

Abstract
The hedgehog pathway, for which sonic hedgehog (Shh) is the most prominent ligand, is highly conserved and is tightly associated with embryonic development in a number of species. This pathway is also tightly associated with development of several types of cancer, including basal cell carcinoma and acute promyelocytic leukemia (APL) among many others. Inactivating mutations in Patched 1 (PTCH1), leading to ligand-independent pathway activation, are frequent in several cancer types but most prominent in basal cell carcinoma. This has led to the development of several compounds targeting this pathway as a cancer therapeutic. These compounds target the inducers of this pathway in Smoothened (SMO) and the GLI transcription factors, although targeting SMO has had the most success. Despite the many attempts at targeting this pathway, there are only three FDA-approved drugs for cancers that affect the Shh pathway. Two of these compounds, vismodegib and sonidegib, target SMO to suppress signaling from either PTCH1 or SMO mutations that lead to upregulation of the pathway. The other approved compound is arsenic trioxide (ATO), which can suppress this pathway at the level of the GLI proteins, although current evidence suggests it also has other targets. This review focuses on the efficacy and safety of these clinically-approved drugs targeting the Shh pathway along with a discussion on other Shh pathway inhibitors being developed.
1. Introduction
The hedgehog pathway is a highly conserved signaling pathway that is linked to many biological processes. This signaling pathway has been linked to development in many species, including humans (1). It has been linked to growth and patterning in many of these multicellular species including the development of the neural system and bone development (2, 3). The hedgehog pathway and its components have also been linked to several diseases, prominently including human cancer (4). Because of the importance of this pathway to human cancer, there have been several attempts to target this pathway for cancer therapies with few successes and many failures. In this review, we aim to provide an update on the successful agents targeting the hedgehog pathway that have been FDA approved for treatment in human cancers. We will also briefly discuss agents that are currently being developed to target this pathway for the treatment of cancer.
2. The Hedgehog Pathway in Cancer
Mammalian hedgehog signaling can be initiated by three unique ligands in Sonic Hedgehog (Shh), Indian hedgehog, and Desert hedgehog. However, Shh is the most widely expressed and also the most potent of these ligands (1, 5). The ligand Shh is expressed as an inactive full-length protein that is proteolytically cleaved to two proteins and the N-terminal 19 kDa fragment is the active Shh ligand (6). The receptor for this active Shh ligand is Patched1 (PTCH1), a 12-transmembrane protein that binds Shh ligand. Binding of Shh to PTCH1 relieves repression of Smoothed (SMO) by PTCH1 thereby activating SMO signaling activity (Figure 1). The activation of SMO ultimately decreases the interaction between suppressor of fused homolog (SUFU) and GLI proteins that allows GLI proteins to enter the nucleus and bind transcriptional targets to regulate cellular gene expression. There are three GLI isoforms in mammals in GLI1-3 wherein gene expression can be induced by GLI1 and repressed by GLI3 whereas GLI2 can regulate expression in either direction. The GLI proteins are the terminal effectors of the Shh signaling pathway and regulate genes that control organismal patterning and development. Many of the genes regulated by GLI proteins are co-opted by cancer cells as they regulate several cancer-related processes including proliferation, migration and invasion, as well as neovascularization (4).
Figure 1.
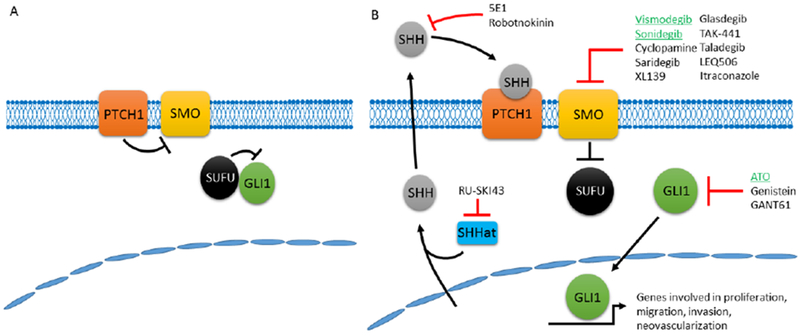
The Sonic Hedgehog Pathway. A) In the absence of Shh ligand, PTCH1 suppresses SMO allowing for SUFU suppression of GLI1. B) In the presence of Shh ligand, PTCH1 repression of SMO is removed allowing for SMO to repress SUFU leading to the release and nuclear translocation of GLI1. GLI1, and the other GLI proteins then promote a gene expression program that promotes multiple cancer phenotypes. The inhibitors to this pathway, the FDA-approved inhibitors highlighted in green, primarily have targeted SMO with some attempts to target Shh itself and the GLI proteins, but with little success.
There have been numerous reports of genetic alterations in key components of the Shh pathway in different tumor types that leads to constitutive signaling of this pathway and that paracrine signaling of Shh may be an important factor in multiple tumor types (7, 8). While there are reports of the Shh pathway being modified in several tumor types such as breast, pancreatic, colorectal, and rhabdomyosarcoma among several, genetic alterations in this pathway are most consistently seen in basal cell carcinomas (BCCs) and medulloblastomas (9–19). The genetic alterations in this pathway are commonly loss-of-function changes to suppressors of the pathway (e.g. PTCH1, SUFU) or gain-of-function changes to promotors of the pathway (e.g. SMO, GLI). This is very prevalent in BCC as PTCH1 has a gene inactivating alteration in 73% of these tumors while SMO has a genetic activation in 20% (20). Therapies targeted to the Shh pathway primarily inhibit the components that promote signaling flux through the pathway including Shh ligand itself, SMO, and GLI proteins. The most successful strategy has been to target SMO with small molecule compounds and the two FDA-approved drugs targeting this pathway use this strategy. Targeting SMO in BCC, for instance, has the potential to target the large percentage of these tumors that harbor inactivating alterations to PTCH1 or activating mutations to SMO. There are also inactivating alterations to SUFU in 8% of BCCs (20) and GLI1 is amplified in several tumor types (4) but SMO inhibitors are unlikely to show efficacy against these populations. There have been attempts to develop inhibitors to Shh and GLI1 but these have yet to make it past clinical trials.
3. Currently Approved Shh Pathway Inhibitors
The first FDA-approved drug for cancer that targeted the Shh pathway was arsenic trioxide (ATO) in 2000, which was approved for treatment of acute promyelocytic leukemia (APL) (21). While it has been proven to have significant effects suppressing the Shh pathway, it likely also targets other mechanisms promoting APL development and progression. Despite, ATO being the first FDA-approved drug that does have effects on the Shh pathway, the first FDA-approved agent that was specifically designed to target the Shh pathway was vismodegib (GDC-449), which was originally discovered in 2009 and later approved for treatment of basal cell carcinoma in 2012 (22, 23). A year later in 2010, sonidegib (LDE225) was discovered and was later also approved for treatment of basal cell carcinoma in 2015 (24, 25). These are currently the only approved agents targeting the Shh pathway with indications for cancer (Table 1).
Table 1.
Approved Shh Pathway Targeting Agents
| Drug | Target | FDA Approval Date | EU Approval Date | Disease Indication | Site of Metabolism | Enzymes of Metabolism |
|---|---|---|---|---|---|---|
| ATO | Glil, PML-RARα | 9/25/2000 | 3/5/2002 | Acute Promyelocytic Leukemia | Primarily liver, oxidation in various tissues | Primarily methyltransferases |
| Vismodegib | SMO | 1/30/2012 | 7/12/2013 | Locally advanced basal cell carcinoma | Primarily liver | CYP enzymes, in particular CYP2C9, CYP3A4, CYP3A5 |
| Sonidegib | SMO | 7/24/2015 | 8/14/2015 | Metastatic or locally advanced basal cell carcinoma basal cell carcinoma | Primarily liver | CYP enzymes, in particular CYP3A |
3.1. Arsenic Trioxide (ATO)
Arsenic formulations have been used for their beneficial therapeutic effects as far back as the 17th century (21). However, chronic exposure to arsenic is also labeled as a carcinogen and has been shown to promote solid tumors (26, 27). As an anti-cancer therapeutic, ATO has been shown to suppress growth in preclinical models in many tumor types including breast cancer, pancreatic cancer, colon cancer, acute promyelocytic leukemia (APL), melanomas, glioblastoma, and medulloblastoma among others (28–32).
Arsenic trioxide (ATO) was initially approved as a therapy for patients with acute promyelocytic leukemia (APL) who are refractory or have relapsed on retinoid and anthracycline chemotherapy (21). This approval came after two landmark trials wherein APL patients had become resistant to standard therapies of chemotherapy or all-trans retinoic acid (ATRA) (33, 34). ATO treatment increased the complete response rate from <40% to >90% and extended the time of this complete response (33, 34). ATO has since been approved for patients with low- and intermediate-risk APL by the EU and approved by the US FDA in combination with ATRA for newly-diagnosed low-risk APL with the t(15; 17) translocation of PML-RARA. ATO has been investigated in several other tumor types, including those with increased Shh dependence, but APL is currently the only indication approved for ATO.
3.1.1. Mechanism of action:
APL develops in 95% of cases due to fusion of the promyelocytic gene (PML) on chromosome 15 with the retinoic acid receptor alpha gene (RARA) on chromosome 17 resulting in the PML-RARA t(15; 17) fusion protein (35). The PML-RARA fusion protein acts a transcriptional repressor to block myeloid differentiation. Early studies indicated ATO was effective against PML as exposure to ATO led to decreased levels of PML-RARA protein, as well as other cell survival proteins, and differentiation (29, 36–38). These results led to studies that found significant clinical benefit for APL patients receiving ATO and eventual FDA approval (37, 39). Despite this obvious link of ATO affecting the precise mechanism leading to APL, recent studies have also indicated that ATO has an effect in suppressing the Shh pathway. One of these early findings indicated that ATO suppressed GLI transcriptional activity that was not linked to cell viability, suggesting ATO was specifically targeting the Shh pathway (40). Further confirmation was found when ATO suppressed GLI activity in the presence of SMO agonists or SMO activating mutants (40). These studies also indicated ATO suppressed GLI2 trafficking and other studies with similar results indicated ATO directly bound to GLI1, suggesting ATO has inhibitory effects on several aspects of the Shh pathway (28, 40, 41). These studies, and others showing similar suppression of the Shh pathway by ATO, have been shown in several tumor types (28, 40–51). Studies have also shown that ATO suppresses the Shh pathway in APL including clinical studies that showed ATO treatment led to clinical response and significant suppression of the Shh pathway (50). Thus, it appears that ATO has pleiotropic effects in the suppression of APL that likely include both suppression of PML-RARA and the Shh pathway, among several other possible mechanisms (52).
3.1.2. Metabolic profile:
ATO hydrolyzes to arsenious acid (AsIII) in solution. After administration of the instructed dose at 0.15mg/kg daily for 5 days a week, AsIII reaches peak concentrations in the plasma in approximately 2 hours. AsIII has a half-life of 10-14 hours (52). AsIII metabolizes to two main products, monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV). It can also oxidize to arsenic acid, although at low levels. The half-lives of MMAV and DMAV are 32 and 72 hours, respectively. Arsenious acid has a high volume of distribution, although not as high as sonidegib, and is present in tissues. AsIII metabolism to MMA and DMA occurs in the liver but oxidation to arsenic acid occurs in various tissues utilizing various processes. About 15% of AsIII is excreted through the urine without being metabolized (52). The total and renal clearance of AsIII is 49L/h and 9L/h, respectively. The metabolic breakdown of ATO, the pharmacodynamics, and the pharmacokinetics has led to approval for treatment of ATO using a dose of approximately 0.15 mg/kg via intravenous administration with tretinoin and the half-life of 10-14 hours has led to daily administration according to these recommendations.
3.1.3. Serious and life-threatening adverse reactions:
Perhaps the most serious life-threatening adverse reaction to ATO is QT interval disruption. For example, 38 out of 99 patients treated with ATO for cancers developed prolonged QTc intervals (53). Further, several studies have also observed, although rare, development of torsade de pointes after treatment with ATO (54–56). The ongoing management of these complications is accomplished by regular EKG monitoring, discontinuation of other QT interval prolonging drugs, and ensuring sufficiently high levels of serum magnesium and potassium (57).
In addition to QT elongation, ATO can also cause differentiation syndrome which can be fatal. When used in combination with tretinoin, ATO can lead to elevated levels of hepatic transaminase which can be toxic to the liver. In addition, ATO is a carcinogen, teratogen, and toxic to fetuses and embryos (27).
3.1.4. Most frequent other adverse reactions:
In addition to QT interval elongation and tachychardia, ATO has numerous adverse effects on the circulatory system. Many of these involve levels of cells or substances in the blood including leukocytosis, neutropenia, thrombocytopenia, hyperglycemia, hypokalemia, and hypomagnesemia. ATO also affects the GI system and has caused vomiting, diarrhea, and abdominal pain. Reactions affecting the respiratory system include dyspnea, cough, and sore throat. Other general adverse reactions such as nausea, fever, rigor, fatigue, insomnia, edema, rash or itching, arthralgia, headaches, paresthesia and dizziness can also occur (Table 2).
Table 2.
Adverse reactions of FDA Approved Shh Targeting Agents
| Drug | Adverse Reactions |
|---|---|
| ATO | Differentiation syndrome, elevated hepatic transaminase, QT interval elongation, tachycardia, leukocytosis, neutropenia, thrombocytopenia, hyperglycemia, hypokalemia, hypomagnesemia, vomiting, diarrhea, abdominal pain, dyspnea, cough, sore throat, nausea, fever, rigor, fatigue, insomnia, edema, rash, itching, arthralgia, headache, paresthesia, dizziness |
| Vismodegib | Vomiting, diarrhea, constipation, dysgeusia, muscle spasms, arthralgias, alopecia, weight loss, fatigue, nausea, decreased appetite |
| Sonidegib | Elevated creatine kinase, muscle spasms, musculoskeletal pain, myalgia, diarrhea, abdominal pain, vomiting dysgeusia, alopecia, pruritus, nausea, decreased weight, decreased appetite, headache, pain |
There are various treatments available to manage these adverse events. Leukocytosis, neutropenia, and thrombocytopenia can be managed with specific medications for these conditions as well as transfusions. Hyperglycemia can be managed with insulin injection and nutrition counseling. Potassium and magnesium supplements have been sued for hypokalemia and hypomagnesemia, respectively. Dyspnea can be treated using opioids with morphine a common treatment for dyspnea in cancer patients (58).
3.1.5. Drug interactions:
ATO should not be taken with drugs that prolong the QT interval, alter electrolyte levels, or are hepatotoxic. The adverse reactions to ATO, especially QT prolongation, dictate avoiding these interactions for the safety of the patients.
3.1.6. Post-marketing spontaneous reports of adverse reactions:
Many additional adverse reactions to ATO have been reported post-marketing. These include various cardiac disorders, nerve damage, seizure, confusion, deficiency of white and red blood cells and platelets, herpes zoster infection, muscle and bone pain, rhabdomyolysis, deafness, and toxic epidermal necrolysis. Development of other cancers, melanoma, pancreatic cancer, and squamous cell carcinomas have also been reported.
3.2. Vismodegib (GDC-0449)
Vismodegib was discovered in 2009 from a compound screen with GLI-responsive luciferase cells and was observed to have suppression of medulloblastoma allografts (23). There was clear evidence that supported vismodegib inhibiting SMO leading to tumor suppression in Shh-dependent preclinical models including medulloblastoma, pancreatic cancer, colorectal cancer, lung cancer, prostate cancer, leukemia, and cholangiosarcoma among others (23, 59–73). Clinical trials have been undertaken in many of these tumor types, as well as other diseases with relevance to Shh signaling, but currently it has only been FDA-approved for basal cell carcinoma.
Despite the potential efficacy in a myriad of tumor types, BCC was the obvious setting for the greatest clinical utility as BCC has constitutive activation of the Shh pathway in >85% of cases (74). This overwhelming majority of cases involving the Shh pathway is primarily due to mutations in PTCH1 that leads to ligand-independent activation of SMO (11, 20). The first phase I clinical trial for vismodegib in BCC resulted in 18 out of 33 patients having an objective response (75, 76). The primary clinical evidence of efficacy for vismodegib in BCC was from a multicenter, two-cohort, non-randomized clinical trial (22, 77). In this trial, metastatic BCC patients showed a 33% objective response rate whereas locally advanced BCC had an objective response rate of 43% (77). Furthermore, stable disease was observed in 64% of metastatic BCC patients and 38% of locally advanced BCC patients (77). These outcomes resulted in a median response duration of >7 months and progression-free survival of >9 months (77). Additionally, there has been an overall survival increase from 24 months to 33.4 months for BCC patients since vismodegib became available (78, 79). To further this point, the historical median survival of BCC prior to 1990 was approximately 8 months but is now approximately 7 years due to the introduction of agents targeting the sonic hedgehog pathway such as vismodegib and sonidigib (80–83). The summation of these encouraging clinical results led to FDA approval of vismodegib for metastatic or locally advanced BCC in 2012.
3.2.1. Mechanism of action.
Vismodegib was discovered from a screening of cyclopamine derivatives on a GLI luciferase reporter cell line and it was found to inhibit SMO (23). Further studies confirmed that vismodegib directly bound to SMO via competitive binding assays and molecular docking prediction studies (59, 63, 65, 69, 70, 72). In confirming that vismodegib binds to SMO, it was also observed that vismodegib exposure could induce a mutations in SMO that ablated the interaction of the drug with SMO (72, 84). This mutation was observed in patient samples and animal models that led to vismodegib adaptive resistance (72).
3.2.2. Metabolic profile.
Vismodegib has a bioavailability of 31.8% upon oral administration (85). At steady-state levels, which is achieved unusually fast in 7-14 days, more than 99% of the drug binds to proteins in the plasma, including AAG and human serum albumin and is unaffected by concentration until 100 uM (85). It is metabolized primarily by CYP enzymes with CYP3A4, CYP3A5, and CYP2C9 producing most of the metabolites (86). Vismodegib has an unusually long half-life of about 12 days after a single dose of 150 mg (87). However, its half-life with continuous administration on a daily basis is about 4 days (22).
3.2.3. Serious and life-threatening adverse reactions.
Vismodegib, similar to other drugs targeting the hedgehog pathway, is a teratogen. The hedgehog pathway is an important element in the development of various organs and organ systems (88). A drug that disrupts this pathway is therefore expected to be, and is, detrimental to the development of embryos and fetuses. The FDA recommends verifying pregnancy status within 7 days of the start of treatment with vismodegib and use of contraception during treatment and for 24 months after the end of treatment. It is worth noting that vismodegib not only targets the Shh pathway more specifically than ATO, but also does not have the side effect of QT internal prolongation that comes with ATO (89).
3.2.4. Most frequent other adverse reactions.
Several of the most common adverse reactions involve the GI system and include vomiting, diarrhea, constipation, and dysgeusia (disorder of taste). The drug also affects the musculoskeletal system and can cause muscle spasms and joint pain (arthralgias). Another common adverse reaction is alopecia. Other general adverse effects include weight loss, fatigue, nausea, and decreased appetite (75) (Table 2).
These adverse events do have sufficient approaches to management. Strategies for managing dysgeusia, decreased appetite, and weight loss include nutritional consultation and changes in preparation of foods (90, 91). Furthermore, there are medicinal options as well that range from fish oil supplementation to corticosteroids (92). Attention to other factors that may affect taste, such as oral hygiene, infection, acid reflux, and postnasal drip have also been reported to improve symptoms (93). Proper hydration, stretching and other light physical activity may help in the prevention of muscle spasms and join paint. Calcium blockers, nerve pain medication, and sports drinks are also recommended. For higher than grade 3 muscle spasms, stopping treatment for 2-4 weeks may help as well as addition of other medications such as gabapentin among other options (90, 92). For management of alopecia, 2-5% minoxidil is the most commonly suggested treatment. Strategies for concealing hair loss can also be considered (90, 92, 93). Serotonin inhibitors taken before and during treatment can be used to prevent nausea (90).
3.2.5. Drug interactions.
When administered with fluconazole, a moderate inhibitor of CYP2C9 and CYP3A4, vismodegib was found to have an increase in steady-state concentrations while itraconazole, a strong inhibitor of CYP3A4, and rabeprazol, a proton pump inhibitor was found to have no effect (94). Vismodegib may inhibit CYP2C8, CYP2C9, CYP2C19 and BCRP transporter (86). These drug interactions are important for the safety of vismodegib administration and minimizing the likelihood of occurrence and severity of the adverse events described above.
3.2.6. Post-marketing spontaneous reports of adverse reactions.
Vismodegib has been associated with hepatotoxicity post-marketing. As of January 2013, 23% of adverse event reports on the FDA Adverse Events Reporting system have included liver toxicity (95). Increase in blood phosphocreatine kinase was also reported post-marketing (96).
3.3. Sonidegib (LDE-225)
Sonidegib (LDE-225) was discovered in 2010 using a GLI-responsive luciferase reporter cell line and had efficacy in a medulloblastoma allograft model (25). This study, and others, confirmed sonidegib interacted with and inhibited SMO (25, 59, 63, 69, 70). Sonidegib has shown efficacy in multiple tumor types in preclinical models including medulloblastoma, ovarian cancer, glioblastoma, melanoma, renal cell carcinoma, leukemia, and breast cancer among several others (97–111). As mentioned above, there has been a very large increase in the median survival since these Shh pathway inhibitors were introduced from approximately 8 months to approximately 7 years (80–83). Clinical trials have been undertaken for sonidegib in many of these tumors types but BCC remains the only FDA-approved indication.
Due to the prevalence for the Shh pathway to be activated in BCC, sonidegib was an obvious compound for treatment of BCC. A very early treatment with sonidegib as a topical treatment suggested it may have efficacy in nevoid BCC (112). An initial phase I trial in patients with advanced solid tumors and 37.5% of BCC patients and 33% of medulloblastoma patients showed objective tumor responses (113). This trial established sonidegib as safe with possible efficacy leading to further trials. A phase II multicenter, randomized, double-blind trial was later completed in locally advanced or metastatic BCC with patients receiving either a low (200 mg) or high (800 mg) dose of sonidegib (114). The patients receiving the low dose showed a 36% objective response rate with the high dose group achieving a 43% objective response rate (114). A later update after 12-months of follow-up with these patients indicated a sustained response as the low dose group had a 57.6% objective response rate in locally advanced BCC whereas the high dose group maintained a 43.8% response rate in locally advanced BCC (115). Results of this trial led to FDA approval in 2015 for adult patients with locally advanced BCC that has recurred following surgery or radiation therapy or those who are not candidates for surgery or radiation therapy.
3.3.1. Mechanism of action:
Sonidegib was discovered via a screen in a GLI luciferase reporter cell line and binding to SMO was confirmed via a GLI1 IC50 shift assay (25). Molecular prediction docking studies further predict sonidegib binds to SMO in the “drug binding pocket” (59, 63, 69, 70). The same mutations in the drug binding pocket that led to vismodegib resistance were also observed to cause sonidegib resistance (116), suggesting these drugs share a similar mechanism of action.
3.3.2. Metabolic profile:
Sonidegib has an absorption rate of less than 10% upon oral administration. When taken with a meal high in fat, however, absorption increases 7.4- to 7.8-fold (117). Sonidegib reaches steady state levels after approximately 4 months. It has a very high volume of distribution of 9,166 L suggesting high accumulation in tissues (118). More than 97% of the sonidegib in plasma remains bound to proteins and is unaffected by concentration. Sonidegib has an estimated half-life of 28 days. It is metabolized primarily by CYP3A enzymes in the liver (119). These factors have led to a dosing schedule of 200 mg given once daily via oral administration on an empty stomach.
3.3.3. Serious and life-threatening adverse reactions:
As sonidegib is an inhibitor of the sonic hedgehog pathway, like vismodegib, it also is toxic to fetuses and embryos and is a teratogen. Sonidegib has been shown to elevate creatine kinase levels in several patients and was often accompanied by musculoskeletal complications (113). Thus CK level should be monitored before and periodically throughout the duration of treatment with this drug. Similar to vismodegib, sonidegib is more specific than ATO and does not result in QT interval prolongation (120).
3.3.4. Most frequent other adverse reactions:
Sonidegib affects the musculoskeletal system in various ways with symptoms including muscle spasms, musculoskeletal pain, and myalgia. It can also cause gastrointestinal disturbances including diarrhea, abdominal pain, vomiting, and dysgeusia. Sonidegib can lead to alopecia and pruritus (severe itching of skin). Other common adverse reactions are nausea, decreased weight, decreased appetite, headache and pain (113) (Table 2). The management of these adverse reactions are similar to those listed for vismodegib. An additional factor worth mentioning is that pruritis can be treated with emollients, antipruritc creams, and antihistamines (93).
3.3.5. Drug interactions:
Sonidegib users should avoid concurrently using any strong or moderate CYP3A inhibitors or inducers. Moderate inhibitors may be used if necessary but only for less than 14 days and with close monitoring. Some strong CYP3A inhibitors include saquinavir, telithromycin, ketoconazole, itraconazole, voriconazole, posaconazole and nefazodone. Moderate inhibitors include atazanavir, diltiazem, and fluconazole. Inducers of CYP3A include carbamazepine, efavirenz, modafinil, phenobarbital, phenytoin, rifabutin, rifampin and St. John’s Wort. These drug interaction are of direct importance to the safety of this drug in patients and minimizing as much as possible the likelihood and severity of the adverse reactions described above.
4. Shh Pathway Agents Under Development
As described above, the primary drug targets for the Shh pathway are molecules that promote active signaling. The primary targets that have been attempted to be therapeutically targeted are Shh itself, SMO, and GLI proteins with GLI1 being the primary target (Table 3).
Table 3.
Shh Targeting Agents under Development
| Drug | Cancer Types | Target | Clinical Trial Status | Clinical Trial Numbers | Human Efficacy | Reference for efficacy |
|---|---|---|---|---|---|---|
| 5E1 | Preclinical studies only in various cancer types | Shh | No trials to date | N/A | Unknown | |
| Robotnikinin | Preclinical studies only in various cancer types | Shh | No trials to date | N/A | Unknown | |
| RU-SKI3 | Preclinical studies only in various cancer types | SMO | No trials to date | N/A | Unknown | |
| Cyclopamine | Preclinical studies only in various cancer types | SMO | No trials to date | N/A | Unknown | |
| Saridegib | Chondrosarcoma, head and neck, pancreatic, adenocarcinoma, BCC, myelofibrosis | SMO | Phase 2 |
NCT01371617, NCT01130142*, NCT00761696, NCT01383538*, NCT01255800*, NCT01310816 |
Majority result is stable disease | Jimeno 2013, Sasaki 2015 |
| XL139 | Leukemia, BCC, BCNS, stomach and esophageal neoplasms, other cancers | SMO | Phase 2 |
NCT01413906, NCT00670189, NCT01218477*, NCT00927875*, NCT00909402*, NCT01357655*, NCT00884546*, |
Unknown for XL139 alone, variable with combination therapies | |
| Glasdegib | AML, other cancers | SMO | Phase 2 |
NCT03466450*, NCT02226172, NCT03416179*, NCT02367456*, NCT03390296*, NCT02038777*, NCT01546038*, NCT03529448* |
Majority result is stable disease | Minami 2017 |
| TAK-441 | BCC, advanced nonhematologic malignancies | SMO | Phase 1 | NCT01204073 | Majority had no response, 25% exhibited stable disease | Goldman 2015 |
| Taladegib | Various carcinomas and sarcomas | SMO | Phase 2 |
NCT02530437*, NCT01919398, NCT01722292*, NCT01226485, NCT02784795* |
Unknown, no results yet | |
| LEQ506 | BCC and Medulloblastoma | SMO | Phase 1 | NCT01106508 | Unknown | |
| Itraconazole | Multiple cancer types and other diseases | SMO | Phase 2 | >40 Studies | Variable for cancer type and disease type | |
| Genistein | Multiple cancer types and other diseases | Gli1 | Phase 2 | 30 Studies | Variable for cancer type and disease type | |
| GANT61 | Preclinical studies only in various cancer types | Gli1 | No trials to date | N/A | Unknown |
= Combination studies with other drugs or therapies
4.1. Agents Targeting Shh
5E1 is a monoclonal antibody that binds the Shh ligand preventing its interaction with PTCH1 (121, 122). This antibody has been to suppress growth of esophageal PDX models in combination with radiation (111). 5E1 was also shown to enhance the effect of platinum-based chemotherapy delivered concurrently with radiation RTCT on cervical cancer xenografts but there was no effect of 5E1 as a single agent (98). 5E1 also enhanced the effect of platinum-based therapy in models of gastric cancer (123). 5E1 has been shown in a mouse model of breast cancer to decrease tumor size as well as liver and pancreatic metastases (124). 5E1 was effective as a monotherapy in a mouse model of medulloblastoma in suppressing tumor growth and extending survival time (125). 5E1 also reduced primary tumor growth and metastasis in an orthotopic mouse pancreatic cancer model as a single agent (7).
Robotnikinin is a 12-membered macrocycle discovered screening molecules that suppress the Shh pathway (126). This compound was found to directly bind Shh and prevent its interaction with PTCH1 leading to suppression of GLI1 activity (126). This compound has yet to show any antitumor effects in preclinical models.
During the synthesis of Shh, the enzyme SHHat catalyzes the final steps to attach a palmitate to the Shh protein (127, 128). The compound RU-SKI 43 was a compound found from screening for inhibitors of SHHat that suppressed Shh production and signaling (129). This compound was observed to reduce proliferation and anchorage-independent growth of breast cancer cells (130) as well as pancreatic tumor growth (131).
4.2. Agents Targeting SMO
Cyclopamine is an alkaloid from V. californicum that is one of the early discovered compounds to have significant inhibitory action toward SMO and suppressing signaling through the Shh pathway (132, 133). Cyclopamine was shown to have several antitumor effects in many tumor types (134–137). However, this compound had many adverse effects preventing its widespread clinical use and led to the development of second generation derivatives of cyclopamine such as vismodegib described above.
Another derivative of cyclopamine is Saridegib (IPI-926), which has shown antitumor activity in models of medulloblastoma, ovarian cancer, chondrosarcoma, and osteosarcoma (138–142). There have been early stage clinical trials with Saridegib that have shown favorable pharmacodynamics and pharmacokinetics while having some antitumor activity toward solid tumors (143–146). However, Saridegib was not recommended for further developing in patients with myelofibrosis or pancreatic cancer (145, 147). Clinical trials with Saridegib for other tumor types, such as head and neck cancer (NCT01255800) and chondrosarcoma (NCT01310816), have been completed but not all results have been made public as of the time of this review. No new clinical trials for Saridegib have been initiated.
Another cyclopamine derivative in BMS-833923 (XL139) had shown effectiveness in suppressing SMO (59, 148, 149). Preclinical studies indicate efficacy of BMS-833923 in esophageal, prostate, cholangiosarcoma, and lung cancers (148–151). A number of early phase clinical trials were initiated with BMS-833923 in the early part of this decade but little success has led to Bristol-Myers Squibb, the BMS-833923, to discontinue their research in the area of SMO inhibitors (see NCT01218477).
Another compound, Glasdegib (PF-04449913), was described in 2011 has since made significant progress in preclinical and clinical settings (152). Glasdegib was described to interact with and suppress SMO activity, which was effective in targeting myeloid leukemias in preclinical models (59, 152–155). Several phase I clinical trials with Glasdegib have reported favorable drug profiles with some instances of efficacy suggesting further development (156–163). A phase II trial with Glasdegib plus cytarabine/daunorubicin was well tolerated and showed clinical activity in patients with untreated acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (MDS) (164). There are a number of phase I and II trials ongoing with Glasdegib in both hematological and solid tumors as well as one phase III trial in AML (NCT03416179). These clinical successes have led to the FDA recently granting Priority Review for Glasdegib in untreated AML.
The compound TAK-441 is another SMO inhibitor discovered in the last decade with some clinical relevance (165). It has a potent ability to inhibit SMO and the Shh pathway and has shown the ability to suppress growth of multiple solid tumor types in preclinical studies (59, 165–169). One unfortunate side effect of SMO inhibition can be adaptive mutations in the SMO gene making them resistant to vismodegib or cyclopamine (170). An encouraging finding is that TAK-441 has been shown in preclinical studies to maintain activity in cells expressing these SMO adaptive mutants, suggesting it may be highly relevant to resistant patients (171, 172). To date, there has only been clinical trial with TAK-441, a phase I trial that found it was well tolerated and showed preliminary antitumor activity in advanced solid tumors (173). There are no further trials registered for TAK-441 so the current status and future plans for development are unknown.
Taladegib (LY2940680) is another inhibitor developed in recent years with potent SMO binding and inhibitory action, including inhibition of the adaptive SMO mutant (70, 174–176). Two separate phase I trials indicated a favorable safety profile for Taladegib in patients with solid tumors, including patients resistant to previous Shh therapies (177, 178). There are several phase I and II trials ongoing with Taladegib in multiple solid tumor types.
The compound LEQ506 was also developed in recent years and has ability to bind and inhibit SMO leading to suppression of Shh pathway signaling (179, 180). This binding to SMO including binding to the adaptive mutant SMO following vismodegib administration (179). This compound was subjected to a phase I trial in patients with advanced solid tumors but results have yet to be posted.
Lastly, Itraconazole is a known anti-fungal drug but has shown the ability to inhibit SMO in basal cell carcinoma (181–183). Itraconazole has seen efficacy in combination with other chemotherapies in phase II trials for castration-resistance prostate cancer, non-small cell lung cancer, and basal cell carcinoma (184–186). There are many ongoing trials that include Itraconazole with the purpose of treating several different solid tumor types, although these are all phase I and II trials. There are higher level phase trials including Itraconazole but these are all for its antifungal properties.
4.3. Agents Targeting GLI1
Genistein is an isoflavone isolated from Genista tinctoria that is widely available in legumes and plant foods. Genistein has been found to suppress GLI1 in recent years leading to an ability to suppress several tumor types and, in particular, the cancer stem cell niche (187–191). Exactly how Genistein suppresses GLI1 remains to be understood but many of its antitumor properties have been attributed to suppression of the Shh pathway despite Genistein also suppressing several other important tumor-related molecules. Phase I trials with Genistein have shown a very favorable safety profile leading to several phase II trials wherein Genistein showed some efficacy in prostate cancers but had mixed results in pancreatic cancers (192–197). Genistein is being evaluated in several ongoing trials, most in phase I or II, and as is often the case with natural compounds, it is frequently being evaluated as a prevention agent.
GANT61 is a compound discovered from a GLI-luciferase drug screen that effectively reduced GLI1/2 DNA-binding (198). GANT61 has shown to have inhibition of GLI activity in multiple preclinical models that also leads to suppression of tumor growth and proliferation (198–212). However, this lone GLI-specific inhibitor does not have any clinical trials registered as the writing of this review.
4.4. Resistance to Shh Pathway-Targeted Therapies
Several agents that target the Shh pathway have been shown to develop resistance, including FDA-approved agents as described above (72, 116, 139). This is highly related to drug safety as patients develop resistance to agents, the dose escalation required to maintain tumor suppression can become toxic or cause the patients to be entirely removed from such therapies. As mentioned above, resistance to vismodegib and sonidegib occurs due to an adaptive mutation in SMO causing ineffectiveness of these compounds to inhibit SMO activity (72, 116). It seems there have been two primary strategies to attempt to over the resistance of these FDA-approved therapies in a) developing alternative inhibitors that can inhibit vismodegib-resistant SMO activity or b) combinatorial therapy with other drugs. Many of the therapies under development that are described above have been tested whether they can sufficiently inhibit SMO in the presence of the adaptive mutation caused by long-term vismodegib or sonidegib treatment. Furthermore, the continued development of new inhibitors is due to the resistance these approved therapies eventually succumb. Clinical trials have been, or are continuing, to occur with combinatorial studies with vismodegib and other agents such as radiation (NCT02956889, NCT01835626), temozolomide (NCT01601184), gemcitabine (NCT01713218, NCT01195415, NCT01064622, NCT00878163, NCT01088815), paclitaxel (NCT02694224), bevacizumab (NCT00636610), oxaliplatin (NCT00982592), and decitabine (NCT02073838). Similarly, sonidegib is also currently in clinical trials in combination with other therapies such as everolimus (NCT02138929), docetaxel/paclitaxel (NCT02027376, NCT01954355, NCT02182622), gemcitabine (NCT01487785, NCT01431794, NCT02358161, NCT03434262), cisplatin (NCT01579929), fluorouracil (NCT01485744), Many of these trials are ongoing and do not have results available, however, the few with obtainable results seem to have mild, if any, enhancement of the efficacy. It is also unclear how these combinatorial studies will affect adverse events as the studies above with available information indicate more events in some cases while others see mild effects. Completion of many of these, and any information on developing resistance, will certainly increase our understanding of how to overcome resistance to these therapies and the correct populations likely to benefit.
In addition to these resistance mechanisms in FDA-approved therapies, there is a small amount of evidence for resistance to agents currently under development. Saridegib, which targets SMO and was shown to inhibit medulloblastoma in a mouse model and increase the lifespan of these animals, but also these animals did develop resistance (139). However, many of these agents have not been tested for long time periods to establish whether they will develop resistance. Aside from the study mentioned above regarding saridegib, there are very few pre-clinical long-term studies that would allow such an observation. Furthermore, while some of these agents have made it to clinical trials, none have currently made it past Phase 2 trials leading to insufficient evidence as to whether any of them will induce an adaptive resistance in patients.
5. Conclusions
There continues to be evidence generated from many laboratories indicating the importance of the Shh pathway in tumor initiation and progression. In particular, this pathway seems highly important to brain tumors, especially medulloblastoma, and skin cancers, especially basal cell carcinoma. The primary target of this pathway that has shown any successful efficacy is SMO. However, targeting this molecule can lead to adaptive mutations that induce resistance. Development of next generation SMO inhibitors should take into account this adaptive mechanism. Due to the lack of an enzymatic domain in the GLI transcription factors, these proteins are likely going to be difficult to directly target with future drug development. Despite this drawback, there continues to be attempts to therapeutically target transcription factors and a breakthrough in this area could lead to new vigor in the attempt to target GLI proteins. Overall, the Shh pathway appears to be important for many types of tumors and at different stages of the disease. Therefore, it is in the interest of future patients to continue basic research on this pathway and continue drug development towards viable targets in the Shh pathway.
Key Points.
There are three FDA-approved inhibitors to the sonic hedgehog pathway for use in cancers in arsenic trioxide, vismodegib, and sonidegib
All of these approved inhibitors have common effects on safety of patients but these adverse events are manageable
Patients often develop resistance to vismodegib and sonidegib leading to a long list of agents under development to target the sonic hedgehog pathway
Acknowledgements
We would like to acknowledge our funding source in the National Cancer Institute K22CA207575 (RLC) and Indiana University School of Medicine.
Funding: We would like to acknowledge our funding source in the National Cancer Institute K22CA207575 (RLC) and Indiana University School of Medicine.
Footnotes
Conflict of Interest: Richard L Carpenter and Haimanti Ray have no conflicts of interests to declare.
References
- 1.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nature reviews Genetics. 2011;12(6):393–406. [DOI] [PubMed] [Google Scholar]
- 2.Alman BA. The role of hedgehog signalling in skeletal health and disease. Nature reviews Rheumatology. 2015;11(9):552–60. [DOI] [PubMed] [Google Scholar]
- 3.Choudhry Z, Rikani AA, Choudhry AM, Tariq S, Zakaria F, Asghar MW, et al. Sonic hedgehog signalling pathway: a complex network. Annals of neurosciences. 2014;21(1):28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter RL, Lo HW. Identification, functional characterization, and pathobiological significance of GLI1 isoforms in human cancers. Vitamins and hormones. 2012;88:115–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathi S, Pagan-Westphal S, Baker DP, Garber EA, Rayhorn P, Bumcrot D, et al. Comparative biological responses to human Sonic, Indian, and Desert hedgehog. Mechanisms of development. 2001;106(1-2):107–17. [DOI] [PubMed] [Google Scholar]
- 6.Porter JA, von Kessler DP, Ekker SC, Young KE, Lee JJ, Moses K, et al. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature. 1995;374(6520):363–6. [DOI] [PubMed] [Google Scholar]
- 7.Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28(40):3513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–10. [DOI] [PubMed] [Google Scholar]
- 9.Gailani MR, Bale SJ, Leffell DJ, DiGiovanna JJ, Peck GL, Poliak S, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69(1):111–7. [DOI] [PubMed] [Google Scholar]
- 10.Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nature genetics. 1996;14(1):78–81. [DOI] [PubMed] [Google Scholar]
- 11.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–51. [DOI] [PubMed] [Google Scholar]
- 12.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science (New York, NY). 1996;272(5268):1668–71. [DOI] [PubMed] [Google Scholar]
- 13.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nature genetics. 2009;41(4):465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer research. 1997;57(11):2085–8. [PubMed] [Google Scholar]
- 15.Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer research. 1997;57(5):842–5. [PubMed] [Google Scholar]
- 16.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nature genetics. 2002;31(3):306–10. [DOI] [PubMed] [Google Scholar]
- 17.Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. The Journal of pathology. 2006;208(1):17–25. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer research. 1997;57(12):2369–72. [PubMed] [Google Scholar]
- 19.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–2. [DOI] [PubMed] [Google Scholar]
- 20.Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nature genetics. 2016;48(4):398–406. [DOI] [PubMed] [Google Scholar]
- 21.Cohen MH, Hirschfeld S, Flamm Honig S, Ibrahim A, Johnson JR, O’Leary JJ, et al. Drug approval summaries: arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. The oncologist. 2001;6(1):4–11. [DOI] [PubMed] [Google Scholar]
- 22.Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(9):2289–93. [DOI] [PubMed] [Google Scholar]
- 23.Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorganic & medicinal chemistry letters. 2009;19(19):5576–81. [DOI] [PubMed] [Google Scholar]
- 24.Casey D, Demko S, Shord S, Zhao H, Chen H, He K, et al. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(10):2377–81. [DOI] [PubMed] [Google Scholar]
- 25.Pan S, Wu X, Jiang J, Gao W, Wan Y, Cheng D, et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS medicinal chemistry letters. 2010;1(3):130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter RL, Jiang Y, Jing Y, He J, Rojanasakul Y, Liu LZ, et al. Arsenite induces cell transformation by reactive oxygen species, AKT, ERK1/2, and p70S6K1. Biochemical and biophysical research communications. 2011;414(3):533–8. [DOI] [PubMed] [Google Scholar]
- 27.IARC. Some drinking-water disinfectants and contaminants, including arsenic. IARC monographs on the evaluation of carcinogenic risks to humans. 2004;84:1–477. [PMC free article] [PubMed] [Google Scholar]
- 28.Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. The Journal of clinical investigation. 2011;121(1):148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88(3):1052–61. [PubMed] [Google Scholar]
- 30.Li X, Ding X, Adrian TE. Arsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cells. Cancer investigation. 2004;22(3):389–400. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Li B, Zhang H, Shi G, Li W, Jonas JB. Effect of arsenic trioxide on uveal melanoma cell proliferation in vitro. Ophthalmic research. 2007;39(6):302–7. [DOI] [PubMed] [Google Scholar]
- 32.Zhao S, Tsuchida T, Kawakami K, Shi C, Kawamoto K. Effect of As2O3 on cell cycle progression and cyclins D1 and B1 expression in two glioblastoma cell lines differing in p53 status. International journal of oncology. 2002;21(1):49–55. [PubMed] [Google Scholar]
- 33.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89(9):3354–60. [PubMed] [Google Scholar]
- 34.Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. The New England journal of medicine. 1998;339(19):1341–8. [DOI] [PubMed] [Google Scholar]
- 35.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93(10):3167–215. [PubMed] [Google Scholar]
- 36.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89(9):3345–53. [PubMed] [Google Scholar]
- 37.Ghavamzadeh A, Alimoghaddam K, Ghaffari SH, Rostami S, Jahani M, Hosseini R, et al. Treatment of acute promyelocytic leukemia with arsenic trioxide without ATRA and/or chemotherapy. Annals of oncology : official journal of the European Society for Medical Oncology. 2006;17(1):131–4. [DOI] [PubMed] [Google Scholar]
- 38.Lallemand-Breitenbach V, Guillemin MC, Janin A, Daniel MT, Degos L, Kogan SC, et al. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. The Journal of experimental medicine. 1999;189(7):1043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathews V, George B, Lakshmi KM, Viswabandya A, Bajel A, Balasubramanian P, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006;107(7):2627–32. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(30):13432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han JB, Sang F, Chang JJ, Hua YQ, Shi WD, Tang LH, et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. OncoTargets and therapy. 2013;6:1129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ally MS, Ransohoff K, Sarin K, Atwood SX, Rezaee M, Bailey-Healy I, et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA dermatology. 2016;152(4):452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai X, Yu K, Zhang L, Li Y, Li Q, Yang Z, et al. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets and therapy. 2015;8:877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang KJ, Yang MH, Zheng JC, Li B, Nie W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. American journal of translational research. 2016;8(2):1133–43. [PMC free article] [PubMed] [Google Scholar]
- 45.Kerl K, Moreno N, Holsten T, Ahlfeld J, Mertins J, Hotfilder M, et al. Arsenic trioxide inhibits tumor cell growth in malignant rhabdoid tumors in vitro and in vivo by targeting overexpressed Gli1. International journal of cancer. 2014;135(4):989–95. [DOI] [PubMed] [Google Scholar]
- 46.Meister MT, Boedicker C, Graab U, Hugle M, Hahn H, Klingebiel T, et al. Arsenic trioxide induces Noxa-dependent apoptosis in rhabdomyosarcoma cells and synergizes with antimicrotubule drugs. Cancer letters. 2016;381(2):287–95. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura S, Nagano S, Nagao H, Ishidou Y, Yokouchi M, Abematsu M, et al. Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PloS one. 2013;8(7):e69466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neumann JE, Wefers AK, Lambo S, Bianchi E, Bockstaller M, Dorostkar MM, et al. A mouse model for embryonal tumors with multilayered rosettes uncovers the therapeutic potential of Sonic-hedgehog inhibitors. Nature medicine. 2017;23(10):1191–202. [DOI] [PubMed] [Google Scholar]
- 49.Tang CM, Lee TE, Syed SA, Burgoyne AM, Leonard SY, Gao F, et al. Hedgehog pathway dysregulation contributes to the pathogenesis of human gastrointestinal stromal tumors via GLI-mediated activation of KIT expression. Oncotarget. 2016;7(48):78226–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang D, Cao F, Ye X, Zhao H, Liu X, Li Y, et al. Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta haematologica. 2013;130(4):260–7. [DOI] [PubMed] [Google Scholar]
- 51.Zhang KZ, Zhang QB, Zhang QB, Sun HC, Ao JY, Chai ZT, et al. Arsenic trioxide induces differentiation of CD133+ hepatocellular carcinoma cells and prolongs posthepatectomy survival by targeting GLI1 expression in a mouse model. Journal of hematology & oncology. 2014;7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathews V, Chendamarai E, George B, Viswabandya A, Srivastava A. Treatment of acute promyelocytic leukemia with single-agent arsenic trioxide. Mediterranean journal of hematology and infectious diseases. 2011;3(1):e2011056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbey JT, Pezzullo JC, Soignet SL. Effect of arsenic trioxide on QT interval in patients with advanced malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21(19):3609–15. [DOI] [PubMed] [Google Scholar]
- 54.Hai JJ, Gill H, Tse HF, Kumana CR, Kwong YL, Siu CW. Torsade de Pointes during oral arsenic trioxide therapy for acute promyelocytic leukemia in a patient with heart failure. Annals of hematology. 2015;94(3):501–3. [DOI] [PubMed] [Google Scholar]
- 55.Naito K, Kobayashi M, Sahara N, Shigeno K, Nakamura S, Shinjo K, et al. Two cases of acute promyelocytic leukemia complicated by torsade de pointes during arsenic trioxide therapy. International journal of hematology. 2006;83(4):318–23. [DOI] [PubMed] [Google Scholar]
- 56.Yamazaki K, Terada H, Satoh H, Naito K, Takeshita A, Uehara A, et al. Arrhythmogenic effects of arsenic trioxide in patients with acute promyelocytic leukemia and an electrophysiological study in isolated guinea pig papillary muscles. Circulation journal : official journal of the Japanese Circulation Society. 2006;70(11):1407–14. [DOI] [PubMed] [Google Scholar]
- 57.Roboz GJ, Ritchie EK, Carlin RF, Samuel M, Gale L, Provenzano-Gober JL, et al. Prevalence, management, and clinical consequences of QT interval prolongation during treatment with arsenic trioxide. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32(33):3723–8. [DOI] [PubMed] [Google Scholar]
- 58.Vargas-Bermudez A, Cardenal F, Porta-Sales J. Opioids for the Management of Dyspnea in Cancer Patients: Evidence of the Last 15 Years--A Systematic Review. Journal of pain & palliative care pharmacotherapy. 2015;29(4):341–52. [DOI] [PubMed] [Google Scholar]
- 59.Akare UR, Bandaru S, Shaheen U, Singh PK, Tiwari G, Singare P, et al. Molecular docking approaches in identification of High affinity inhibitors of Human SMO receptor. Bioinformation. 2014;10(12):737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karlou M, Lu JF, Wu G, Maity S, Tzelepi V, Navone NM, et al. Hedgehog signaling inhibition by the small molecule smoothened inhibitor GDC-0449 in the bone forming prostate cancer xenograft MDA PCa 118b. The Prostate. 2012;72(15):1638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katagiri S, Tauchi T, Okabe S, Minami Y, Kimura S, Maekawa T, et al. Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(6):1422–32. [DOI] [PubMed] [Google Scholar]
- 62.Mimeault M, Rachagani S, Muniyan S, Seshacharyulu P, Johansson SL, Datta K, et al. Inhibition of hedgehog signaling improves the anti-carcinogenic effects of docetaxel in prostate cancer. Oncotarget. 2015;6(6):3887–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, et al. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife. 2013;2:e01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, et al. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. Journal of hepatology. 2014;60(3):599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rominger CM, Bee WL, Copeland RA, Davenport EA, Gilmartin A, Gontarek R, et al. Evidence for allosteric interactions of antagonist binding to the smoothened receptor. The Journal of pharmacology and experimental therapeutics. 2009;329(3):995–1005. [DOI] [PubMed] [Google Scholar]
- 66.Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PloS one. 2011;6(11):e27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tian F, Mysliwietz J, Ellwart J, Gamarra F, Huber RM, Bergner A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clinical and experimental medicine. 2012;12(1):25–30. [DOI] [PubMed] [Google Scholar]
- 68.Tian F, Schrodl K, Kiefl R, Huber RM, Bergner A. The hedgehog pathway inhibitor GDC-0449 alters intracellular Ca2+ homeostasis and inhibits cell growth in cisplatin-resistant lung cancer cells. Anticancer research. 2012;32(1):89–94. [PubMed] [Google Scholar]
- 69.Wang C, Wu H, Evron T, Vardy E, Han GW, Huang XP, et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nature communications. 2014;5:4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497(7449):338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong H, Alicke B, West KA, Pacheco P, La H, Januario T, et al. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(14):4682–92. [DOI] [PubMed] [Google Scholar]
- 72.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science (New York, NY). 2009;326(5952):572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuo M, Rashid A, Churi C, Vauthey JN, Chang P, Li Y, et al. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. British journal of cancer. 2015;112(6):1042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. The Annals of pharmacotherapy. 2014;48(1):99–106. [DOI] [PubMed] [Google Scholar]
- 75.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(8):2502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. The New England journal of medicine. 2009;361(12):1164–72. [DOI] [PubMed] [Google Scholar]
- 77.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. The New England journal of medicine. 2012;366(23):2171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCusker M, Basset-Seguin N, Dummer R, Lewis K, Schadendorf D, Sekulic A, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. European journal of cancer (Oxford, England : 1990). 2014;50(4):774–83. [DOI] [PubMed] [Google Scholar]
- 79.Sekulic A, Migden MR, Basset-Seguin N, Garbe C, Gesierich A, Lao CD, et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC cancer. 2017;17(1):332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Danial C, Lingala B, Balise R, Oro AE, Reddy S, Colevas A, et al. Markedly improved overall survival in 10 consecutive patients with metastatic basal cell carcinoma. The British journal of dermatology. 2013;169(3):673–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.von Domarus H, Stevens PJ. Metastatic basal cell carcinoma. Report of five cases and review of 170 cases in the literature. Journal of the American Academy of Dermatology. 1984;10(6):1043–60. [DOI] [PubMed] [Google Scholar]
- 82.Lear JT, Migden MR, Lewis KD, Chang ALS, Guminski A, Gutzmer R, et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. Journal of the European Academy of Dermatology and Venereology : JEADV. 2018;32(3):372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raszewski RL, Guyuron B. Long-term survival following nodal metastases from basal cell carcinoma. Annals of plastic surgery. 1990;24(2):170–5. [DOI] [PubMed] [Google Scholar]
- 84.Sharpe HJ, Wang W, Hannoush RN, de Sauvage FJ. Regulation of the oncoprotein Smoothened by small molecules. Nature chemical biology. 2015;11(4):246–55. [DOI] [PubMed] [Google Scholar]
- 85.Graham RA, Lum BL, Cheeti S, Jin JY, Jorga K, Von Hoff DD, et al. Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(8):2512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wong H, Chen JZ, Chou B, Halladay JS, Kenny JR, La H, et al. Preclinical assessment of the absorption, distribution, metabolism and excretion of GDC-0449 (2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide), an orally bioavailable systemic Hedgehog signalling pathway inhibitor. Xenobiotica; the fate of foreign compounds in biological systems. 2009;39(11):850–61. [DOI] [PubMed] [Google Scholar]
- 87.Ding X, Chou B, Graham RA, Cheeti S, Percey S, Matassa LC, et al. Determination of GDC-0449, a small-molecule inhibitor of the Hedgehog signaling pathway, in human plasma by solid phase extraction-liquid chromatographic-tandem mass spectrometry. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2010;878(9-10):785–90. [DOI] [PubMed] [Google Scholar]
- 88.Tang JY, So PL, Epstein EH Jr., Novel Hedgehog pathway targets against basal cell carcinoma. Toxicology and applied pharmacology. 2007;224(3):257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Graham RA, Chang I, Jin JY, Wang B, Dufek MB, Ayache JA, et al. Daily dosing of vismodegib to steady state does not prolong the QTc interval in healthy volunteers. Journal of cardiovascular pharmacology. 2013;61(1):83–9. [DOI] [PubMed] [Google Scholar]
- 90.Apalla Z, Papageorgiou C, Lallas A, Sotiriou E, Lazaridou E, Vakirlis E, et al. Spotlight on vismodegib in the treatment of basal cell carcinoma: an evidence-based review of its place in therapy. Clinical, cosmetic and investigational dermatology. 2017;10:171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jacobsen AA, Kydd AR, Strasswimmer J. Practical management of the adverse effects of Hedgehog pathway inhibitor therapy for basal cell carcinoma. Journal of the American Academy of Dermatology. 2017;76(4):767–8. [DOI] [PubMed] [Google Scholar]
- 92.Lacouture ME, Dreno B, Ascierto PA, Dummer R, Basset-Seguin N, Fife K, et al. Characterization and Management of Hedgehog Pathway Inhibitor-Related Adverse Events in Patients With Advanced Basal Cell Carcinoma. The oncologist. 2016;21(10):1218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. Journal of the American Academy of Dermatology. 2015;72(2):221–36; quiz 37-8. [DOI] [PubMed] [Google Scholar]
- 94.Malhi V, Colburn D, Williams SJ, Hop CE, Dresser MJ, Chandra P, et al. A clinical drug-drug interaction study to evaluate the effect of a proton-pump inhibitor, a combined P-glycoprotein/cytochrome 450 enzyme (CYP)3A4 inhibitor, and a CYP2C9 inhibitor on the pharmacokinetics of vismodegib. Cancer chemotherapy and pharmacology. 2016;78(1):41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ventarola DJ, Silverstein DI. Vismodegib-associated hepatotoxicity: a potential side effect detected in postmarketing surveillance. Journal of the American Academy of Dermatology. 2014;71(2):397–8. [DOI] [PubMed] [Google Scholar]
- 96.Basset-Seguin N, Hauschild A, Grob JJ, Kunstfeld R, Dreno B, Mortier L, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. The Lancet Oncology. 2015;16(6):729–36. [DOI] [PubMed] [Google Scholar]
- 97.Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120(25):5002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chaudary N, Pintilie M, Hedley D, Hill RP, Milosevic M, Mackay H. Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. British journal of cancer. 2017;116(1):50–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.D’Amato C, Rosa R, Marciano R, D’Amato V, Formisano L, Nappi L, et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. British journal of cancer. 2014;111(6):1168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fan YH, Ding J, Nguyen S, Liu XJ, Xu G, Zhou HY, et al. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene. 2016;35(1):116–24. [DOI] [PubMed] [Google Scholar]
- 101.Fendrich V, Wiese D, Waldmann J, Lauth M, Heverhagen AE, Rehm J, et al. Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms. Annals of surgery. 2011;254(5):818–23; discussion 23. [DOI] [PubMed] [Google Scholar]
- 102.Fu J, Rodova M, Nanta R, Meeker D, Van Veldhuizen PJ, Srivastava RK, et al. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro-oncology. 2013;15(6):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gorojankina T, Hoch L, Faure H, Roudaut H, Traiffort E, Schoenfelder A, et al. Discovery, molecular and pharmacological characterization of GSA-10, a novel small-molecule positive modulator of Smoothened. Molecular pharmacology. 2013;83(5):1020–9. [DOI] [PubMed] [Google Scholar]
- 104.Irvine DA, Zhang B, Kinstrie R, Tarafdar A, Morrison H, Campbell VL, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Scientific reports. 2016;6:25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jalili A, Mertz KD, Romanov J, Wagner C, Kalthoff F, Stuetz A, et al. NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo. PloS one. 2013;8(7):e69064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer cell. 2014;25(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li X, Chen F, Zhu Q, Ding B, Zhong Q, Huang K, et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget. 2016;7(22):33004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, et al. PDGFRalpha up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget. 2014;5(7):1926–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sirkisoon SR, Carpenter RL, Rimkus T, Anderson A, Harrison A, Lange AM, et al. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene. 2018;37(19):2502–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steg AD, Katre AA, Bevis KS, Ziebarth A, Dobbin ZC, Shah MM, et al. Smoothened antagonists reverse taxane resistance in ovarian cancer. Molecular cancer therapeutics. 2012;11(7):1587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Teichman J, Dodbiba L, Thai H, Fleet A, Morey T, Liu L, et al. Hedgehog inhibition mediates radiation sensitivity in mouse xenograft models of human esophageal adenocarcinoma. PloS one. 2018;13(5):e0194809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Skvara H, Kalthoff F, Meingassner JG, Wolff-Winiski B, Aschauer H, Kelleher JF, et al. Topical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitor. The Journal of investigative dermatology. 2011;131(8):1735–44. [DOI] [PubMed] [Google Scholar]
- 113.Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(7):1900–9. [DOI] [PubMed] [Google Scholar]
- 114.Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. The Lancet Oncology. 2015;16(6):716–28. [DOI] [PubMed] [Google Scholar]
- 115.Dummer R, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. Journal of the American Academy of Dermatology. 2016;75(1):113–25.e5. [DOI] [PubMed] [Google Scholar]
- 116.Danial C, Sarin KY, Oro AE, Chang AL. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(6):1325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Einolf HJ, Zhou J, Won C, Wang L, Rebello S. A Physiologically-Based Pharmacokinetic Modeling Approach To Predict Drug-Drug Interactions of Sonidegib (LDE225) with Perpetrators of CYP3A in Cancer Patients. Drug metabolism and disposition: the biological fate of chemicals. 2017;45(4):361–74. [DOI] [PubMed] [Google Scholar]
- 118.Goel V, Hurh E, Stein A, Nedelman J, Zhou J, Chiparus O, et al. Population pharmacokinetics of sonidegib (LDE225), an oral inhibitor of hedgehog pathway signaling, in healthy subjects and in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2016;77(4):745–55. [DOI] [PubMed] [Google Scholar]
- 119.Jain S, Song R, Xie J. Sonidegib: mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. OncoTargets and therapy. 2017;10:1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quinlan M, Zhou J, Hurh E, Sellami D. Exposure-QT analysis for sonidegib (LDE225), an oral inhibitor of the hedgehog signaling pathway, for measures of the QT prolongation potential in healthy subjects and in patients with advanced solid tumors. European journal of clinical pharmacology. 2016;72(12):1427–32. [DOI] [PubMed] [Google Scholar]
- 121.Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87(4):661–73. [DOI] [PubMed] [Google Scholar]
- 122.Maun HR, Wen X, Lingel A, de Sauvage FJ, Lazarus RA, Scales SJ, et al. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. The Journal of biological chemistry. 2010;285(34):26570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Song Z, Yue W, Wei B, Wang N, Li T, Guan L, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PloS one. 2011;6(3):e17687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.O’Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, Schofield P, et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer research. 2011;71(11):4002–14. [DOI] [PubMed] [Google Scholar]
- 125.Coon V, Laukert T, Pedone CA, Laterra J, Kim KJ, Fults DW. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Molecular cancer therapeutics. 2010;9(9):2627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nature chemical biology. 2009;5(3):154–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Buglino JA, Resh MD. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. The Journal of biological chemistry. 2008;283(32):22076–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mann RK, Beachy PA. Novel lipid modifications of secreted protein signals. Annual review of biochemistry. 2004;73:891–923. [DOI] [PubMed] [Google Scholar]
- 129.Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, Resh MD. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nature chemical biology. 2013;9(4):247–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Matevossian A, Resh MD. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Molecular cancer. 2015;14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Petrova E, Matevossian A, Resh MD. Hedgehog acyltransferase as a target in pancreatic ductal adenocarcinoma. Oncogene. 2015;34(2):263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science (New York, NY). 1998;280(5369):1603–7. [DOI] [PubMed] [Google Scholar]
- 133.Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development (Cambridge, England). 1998;125(18):3553–62. [DOI] [PubMed] [Google Scholar]
- 134.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer research. 2007;67(5):2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–12. [DOI] [PubMed] [Google Scholar]
- 136.Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mechanisms of development. 2005;122(2):223–30. [DOI] [PubMed] [Google Scholar]
- 137.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(14):5895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Campbell VT, Nadesan P, Ali SA, Wang CY, Whetstone H, Poon R, et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Molecular cancer therapeutics. 2014;13(5):1259–69. [DOI] [PubMed] [Google Scholar]
- 139.Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(20):7859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lo WW, Wunder JS, Dickson BC, Campbell V, McGovern K, Alman BA, et al. Involvement and targeted intervention of dysregulated Hedgehog signaling in osteosarcoma. Cancer. 2014;120(4):537–47. [DOI] [PubMed] [Google Scholar]
- 141.McCann CK, Growdon WB, Kulkarni-Datar K, Curley MD, Friel AM, Proctor JL, et al. Inhibition of Hedgehog signaling antagonizes serous ovarian cancer growth in a primary xenograft model. PloS one. 2011;6(11):e28077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). Journal of medicinal chemistry. 2009;52(14):4400–18. [DOI] [PubMed] [Google Scholar]
- 143.Bowles DW, Keysar SB, Eagles JR, Wang G, Glogowska MJ, McDermott JD, et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral oncology. 2016;53:74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jimeno A, Weiss GJ, Miller WH Jr., Gettinger S, Eigl BJ, Chang AL, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(10):2766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016;45(3):370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smith S, Hoyt J, Whitebread N, Manna J, Peluso M, Faia K, et al. The pre-clinical absorption, distribution, metabolism and excretion properties of IPI-926, an orally bioavailable antagonist of the hedgehog signal transduction pathway. Xenobiotica; the fate of foreign compounds in biological systems. 2013;43(10):875–85. [DOI] [PubMed] [Google Scholar]
- 147.Sasaki K, Gotlib JR, Mesa RA, Newberry KJ, Ravandi F, Cortes JE, et al. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leukemia & lymphoma. 2015;56(7):2092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Riedlinger D, Bahra M, Boas-Knoop S, Lippert S, Bradtmoller M, Guse K, et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. Journal of hepato-biliary-pancreatic sciences. 2014;21(8):607–15. [DOI] [PubMed] [Google Scholar]
- 149.Zaidi AH, Komatsu Y, Kelly LA, Malhotra U, Rotoloni C, Kosovec JE, et al. Smoothened inhibition leads to decreased proliferation and induces apoptosis in esophageal adenocarcinoma cells. Cancer investigation. 2013;31(7):480–9. [DOI] [PubMed] [Google Scholar]
- 150.Ishiwata T, Iwasawa S, Ebata T, Fan M, Tada Y, Tatsumi K, et al. Inhibition of Gli leads to antitumor growth and enhancement of cisplatin-induced cytotoxicity in large cell neuroendocrine carcinoma of the lung. Oncology reports. 2018;39(3):1148–54. [DOI] [PubMed] [Google Scholar]
- 151.Wilkinson SE, Furic L, Buchanan G, Larsson O, Pedersen J, Frydenberg M, et al. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. The Prostate. 2013;73(16):1810–23. [DOI] [PubMed] [Google Scholar]
- 152.Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS medicinal chemistry letters. 2012;3(2):106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chaudhry P, Singh M, Triche TJ, Guzman M, Merchant AA. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood. 2017;129(26):3465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, et al. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer science. 2016;107(10):1422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sadarangani A, Pineda G, Lennon KM, Chun HJ, Shih A, Schairer AE, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. Journal of translational medicine. 2015;13:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Giri N, Lam LH, LaBadie RR, Krzyzaniak JF, Jiang H, Hee B, et al. Evaluation of the effect of new formulation, food, or a proton pump inhibitor on the relative bioavailability of the smoothened inhibitor glasdegib (PF-04449913) in healthy volunteers. Cancer chemotherapy and pharmacology. 2017;80(6):1249–60. [DOI] [PubMed] [Google Scholar]
- 157.Lam JL, Vaz A, Hee B, Liang Y, Yang X, Shaik MN. Metabolism, excretion and pharmacokinetics of [(14)C]glasdegib (PF-04449913) in healthy volunteers following oral administration. Xenobiotica; the fate of foreign compounds in biological systems. 2017;47(12):1064–76. [DOI] [PubMed] [Google Scholar]
- 158.Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. The Lancet Haematology. 2015;2(8):e339–46. [DOI] [PubMed] [Google Scholar]
- 159.Minami H, Ando Y, Ma BB, Hsiang Lee J, Momota H, Fujiwara Y, et al. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. Cancer science. 2016;107(10):1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(10):2294–303. [DOI] [PubMed] [Google Scholar]
- 161.Shaik MN, Hee B, Wei H, LaBadie RR. Evaluation of the effect of rifampin on the pharmacokinetics of the Smoothened inhibitor glasdegib in healthy volunteers. British journal of clinical pharmacology. 2018;84(6):1346–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shaik MN, LaBadie RR, Rudin D, Levin WJ. Evaluation of the effect of food and ketoconazole on the pharmacokinetics of the smoothened inhibitor PF-04449913 in healthy volunteers. Cancer chemotherapy and pharmacology. 2014;74(2):411–8. [DOI] [PubMed] [Google Scholar]
- 163.Wagner AJ, Messersmith WA, Shaik MN, Li S, Zheng X, McLachlan KR, et al. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(5):1044–51. [DOI] [PubMed] [Google Scholar]
- 164.Cortes JE, Smith BD, Wang ES, Merchant A, Oehler VG, Arellano M, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. American journal of hematology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Tanaka Y, Shibata S, et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: modification of the core skeleton for improved solubility. Bioorganic & medicinal chemistry. 2012;20(18):5507–17. [DOI] [PubMed] [Google Scholar]
- 166.Lubik AA, Nouri M, Truong S, Ghaffari M, Adomat HH, Corey E, et al. Paracrine sonic hedgehog signaling contributes significantly to acquired steroidogenesis in the prostate tumor microenvironment. International journal of cancer. 2017;140(2):358–69. [DOI] [PubMed] [Google Scholar]
- 167.Ibuki N, Ghaffari M, Pandey M, Iu I, Fazli L, Kashiwagi M, et al. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. International journal of cancer. 2013;133(8):1955–66. [DOI] [PubMed] [Google Scholar]
- 168.Kogame A, Tagawa Y, Shibata S, Tojo H, Miyamoto M, Tohyama K, et al. Pharmacokinetic and pharmacodynamic modeling of hedgehog inhibitor TAK-441 for the inhibition of Gli1 messenger RNA expression and antitumor efficacy in xenografted tumor model mice. Drug metabolism and disposition: the biological fate of chemicals. 2013;41(4):727–34. [DOI] [PubMed] [Google Scholar]
- 169.Lauressergues E, Heusler P, Lestienne F, Troulier D, Rauly-Lestienne I, Tourette A, et al. Pharmacological evaluation of a series of smoothened antagonists in signaling pathways and after topical application in a depilated mouse model. Pharmacology research & perspectives. 2016;4(2):e00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer research. 2011;71(2):435–44. [DOI] [PubMed] [Google Scholar]
- 171.Ishii T, Shimizu Y, Nakashima K, Kondo S, Ogawa K, Sasaki S, et al. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. European journal of pharmacology. 2014;723:305–13. [DOI] [PubMed] [Google Scholar]
- 172.Shimizu Y, Ishii T, Ogawa K, Sasaki S, Matsui H, Nakayama M. Biochemical characterization of smoothened receptor antagonists by binding kinetics against drug-resistant mutant. European journal of pharmacology. 2015;764:220–7. [DOI] [PubMed] [Google Scholar]
- 173.Goldman J, Eckhardt SG, Borad MJ, Curtis KK, Hidalgo M, Calvo E, et al. Phase I dose-escalation trial of the oral investigational Hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(5):1002–9. [DOI] [PubMed] [Google Scholar]
- 174.Bai Q, Shen Y, Jin N, Liu H, Yao X. Molecular modeling study on the dynamical structural features of human smoothened receptor and binding mechanism of antagonist LY2940680 by metadynamics simulation and free energy calculation. Biochimica et biophysica acta. 2014;1840(7):2128–38. [DOI] [PubMed] [Google Scholar]
- 175.Bender MH, Hipskind PA, Capen AR, Cockman M, Credille KM, Gao H, et al. Abstract 2819: Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer research. 2011;71(8 Supplement):2819-. [Google Scholar]
- 176.Sinha N, Chowdhury S, Sarkar RR. Deciphering structural stability and binding mechanisms of potential antagonists with smoothened protein. Journal of biomolecular structure & dynamics. 2017:1–21. [DOI] [PubMed] [Google Scholar]
- 177.Bendell J, Andre V, Ho A, Kudchadkar R, Migden M, Infante J, et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naive and Previously Treated Basal Cell Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(9):2082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ueno H, Kondo S, Yoshikawa S, Inoue K, Andre V, Tajimi M, et al. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Investigational new drugs. 2018;36(4):647–56. [DOI] [PubMed] [Google Scholar]
- 179.Peukert S, He F, Dai M, Zhang R, Sun Y, Miller-Moslin K, et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem. 2013;8(8):1261–5. [DOI] [PubMed] [Google Scholar]
- 180.Tu J, Li JJ, Song LT, Zhai HL, Wang J, Zhang XY. Molecular modeling study on resistance of WT/D473H SMO to antagonists LDE-225 and LEQ-506. Pharmacological research. 2018;129:491–9. [DOI] [PubMed] [Google Scholar]
- 181.Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer cell. 2013;23(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science (New York, NY). 1996;274(5285):255–9. [DOI] [PubMed] [Google Scholar]
- 183.Wahid M, Jawed A, Mandal RK, Dar SA, Khan S, Akhter N, et al. Vismodegib, itraconazole and sonidegib as hedgehog pathway inhibitors and their relative competencies in the treatment of basal cell carcinomas. Critical reviews in oncology/hematology. 2016;98:235–41. [DOI] [PubMed] [Google Scholar]
- 184.Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC, et al. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. The oncologist. 2013;18(2):163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kim DJ, Kim J, Spaunhurst K, Montoya J, Khodosh R, Chandra K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32(8):745–51. [DOI] [PubMed] [Google Scholar]
- 186.Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R, et al. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8(5):619–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fan P, Fan S, Wang H, Mao J, Shi Y, Ibrahim MM, et al. Genistein decreases the breast cancer stem-like cell population through Hedgehog pathway. Stem cell research & therapy. 2013;4(6):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li W, Frame LT, Hoo KA, Li Y, D’Cunha N, Cobos E. Genistein inhibited proliferation and induced apoptosis in acute lymphoblastic leukemia, lymphoma and multiple myeloma cells in vitro. Leukemia & lymphoma. 2011;52(12):2380–90. [DOI] [PubMed] [Google Scholar]
- 189.Slusarz A, Shenouda NS, Sakla MS, Drenkhahn SK, Narula AS, MacDonald RS, et al. Common botanical compounds inhibit the hedgehog signaling pathway in prostate cancer. Cancer research. 2010;70(8):3382–90. [DOI] [PubMed] [Google Scholar]
- 190.Yu D, Shin HS, Lee YS, Lee D, Kim S, Lee YC. Genistein attenuates cancer stem cell characteristics in gastric cancer through the downregulation of Gli1. Oncology reports. 2014;31(2):673–8. [DOI] [PubMed] [Google Scholar]
- 191.Zhang L, Li L, Jiao M, Wu D, Wu K, Li X, et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer letters. 2012;323(1):48–57. [DOI] [PubMed] [Google Scholar]
- 192.El-Rayes BF, Philip PA, Sarkar FH, Shields AF, Ferris AM, Hess K, et al. A phase II study of isoflavones, erlotinib, and gemcitabine in advanced pancreatic cancer. Investigational new drugs. 2011;29(4):694–9. [DOI] [PubMed] [Google Scholar]
- 193.Lazarevic B, Boezelijn G, Diep LM, Kvernrod K, Ogren O, Ramberg H, et al. Efficacy and safety of short-term genistein intervention in patients with localized prostate cancer prior to radical prostatectomy: a randomized, placebo-controlled, double-blind Phase 2 clinical trial. Nutrition and cancer. 2011;63(6):889–98. [DOI] [PubMed] [Google Scholar]
- 194.Lohr JM, Karimi M, Omazic B, Kartalis N, Verbeke CS, Berkenstam A, et al. A phase I dose escalation trial of AXP107–11, a novel multi-component crystalline form of genistein, in combination with gemcitabine in chemotherapy-naive patients with unresectable pancreatic cancer. Pancreatology : official journal of the International Association of Pancreatology (IAP) [et al]. 2016;16(4):640–5. [DOI] [PubMed] [Google Scholar]
- 195.Messing E, Gee JR, Saltzstein DR, Kim K, diSant’Agnese A, Kolesar J, et al. A phase 2 cancer chemoprevention biomarker trial of isoflavone G-2535 (genistein) in presurgical bladder cancer patients. Cancer prevention research (Philadelphia, Pa). 2012;5(4):621–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pendleton JM, Tan WW, Anai S, Chang M, Hou W, Shiverick KT, et al. Phase II trial of isoflavone in prostate-specific antigen recurrent prostate cancer after previous local therapy. BMC cancer. 2008;8:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Takimoto CH, Glover K, Huang X, Hayes SA, Gallot L, Quinn M, et al. Phase I pharmacokinetic and pharmacodynamic analysis of unconjugated soy isoflavones administered to individuals with cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2003;12(11 Pt 1):1213–21. [PubMed] [Google Scholar]
- 198.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(20):8455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Benvenuto M, Masuelli L, De Smaele E, Fantini M, Mattera R, Cucchi D, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. 2016;7(8):9250–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen Q, Xu R, Zeng C, Lu Q, Huang D, Shi C, et al. Down-regulation of Gli transcription factor leads to the inhibition of migration and invasion of ovarian cancer cells via integrin beta4-mediated FAK signaling. PloS one. 2014;9(2):e88386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Geng L, Lu K, Li P, Li X, Zhou X, Li Y, et al. GLI1 inhibitor GANT61 exhibits antitumor efficacy in T-cell lymphoma cells through down-regulation of p-STAT3 and SOCS3. Oncotarget. 2017;8(30):48701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gonnissen A, Isebaert S, McKee CM, Dok R, Haustermans K, Muschel RJ. The hedgehog inhibitor GANT61 sensitizes prostate cancer cells to ionizing radiation both in vitro and in vivo. Oncotarget. 2016;7(51):84286–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gonnissen A, Isebaert S, McKee CM, Muschel RJ, Haustermans K. The Effect of Metformin and GANT61 Combinations on the Radiosensitivity of Prostate Cancer Cells. International journal of molecular sciences. 2017;18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, Moriya T, et al. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast cancer (Tokyo, Japan). 2017;24(5):683–93. [DOI] [PubMed] [Google Scholar]
- 205.Kurebayashi J, Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, et al. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer science. 2017;108(5):918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li J, Cai J, Zhao S, Yao K, Sun Y, Li Y, et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. Journal of experimental & clinical cancer research : CR. 2016;35(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shahi MH, Holt R, Rebhun RB. Blocking signaling at the level of GLI regulates downstream gene expression and inhibits proliferation of canine osteosarcoma cells. PloS one. 2014;9(5):e96593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Srivastava RK, Kaylani SZ, Edrees N, Li C, Talwelkar SS, Xu J, et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget. 2014;5(23):12151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tong W, Qiu L, Qi M, Liu J, Hu K, Lin W, et al. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. Journal of cellular biochemistry. 2018;119(4):3641–52. [DOI] [PubMed] [Google Scholar]
- 210.Vlckova K, Reda J, Ondrusova L, Krayem M, Ghanem G, Vachtenheim J. GLI inhibitor GANT61 kills melanoma cells and acts in synergy with obatoclax. International journal of oncology. 2016;49(3):953–60. [DOI] [PubMed] [Google Scholar]
- 211.Wickstrom M, Dyberg C, Shimokawa T, Milosevic J, Baryawno N, Fuskevag OM, et al. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. International journal of cancer. 2013;132(7):1516–24. [DOI] [PubMed] [Google Scholar]
- 212.Yang H, Hu L, Liu Z, Qin Y, Li R, Zhang G, et al. Inhibition of Gli1-mediated prostate cancer cell proliferation by inhibiting the mTOR/S6K1 signaling pathway. Oncology letters. 2017;14(6):7970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]