrophy | – | ASD, severe RVH | ASD | – | Dilated RA + RV |
| Pulmonary vascular abnormalities | Dilated pul arteries | – | – | – | – | Small 2 pul Veins |
| Karyotype | 46, XX | 46, XX | 46, XY | 46, XY | 46, XX | 46, XX |
| Familial/sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic |
Conclusions
The PDAC syndrome remains a rare disease with different gene mutations reported and variable prognosis. The main feature in reported cases was anophthalmia/microphthalmia. Therefore, screening for the other associated congenital anomalies is highly suggested. Mutations in STRA6 and RARB genes are commonly encountered in this spectrum. However, whole exome sequencing of suspected cases and their parents is recommended to detect possible de novo mutations. Further reports are needed to identify risk factors and prognosis of this rare syndrome.
Footnotes
Conflict of interests.
None.
References:
- 1.Demirkaya M, Sevinir B, Canitez Y, et al. A case with the combination of bilateral microphthalmia, unilateral pulmonary agenesis, diaphragmatic eventration and atrial septal defect: PDAC syndrome. International Journal of Human Genetics. 2009;9(3–4):255–61. [Google Scholar]
- 2.Seller MJ, Davis TB, Fear CN, et al. Two sibs with anophthalmia and pulmonary hypoplasia (the Matthew-Wood syndrome) Am J Med Genet. 1996;62:227–29. doi: 10.1002/(SICI)1096-8628(19960329)62:3<227::AID-AJMG5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 3.Ostor AG, Stillwell R, Fortune DW. Bilateral pulmonary agenesis. Pathology. 1978;10(3):243–48. doi: 10.3109/00313027809063507. [DOI] [PubMed] [Google Scholar]
- 4.Spear GS, Yetur P, Beyerlein RA, et al. Bilateral pulmonary agenesis and microphthalmia. Am J Med Genet. 1987;3:379–82. doi: 10.1002/ajmg.1320280543. [DOI] [PubMed] [Google Scholar]
- 5.Engellenner W, Kaplan C, Van de Vegte GL. Pulmonary agenesis association with nonimmune hydrops. Pediatr Pathol. 1989;9(6):725–30. doi: 10.3109/15513818909022379. [DOI] [PubMed] [Google Scholar]
- 6.Berkenstadt M, Lev D, Achiron R, et al. Pulmonary agenesis, microphthalmia, and diaphragmatic defect (PMD): New syndrome or association? Am J Med Genet. 1999;86:6–8. [PubMed] [Google Scholar]
- 7.Priolo M, Casile G, Lagana C. Pulmonary agenesis/hypoplasia, microphthalmia, and diaphragmatic defects: Report of an additional case. Clin Dysmorph. 2004;13:45–46. doi: 10.1097/00019605-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Wei J. A newborn with anophthalmia and pulmonary hypoplasia (the Matthew-Wood syndrome) Am J Med Genet. 2006;140A:1564–66. doi: 10.1002/ajmg.a.31298. [DOI] [PubMed] [Google Scholar]
- 9.Lee SYR, Shiu YK, Ng WF, Chow CB. Another patient with pulmonary hypoplasia, microphthalmia and diaphragmatic hernia. Clin Dysmorphol. 2006;15:43–44. doi: 10.1097/01.mcd.0000181604.78252.29. [DOI] [PubMed] [Google Scholar]
- 10.Chitayat D, Sroka H, Keating S, et al. The PDAC syndrome (pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect) (Spear syndrome, Matthew-Wood syndrome): Report of eight cases including a living child and further evidence for autosomal recessive inheritance. Am J Med Genet A. 2007;143A(12):1268–81. doi: 10.1002/ajmg.a.31788. [DOI] [PubMed] [Google Scholar]
- 11.Steiner RD, St J Dignan P, Hopkin RJ, et al. Combination of diaphragmatic eventration and microphthalmia/anophthalmia is probably nonrandom. Am J Med Genet. 2002;108:45–50. doi: 10.1002/ajmg.10167. [DOI] [PubMed] [Google Scholar]
- 12.Chassaing N, Golzio C, Odent S, et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum Mutat. 2009;30:E673–81. doi: 10.1002/humu.21023. [DOI] [PubMed] [Google Scholar]
- 13.Pasutto F, Sticht H, Hammersen G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007;80:550–60. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srour M, Chitayat D, Caron V, et al. Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am J Hum Genet. 2013;93:765–72. doi: 10.1016/j.ajhg.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segel R, Levy-Lahad E, Pasutto F, et al. Pulmonary hypoplasia-diaphragmatic hernia-anophthalmia-cardiac defect (PDAC) syndrome due to STRA6 mutations – What are the minimal criteria? Am J Med Genet Part A. 2009;149A:2457–63. doi: 10.1002/ajmg.a.33038. [DOI] [PubMed] [Google Scholar]