

Summary
Mesenchymal Stromal Cells (MSCs) have been the subject of clinical trials for more than a generation and the outcomes of advanced clinical trials have fallen short of expectations raised by encouraging pre-clinical animal data in a wide array of disease models. In this perspective, important biological and pharmacological disparities in pre-clinical research and human translational studies are highlighted, and analysis of clinical trial failures and recent successes provide a rational pathway to MSC regulatory approval and deployment for disorders with unmet medical needs.
Introduction
Mesenchymal Stromal Cells (MSCs) were first tested as a cellular pharmaceutical in human subjects in 1995 by Hillard Lazarus(Lazarus et al., 1995) and have since become the most clinically studied experimental cell therapy platform worldwide for which there is no marketing approval in the USA (Fung et al., 2017). The enthusiasm for these mostly early phase clinical trials reflects the considerable ease for the enthusiast to manufacture culture-adapted MSCs from readily accessible tissue sourcing from normal volunteers and the use of tissue culture techniques dating back more than a quarter century. In the US in particular, industrial sponsors have led virtually all advanced phase III clinical trials of MSCs, and the field as a whole has been severely criticized for its ill-informed irrational exuberance(Bianco et al., 2013). This criticism often reflects angst arising from the egregiously predatory business activities of unregulated stem cell clinics in the USA and worldwide riding the unproven promise of regenerative therapies, including MSCs, as a cure-all(Turner and Knoepfler, 2016).
The travails of MSC marketing approval worldwide – The MSCs for GvHD paradigm
MSCs have secured conditional approval in 2012 to treat children with GvHD in Canada and New Zealand, and latterly approval in Japan was obtained as well. The historic road map and outcomes of these approvals and how they informed a clinical trial strategy to secure USA approval of MSCs for GvHD can provide some insights on how best to adapt advanced clinical trial designed for regulatory approval.
In May 2009, Osiris Therapeutics (USA) completed the first major industry-sponsored phase III trial of allogeneic, marrow-derived MSCs for treatment of steroid-refractory Graft vs Host Disease (GvHD) (NCT00366145). The MSCs were sourced from normal volunteers from whom up to 10,000 doses were manufactured per donor and the ensuing cryobanked product (Prochymal™) was thawed and transfused at point of care in eligible patients with steroid refractory GvHD (Table 2). The overall response rate with Prochymal™ was 82% vs 73% for placebo (p=0.12). The clinical trial results have been presented as part of press releases and society meeting abstracts (Martin et al.). Subset analysis suggested that children with GvHD were responsive to MSCs(Kurtzberg et al., 2010). On this basis, on May 17, 2012, Health Canada issued marketing approval for Prochymal™ to treat children with acute Graft versus Host Disease (Reicin, 2012). Health Canada approved the drug via a Notice of Compliance with Conditions (NOC/c), which allows certain drugs into the market without full efficacy data. Under these terms, Health Canada subjected Osiris Therapeutics to heightened post-market surveillance. In addition to long-term monitoring, the NOC/c requirements restricted treatment to children with refractory GvHD and restricted access of Prochymal™ to physicians who have experience treating GvHD patients. As of 2018, 6 years after Health Canada approval, Osiris – or latterly Mesoblast – have not marketed Prochymal™ in Canada. Debate at the Canadian House of Commons Standing Committee on Health held on May 30, 2016 spoke to the conundrum at hand, where testimony described the case of Prochymal™, which was “probably the first truly innovative technology to be put forward for a conditional licensing approval” [in Canada], and it “went nowhere because it couldn’t get reimbursed” (Minutes of Proceedings of House of Commons (Canada) Standing Committee on Health, 2016). There is no publicly available data showing that Prochymal™ has been distributed outside of clinical trials on the terms of marketing following conditional approvals in Canada or New Zealand for the indication of pediatric steroid-refractory GvHD.
TABLE 2:
Clinical trial adaptive design for study of Prochymal/Remestemcel-L for GvHD
| Study | NCT00366145 | NCT02336230 |
|---|---|---|
| Product/ Sponsor | Prochymal™/ Osiris Therapeutics | Remestemcel-L™/ Mesoblast |
| Cell dosing | 2 million cells/kg twice a week for 4 weeks | 2 million cells/kg twice a week for 4 weeks |
| Primary outcome | Complete GvHD Response of greater than or equal to 28 days duration | Overall GvHD Response Rate at Day 28 as to include both complete response and partial response: • Complete response (CR) - Resolution of GVHD in all involved organs. • Partial response (PR) - Organ improvement by at least one stage without worsening of any other organ. |
| Target Enrollment | 240 | 60 |
| Ages eligible | 6 Months to 70 Years | 2 months and 17 years |
| GvHD grade at enrollment | Any grade B-D (IBMTR grading) of acute GvHD | • Grade C or D GvHD involving the skin, liver, and/or gastrointestinal (GI) tract • Grade B GvHD involving the liver and/or GI tract, with or without concomitant skin disease • Exclusion: Grade B GvHD with skin-only involvement. |
| Steroid refractory definition | No improvement after 3 days and a duration of no greater than 2 weeks | Progression within 3 days or no improvement within 7 consecutive days |
| Study sites | 70 | 23 |
| Completed | May 2009 | February 2018 |
Meaningful study design adaptations in NCT02336230 are in bold characters.
In Japan, the Act on the Safety of Regenerative Medicine and the Pharmaceuticals, Medical Devices and Other Therapeutic Products Act were enacted in November 2014, creating a new framework for clinical research and products related to regenerative medicine(Sipp, 2015). This act set the stage for approval of MSCs as the first allogeneic regenerative medicine product in Japan. Utilizing the technology licensed-in from Osiris Therapeutics, Inc. in 2003, JCR Pharmaceuticals Co., Ltd. (JCR) developed TEMCELL® in Japan for the treatment of acute GvHD. JCR announced on September 15 2015 that the Japanese Ministry, Labour and Welfare has approved TEMCELL® for acute GvHD. Reimbursement for TEMCELL® was authorized by Japanese National Health Insurance at ¥868,680 (US$7,079) per bag of 72 million cells. In Japan, the average adult patient will receive 16 or up to 24 bags of 72 million cells. On this basis, a treatment course of TEMCELL® in an adult Japanese patient to be reimbursed up to ¥20,848,320 (US$170,000). TEMCELL® generated ¥0.7 billion sales for JCR in FY2016 (JCR Pharmaceuticals Co., Ltd. Annual Report, 2017).
Therefore, with the exception of Japan and its approval of MSCs for acute GvHD, MSCs have remained available solely through clinical trial mechanisms for all indications in other major regulatory jurisdictions, including North America and Europe. Notwithstanding, rigorous, peer-reviewed scientific inquiry and well-designed, regulator-compliant clinical trials can provide mechanistic and translational insights which may well demonstrate a useful role for MSCs in disorders with unmet medical needs including GvHD and others(Phinney et al., 2013, Galipeau, 2013, Fibbe et al., 2013). Indeed in March 2018, the European Commission has approved the first MSC pharmaceutical (Alofisel®) to treat Crohn’s related enterocutaneous fistular disease, and this progress foreshadows such developments.
MSC beginnings
Prior to their branding as mesenchymal stem cells(Caplan, 1991), marrow-derived fibroblasts were exploited in the Dexter assay as feeder cells to allow for long-term study of murine hematopoietic stem cells in a reductionist in vitro system(Dexter et al., 1977). This system spoke to the niche-like properties of marrow fibroblasts, at least in regards to sustaining primitive hematopoietic elements. The advent of recombinant growth factors relegated the Dexter assay to the trash bin of scientific history, but culture adapted marrow fibroblasts themselves were found to serve as a powerful surrogate in vitro model system to study human hematopoiesis, bone developmental biology, and related mesenchymal structural elements(Friedenstein et al., 1974, Kfoury and Scadden, 2015). Based on their niche properties and mesodermal structural capacity, their ability to profoundly affect the functionality of bystander innate and adaptive immune cells was discovered (Bernardo and Fibbe, 2013). These insights led to first-in-human clinical trials, where transfusion of MSCs were successful in accelerating hematopoietic recovery following high-dose myeloablative chemotherapy(Koc et al., 2000) and reverse steroid resistant GvHD(Le Blanc et al., 2008).
From these pioneering translational studies, the exploitation of MSCs’ niche-like regenerative properties and their anti-inflammatory action have spurned the use of both autologous and allogeneic MSCs for acute tissue injury syndromes, chronic degenerative disorders, and inflammatory disease. By far, the most prevalent source of MSCs in clinical trials is adult bone marrow, followed by adipose tissue with an emergence of puerperal discards such as umbilical cord tissue and placental cells (Supplemental Table 1). Industrial-sponsored manufactured allogeneic MSC cells allow for manufacture of up to 1 million doses per donor for mass deployment, whereas a mix of autologous or allogeneic MSCs are used by academic centers, which typically manufacture no more than 10 doses per donor(Galipeau, 2013). The relative merits of allogeneic vs autologous and of distinct tissue MSC sourcing are typically driven by proprietary concerns rather than compelling biological superiority of a MSC platform in regards to potency and outcomes. Notwithstanding, after more than 20 years of clinical research in the translational use of MSCs, the question remains whether MSCs can fulfill the therapeutic promise foreshadowed by pre-clinical animal research.
Cognitive dissonance between pre-clinical murine and human MSC clinical trials outcomes
The knowledge of MSC mechanisms of action are derived predominantly from pre-clinical work in murine systems and in vitro analysis of human MSCs. A typical experimental scenario involves the harvest of mouse bone marrow and culture expansion of plastic adherent MSC progenitors in two dimensional culture flask systems in room air humidified incubators akin to what was described by Alexander Freidenstein in 1974. The resultant polyclonal mix of MSCs are harvested during the log phase of growth, and these MSCs are adoptively transferred to immune-competent experimental mice that are nearly always syngeneic to the MSC product. Using similar manufacturing methods, human MSCs – typically allogeneic – are culture expanded to their replicative limit and cryobanked for later use. Human subjects enrolled in advanced clinical trials designed for marketing approval typically receive one or multiple doses of allogeneic MSCs directly retrieved from the freezer or following culture rescue. Indeed, there are numerous peer-reviewed scientific reports providing unambiguous demonstration of the positive effect of MSC adoptive transfer in pre-clinical mouse models of disease. However, the impact of MSCs on murine outcomes has not readily translated to equivalency in human phase III studies [Table 1]. The dissonance between mouse and human clinical outcomes may be best explained by the apparent discrepancies of: (i) immune compatibility, (ii) dosing, and (iii) fitness of culture adapted MSCs.
TABLE 1:
Comparative analysis of pre-clinical murine data and human clinical trial use of MSC
| Murine pre-clinical studies | Industry Sponsored Phase III MSC clinical trialsa | |
|---|---|---|
| MSC immune compatibility | Overwhelmingly syngeneic | 11:8 allogeneic:autologous |
| MSC fitness | Typically “fresh” | Mostly “thawed”b |
| Cell dose | Typically 50 million cells/kg IV | 2 million cells/kg IV, up to 120-600 million cells subdermal or endomyocardial. |
| Clinical outcomes | Predominantly positive | 1st European commission marketing approval on record for MSC product: Alofisel® (darvadstrocel) granted March 2018 |
Supplemental Table 1 for detailed description of 19 studies
Notable exception of Cx401 and Cx601 (aka: Alofisel®/darvadstrocel - NCT01541579) and Mesenchymal Cardiopoietic Cells (NCT01768702) which are live cell products that were culture rescued prior to administration to human subjects (See Supplemental Table 1)
MSC Immune Compatibility
In the early days of MSC translational development, it was part of lore that MSCs possessed unique - yet unproven – immune privilege allowing for adoptive transfer in allogeneic immune competent recipients without risk of transferred cell rejection. This one size fits all narrative informed the development of mass produced MSCs from a few donors for widespread use in allogeneic unrelated recipients for an array of ailments. Yet, nearly all the pre-clinical mouse data supporting the use of MSCs examined the use of syngeneic, MHC-matched cells when examining efficacy endpoints. It has since been demonstrated that MSCs – like all somatic tissue – express MHC I molecules constitutively and have the ability to express MHC II when exposed to inflammatory cues such as Interferon-γ. Though MSCs have the capacity to express potent inhibitory molecules of both innate and adaptive immune effectors, these may not suffice to fend off acquired alloimmunization. Indeed, the notion of MSC being immunoevasive and not immune privileged has been proposed(Ankrum et al., 2014). Admittedly, the use of pre-banked allogeneic MSCs is the only feasible deployment strategy for use in acute tissue injury syndromes like stroke, sepsis, or myocardial infarction, where the delays in manufacturing autologous MSCs would forfeit their utility in affecting outcomes. Independently from biological concerns for immune compatibility, a business case can be made that only mass produced allogeneic MSCs will ever sustain a margin driven marketing model where cost of goods is the main headwind to commercial success. However, immunological compatibility between donor MSCs and recipient may be critically vital in certain situations such as enhancing engraftment in bone marrow transplantation(Nauta et al., 2006) as well as in a clinical scenario where long-term repeated administration would be required to affect outcomes for a chronic disorder. When sought, measurable humoral alloimmunization in human subjects receiving mismatched MSCs can be detected(Reinders et al., 2015), though its role in failure to meet primary efficacy endpoints in advanced clinical trials using allogeneic MSCs intravenously remain unknown. It is reasonable to assume that for acute tissue injury syndromes, such as myocardial infarction and stroke where long term outcomes are dictated by short term biological recovery (typically in the first week post injury), allogeneic MSCs may still provide a benefit prior to an adaptive immune rejection response. Indeed, secondary analysis of post stroke outcomes at one year following MSC therapy (NCT01436487) are suggestive of a possible benefit(Hess et al., 2017).
Dosing
Following tail vein intravenous transfusion in mice, the bulk of MSCs are immediately trapped in lung microvasculature and a small subset may redistribute to sites of injury or damage(Sensebe and Fleury-Cappellesso, 2013), though there is no evidence that the latter property is required for their clinical effect. Indeed, administration of MSCs in extravascular compartments (subcutaneous, intramuscular, or peritoneal) also affects outcomes of distant organs (e.g. inflamed brain) without meaningful diseased tissue tropic MSC redistribution, speaking to their systemic effect. When MSCs are given intravenously to rodents, it is often at a dose of 50 million MSC/kg. In most clinical indications, human MSCs are transfused intravenously at doses typically in the 1-2 million cells/kg and never more than 12 million cells/kg. From a comparative therapeutics perspective, an argument can be made that body weight adjusted dosing of a cell drug in rodents may not precisely predict human pharmacology or response(Shanks et al., 2009). However, the order of magnitude difference in MSC dosing between species would foreshadow a negative bias in outcomes for humans if the operating biological mechanisms are comparable between species and are dose dependent. Considering these variables, we can surmise that methods and scale of manufacture, point-of-care deployment, and dosing trump MSC paracrine potency independent of their tissue sourcing. Despite the challenges in demonstrating unequivocal efficacy, there is little controversy that intravenous administration of MSCs (mostly marrow derived, but also adipose) is clinically safe, with post infusion febrile reaction being the sole adverse event likely associated with their use(Lalu et al., 2012). Whilst murine culture adapted MSCs are prone to spontaneous immortalization and latter transformation – a feature observed nearly 50 years ago(Franks et al., 1970), claims of similar genetic instability and tumorigenicity for human culture expanded MSCs have been debunked(Sensebe et al., 2012).
Fitness
Using mouse models of tissue injury or inflammatory pathology, it is apparent that MSCs impact outcomes through paracrine secretion of multiple cytokines, morphogens, small molecules, and cargo-bearing exosomes, including contact factors, which affect the biology of adjacent and distant responder cells and tissue(Wang et al., 2014). The vast majority of these mouse studies examine the use of culture adapted murine MSCs harvested during the log phase of growth with optimal metabolic fitness, high replication capacity, and syngeneic status [eg: autologous] to the recipient. The usual disclaimers and biases of using in-bred mouse strains as experimental systems predictive for the human condition are valid, but not specific to MSC science. Reductionist analysis of the MSC secretome in animal disease models clearly identify an array of candidate molecule pathways, which as an aggregate, provide a cogent matrix of mechanisms of action, which may provide a road map for human MSC functionalities where comparable(Chinnadurai et al., 2018).
In contrast, the vast majority of human clinical trials use allogeneic cryobanked MSCs that are thawed immediately prior to transfusion. MSCs display molecular signatures of cell injury in the first 24 hours following retrieval from cryostorage, and these correlate with defects in suppression function in vitro, increased susceptibility to lysis by immune cells, and complement as well as shortened persistence in vivo following intravenous transfusion(Moll et al., 2016). Furthermore, there is emerging data that human MSCs attaining replicative exhaustion [aka senescence] are impaired in their ability to suppress inflammation(Sensebe and Fleury-Cappellesso, 2013), a feature likely to occur in industrial-scale MSC expansion methodologies. Considering that MSCs are typically transfused in patients within a few hours post thaw, we can hypothesize that allogeneic, likely senescent human MSCs directly retrieved from cryostorage, with its associated effect on viability, functionality, and in vivo persistence, are less optimal than metabolically fit materials routinely used in analogous murine systems. Distinct from metabolic fitness arising from culture methods, is the issue of donor fitness, in particular when manufacturing thousands of allogeneic doses from a single human volunteer. As an aggregate, culture adapted MSCs from otherwise healthy volunteer donors invariably express canonical cell surface markers of identity for MSCs and maintain progenitor properties to varying degrees. However, when interrogating the functional response of MSCs to stimulatory cues, there can be substantial variance in the magnitude of expression of key effector pathways – such as IDO upregulation following interferon-γ stimulation(Francois et al., 2012) – suggesting that biomarker-driven donor screening (or the lack thereof) may impact potency of a mass manufactured product.
Efferocytosis as a theorem for cell autonomous MSC effect in vivo
In 2009, Eva Mezey and colleagues at NIH unequivocally demonstrated that intravenously transfused mouse MSCs accumulate in the lung and transmigrate outside to the vascular space, where more than half of lung trapped MSCs are rapidly phagocytosed by lung resident tissue macrophages(Nemeth et al., 2009). They went on to show that MSCs led to a protective effect against lethal sepsis due to IL-10 produced by endogenous macrophages. Since this seminal report, there have been a plurality of reports validating a unique cross-talk between exogenous MSCs and recipient monocyte/macrophages as part of the anti-inflammatory effect of MSCs(Carty et al., 2017). Intriguingly, xenotransplantation of human MSCs into mice teaches us that there are also likely cell-function-autonomous effects of MSCs. Namely, intravenous infusion of live or dead human MSCs evoke a similar molecular genetic response in murine host lungs(Luk et al., 2016). Taking into consideration the inherent biases in studying the immune physiology of interspecies cell transfer, these data speak to the possibility that MSCs may elicit a non-specific immune suppressive effect through their phagocytosis by the host reticuloendothelial system(Poon et al., 2014). Furthermore, the suppressive effect can take place independently of MSC viability. In contrast to blood-borne lymphomyeloid cells, MSCs are the only anchorage-dependent nucleated cellular product ever examined as a transfusion pharmaceutical.
Plazentallen as Rosetta stone of MSC function
Intriguingly, pregnancy is a normal physiological circumstance where a large bolus of circulating nucleated allogeneic non-blood elements give us biological guidance on the pharmacology of transfused MSCs. First described in 1893 as plazentallen, it was observed that pregnant women having succumbed from the complications of eclampsia had fetal trophoblast-derived stromal cellular elements shed in uterine veins and which embolised to lung microvasculature. It was later discovered that this phenomena is part of normal pregnancy and it was estimated that several grams of apoptotic fetal stromal cellular elements travel from the uterus to the lung on a daily basis. These fetal stromal elements are cleared by maternal lung resident phagocytes within a few days and may lead to an upregulation of IL-10 and IDO by these same phagocytes(Abumaree et al., 2006). It is now believed that this phenomena may be an important component of foeto-maternal tolerance and that efferocytosis of fetal apoptotic stromal trophoblastic cells by maternal phagocytic lung macrophages may provide a mechanism to provide tolerance to the semi-allogeneic fetus(Askelund and Chamley, 2011). Herein lies a plausible mechanism that conciliates the observation that intravenous administration of MSCs and their subsequent lung entrapment may recapitulate a foeto-maternal tolerance strategy evolved by placental vertebrates. Indeed, it may well be that the phagocytic clearance of MSCs elicit an IL-10, IDO, and TGFβ suppressive response akin to that observed when recycling apoptotic cellular debris by efferocytosis(Elliott et al., 2017).
Limits to the potency of apoptotic MSCs
The concept of efferocytosis as a means by which MSC can drive immune suppression in a cell function-autonomous manner was first suggested by Moll et al.(Moll et al., 2016) and provides further credence to the observation that functionally compromised, apoptotic, or dead MSCs may drive a suppressive response albeit bereft of added benefit of living MSC functional properties. This unifying theorem may also explain how xenogenic, allogeneic, and syngeneic MSCs – especially if they display cell surface markers of cell injury such as phophatidylserine – may trigger a physiological mechanism of tissue clearance that promotes immune suppression or tolerance. Intriguingly, it has been demonstrated that the infusion of MSCs rendered apoptotic in vitro can deliver some immunosuppressive activity in GvHD mice. However, there are limitations of using apoptotically-rendered MSCs for the treatment of GvHD. In contrast to live MSCs, apoptotic MSCs are completely ineffective when administered intravenously in mice. However, apoptotic MSCs only improved GvHD when infused intraperitoneally and with a therapeutic efficacy substantially reduced in comparison to that of comparably sourced live MSCs (Galleu et al., 2017). Considering that the potency of apoptotic MSCs is substantially less than that of live MSCs suggests that in addition to efferocytotic clearance, biological fitness of MSCs and their functionalities play an important role in their utility as a cell pharmaceutical (Figure 1). The phenomenon of MSC efferocytosis is not in itself undesirable per se, but it certainly mightily complicates the interpretation of pre-clinical animal data where xenotransfer of human MSCs is examined as a primary method of analysis. It also adds a layer of complexity when attempting to distinguish MSC effects arising from cell metabolic activity from non-specific immune modulation, arising from clearance of immune mismatched or functionally marginal MSCs.
Figure 1.
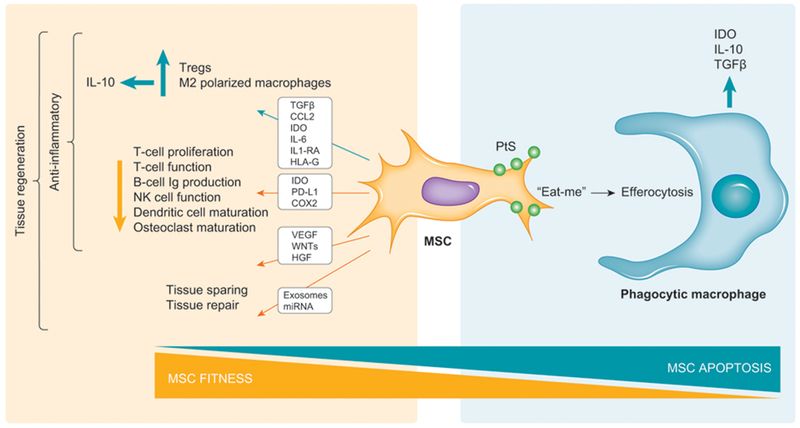
MSC fitness, function, and fate theorem. Culture adapted “fit” MSCs express inflammation-suppressing paracrine factors that augment Treg function and M2-macrophage polarization as well as suppress effector lymphoid cell function. MSCs are also habilitated to produce morphogens and exosomes that promote tissular repair. The sum effect of these additive functionalities is to drive tissue regeneration. MSCs progressing to apoptosis express “eat-me” signals such as phosphatidylserine (PtS) and are susceptible to the alternate efferocytosis pathway where their engulfment by phagocytic macrophages leads to expression of immune tolerance factors. The reciprocal relationship between fitness and apoptosis dictates whether MSC metabolism or their efferocytosis, respectively, is responsible for their in vivo biological effects.
Phase III Industry-sponsored MSC clinical trials - Adaptive Clinical trial design and outcomes
A query of the clincaltrials.gov international database for self-reported industry-sponsored phase III clinical trials provides important insights on the state of the field (Supplemental Table 1). Amongst these 19 studies, ten are completed of which there are three terminal studies that have outcomes that have been disclosed in a public forum that allows for analysis: allogeneic marrow MSCs for GvHD, autologous marrow MSCs for heart disease, and allogeneic adipose MSCs for Crohn’s fistular disease.
Allogeneic marrow MSCs for GvHD
The first major industry-sponsored phase III trial of MSCs (aka: Prochymal™) was for treatment of steroid-refractory GvHD (NCT00366145) and completed in 2009. The primary endpoint of GvHD, complete remission at day 28 post infusion, was not significantly increased relative to placebo (Martin et al.). In 2013, the Prochymal™ assets were divested from Osiris Therapeutics to Mesoblast Inc (Australia), who is now the sponsor on more than four active Phase III studies examining the use of Prochymal™ for Crohn’s disease (NCT00482092), Chronic Heart failure (NCT02032004), back pain (NCT02412735), and pediatric GvHD (NCT02336230). The common denominator in all these studies is the focused use of banked allogeneic, marrow derived MSCs, thawed and infused. The adaptive clinical trial design from the original use of MSC for GvHD (NCT00366145) to the recently completed study of MSC in pediatric GvHD (NCT02336230) provides important insights on the importance of empirical clinical observation informing selective patient enrolment to meet primary clinical endpoints. In the original unsuccessful industry-sponsored placebo-controlled study Prochymal™ for treatment of steroid-resistant GvHD (NCT00366145), both children and adults with any grade B-D GvHD were treated if steroid resistant for at least 3 days up to 14 days(Martin et al.). Since then, some of the best empirical insights on patient selection for MSCs therapy of GvHD have been obtained from the large number of GvHD patients having received MSCs either through clinical trials in the US, Europe, or through hospital exemption in Europe. In GvHD, it has been observed that children respond better to allogeneic MSCs than do adults overall(Kurtzberg et al., 2010) and treating patients early on is better than delaying therapy after onset acute GvHD(Ball et al., 2013). Gut and liver GvHD are more responsive than skin GvHD. In the absence of robust predictive biomarkers, these observations provide guidance in clinical trial design biased towards subject selection likely to be responders. These data likely informed an adaptive clinical trial design for NCT02336230 where identical MSC product and dosing scheme between both studies was maintained but where the definition of response, age of inclusion, severity of disease and exclusion of skin-only GvHD, as well as a more aggressive start time for MSC transfusion were implemented [Table 2]. The latter Mesoblast-sponsored study of marrow MSCs in pediatric GvHD completed recruitment in December 2017. In February 2018, it was announced by press release that the study had successfully met the primary endpoint of improved Day 28 Overall Response (OR) in steroid-refractory pediatric subjects with severe disease. Day 28 OR was 69% and was significantly improved (p=0.0003) compared to protocol-defined historical control rate of 45%. This outcome likely foreshadows the first FDA approved MSC product in the USA. In the absence of robust predictive biomarkers of response, the judicious use of clinical observation and subgroup analysis of responders and non-responders in early phase clinical trials may inform the rational selection of patients enrolled in advanced clinical trials so as to bias towards outcomes meeting primary clinical endpoints of success.
Autologous marrow MSCs for heart failure
Marrow-derived MSCs have also been tested in a phase III trial by Celyad S.A. (Belgium) for treatment of chronic advanced ischemic heart failure (NCT01768702). A critical distinction with the identically marrow-sourced sourced Prochymal™ product is that the Celyad protocol utilized autologous marrow-sourced MSCs that were subsequently polarized towards a “cardiopoietic” phenotype and were administered without interval freezing between culture and endoventricular delivery of 600 million MSCs in subjects. An earlier Celyad-sponsored study performed with the same MSC product in subjects with heart failure secondary to ischemic cardiomyopathy (NCT00810238) was suggestive of improved cardiac outcomes(Bartunek et al., 2013). However, results published in 2016 of an adequately powered clinical trial (NCT01768702) was unable to demonstrate a significant change between MSC and placebo groups from baseline and 39 weeks in a hierarchical composite primary endpoint(Bartunek et al., 2016). Distinct from prior studies in this space, these trials utilized metabolically fit and autologous marrow MSC products, thereby mitigating any negative functional bias arising from immune incompatibility between MSC product and recipient as well as optimizing functionality. Here again issues arise due to practical limits in cell dosing. Subjects received 600 million to 1.2 billion autologous MSCs resuspended in 10 mLs and delivered by an average 18 separate 0.5 mL endoventricular injections at least 1 cm apart. The technical challenge in delivering MSCs in this study is likely an important headwind to achieving biological impact. The delivery of multiple endoventricular injections to a heart of 0.5 ml each whilst circumventing areas of thin myocardium [< 8 mm thickness] to avoid perforation of the ventricle would introduce substantial patient-to-patient variability considering that for a disease like ischemic cardiomyopathy with patchy scar scattered often all around the left ventricle, this could have been a challenge. Thus, there is the concern that the injections are concentrated in areas of good myocardium that may increase the risk of disruption of the tissue and worsening outcomes. Indeed, post hoc analysis of ventricular remodeling at 52 weeks post treatment for NCT01768702 study participants revealed that the largest reverse remodeling was evident in the patients receiving a moderate number of injections (eg: <20). These data suggest an inverted U-shaped dose/response curve with worse outcomes at higher dose delivery attempts. This counterintuitive observation informs that the number of injections may well be an important factor in improving outcomes, as more injections may cause potential myocardial damage, through multiple mechanisms both mechanical and biological(Teerlink et al., 2017). A path forward for this approach may well be to consider alternate means of augmenting the biology of infused MSCs as a method to enhance potency considering the practical limits on dosing via the endoventricular route.
“Fit” adipose MSCs for enterocutaneous fistular disease
Akin to the Celyad studies that utilized metabolically fit autologous MSCs, Cellerix, S.A. sponsored a study (NCT00475410) of autologous adipose MSCs (aka: Adipose stomal cells – ASCs) that were culture revived for 48 hours prior to local injection for treatment of complex perianal fistulas in patients without inflammatory bowel disease. Up to 60 million ASCs were admixed with fibrin glue (or not) and administered as multiple local injections in and around the fistula and compared to fibrin glue alone as the control. The study completed enrollment of 214 subjects allocated to the three arms in 2009 and the ASC treatment groups were non-superior to fibrin glue alone when examining the primary endpoint of sustained closure and healing of fistulas at 6 months post treatment(Herreros et al., 2012). Cellerix, S.A. was acquired by TiGenix, N.V. in May 2011. The data obtained from NCT00475410 informed the adaptive design of a distinct, TiGenix-sponsored, phase III trial of culture-rescued ASCs for enterocutaneous fistular disease, but with four meaningful changes: (i) allogeneic ASCs were used rather than autologous; (ii) cell dose was substantially increased to 120 million cells; (iii) no fibrin glue matrix was used for intrafistular injection and (iv) only patients with Crohn’s disease were enrolled whereas these patients were excluded in the previous trial (Table 3). The resultant TiGenix-sponsored trial NCT01541579 was completed in 2015 and represents the first unambiguously successful use of MSCs/ASCs in an advanced clinical trial. Results were published in 2016 where it was shown that allogeneic ASCs were significantly and substantially superior to placebo in treating Crohn’-associated perianal fistulas. Indeed, a significantly greater proportion of patients treated with ASCs versus placebo achieved remission of fistular disease at 24 weeks post treatment (50% vs 34%, p=0.24) and suffered less treatment related adverse events (17%) when compared to placebo (29%) (Panes et al., 2016). These results were sustained for at least one year after treatment as well(Panes et al., 2018).
TABLE 3:
Clinical trial adaptive design for study of Adipose Stromal Cells for enterocutaneous perianal fistular disease
| Study | NCT00475410 | NCT01541579 |
|---|---|---|
| Product/Sponsor | Cx401/Cellerix | Cx601/TiGenix |
| Source | Autologous ASCs | Allogeneic ASCs |
| Clinical indication | Adults with cryptoglandular complex fistula-in-ano excluding Crohn’s disease | Adults with treatment-refractory complex perianal fistulas in patients with Crohn’s disease. |
| Cell dosing | 20 million cells + optional repeat of 40 million cells intralesional (with and without fibrin glue) |
120 million cells intralesional in a single course of treatment (24 mLs in and around lesion). No fibrin glue. |
| Primary outcome | Closure of fistula at 24 weeks and 1 year | Closure of fistula at 24 weeks and 2 years |
| Target Enrollment | 214 | 278 |
| Study sites | 19 | 49 |
| Completed | August 2009 | July 2015 |
Meaningful study design adaptations in NCT01541579 are in bold characters.
There are three meaningful distinctions that sets this study apart from precedent discussed negative MSC trials: MSC fitness, route of delivery, and dose intensity. First, batch manufactured allogeneic, adipose-derived MSC cell doses were retrieved from cryostorage and subsequently allowed to fully recover in tissue culture for a few days before being shipped to point-of-care and administered to patients, and secondly, 120 million MSCs were formulated in a total volume of 24 mLs and delivered at cutaneous site of disease – intrafistular – rather than transfused IV, allowing for a local mass effect not otherwise achievable. Though the study made a pivot from autologous ASCs used in NCT00475410 to allogeneic ASCs used in NCT01541579, likely to mitigate cost-of-goods as part of a marketing deployment strategy, the trial’s use of replication fit MSCs parallels the approach used in murine pre-clinical models (Table 1) and also biases towards a local anti-inflammatory effect through a cell dosing strategy which maximizes a locoregional paracrine MSC biology. Indeed, a substantial number of European-based academic MSC clinical trials are testing the use of fit – rather than thawed – MSCs for systemic and local use for osteoarticular and inflammatory disorders. Therein lies a pathway for clinical utility of MSCs where practical remedies maintain fitness and address dose intensity required to achieve a biological endpoint.
The importance of MSC potency assays and development of biomarkers predictive of response
MSC identity and potency assays are a mandatory component for advanced clinical trials and are required for securing a FDA biologics license application (BLA) for marketing in the USA(Mendicino et al., 2014). The core notion is that these assays serve as means to validate the potency of distinct MSC manufactured lots and are a guarantor of functionality predictive of effectiveness in clinical practice. The extensive discovery pre-clinical literature informs us that MSCs – similar to many cell platforms dependent upon metabolic fitness for optimal utility – affect outcomes in a multifunctional manner. Culture adapted MSCs likely mirror the homeostatic functionality of their endogenous counterparts and likely deploy pharmaceutical properties due to their interaction with host tissues and cells following their bolus delivery in vivo. No singular physiological property is entirely predictive of mechanism of action, especially since most MSCs post intravenous infusion are likely subjected to phagocytosis and apoptosis and are cleared by tissue resident perivascular macrophages within no more than a few days. Within that window of time, metabolically active MSCs, possibly responsive to environmental cues, produce cytokines, chemokines, morphogens, and microparticles that alter the biology of host bystander lymphomyeloid cells, and these likely mitigate tissue damage and loss (Figure 1). It is anybody’s guess which combination of factors are most critical to MSC regenerative functionality in humans, and the issue cannot be ethically resolved in human subjects by the type of reductionist experiments readily feasible in rodents.
The effort to map such using in vitro potency assays – even in reductionist systems – may not provide clear guidance on predictive potency assays. At best, potency assays mirror hypothesis-driven informed guesses that are in part cognizant of pre-clinical animal systems and in vitro cell biology(Galipeau et al., 2016). Their utility lies in defining a cell function signature that serves as a yardstick against which distinct MSC manufacturing runs can be compared. A cogent argument can be made that the focus on MSC cell biology provides only part of the answer of predictive potency testing and that measuring patient parameters that would be predictive of responsiveness to MSC therapy is likely as important. In industry-sponsored trials, where a large number of patients are treated with an identical lot of allogeneic cells, outcomes – not surprisingly – vary between subjects. Variables such as severity of disease, co-morbidities, and other clinical parameters always impact outcomes of any therapeutic intervention. Considering that MSCs act in part indirectly through the in vivo licensing of host immune bystander cells, the responsiveness of individuals to MSCs may be predicated by their idiomatic immune status at time of infusion. It may well be that the examination of donor MSC and recipient leukocyte interaction in itself may serve as a biomarker of response. Similar assays have been examined in solid organ transplantation so as to predict functional immune compatibility. Indeed, the advent of modern bone marrow transplantation was informed by the development of the mixed lymphocyte reaction by Fritz Bach in 1964 that paved the way for donor/recipient pairing permissive for good BMT outcomes(Bach and Hirschhorn, 1964). A similar approach could be developed where the MSC biological interplay on recipient lymphoid or myeloid cells serves as a surrogate of host response to MSC products. These latter data can only be inferred by post hoc analysis of select biomarkers from “responders” and “non-responders” enrolled in clinical trials that would be predictive of response to MSCs. The group led by Francesco Dazzi at King’s College London recently provides compelling evidence that such an analysis may provide robust pre-treatment predictive value of response to MSC therapy(Galleu et al., 2017). Indeed, in patients with GvHD, it was found that if recipient-derived CD8+ T-cells and CD56+ NK cells were able to induce perforin and granzyme-dependent apoptosis of MSCs in vitro, that it was predictive of at least a partial clinical response or better following transfusion of MSCs. These data suggest that at least in GVHD, subjects poised to be responsive to MSC therapy display an enhanced innate cytotoxic reactivity to allogeneic MSCs. Whether such an assay system would be predictive of MSC therapeutic utility for other inflammatory or tissue injury syndromes remains to be determined.
Headwinds to USA and EU marketing approval and sustainable deployment of MSC therapies
Clinical trials demonstrating an impact on clinical outcomes are required to eventually secure FDA marketing approval and Biologic License application (BLA) for reimbursement and sustainable large scale clinical deployment of MSCs. Whereas any accredited clinical center can readily adopt practice-of-medicine “minimal” cell therapy technologies without FDA or EMA licensing requirements, the same is not true for adoption of more-than-minimally-manipulated cell therapies such as culture adapted MSCs. This regulation of cells as drugs favors a development model driven by industry where mass produced universal products amenable to batch manufacture with economies of scale is compatible with a sustainable business model. In contrast, there are many cell technologies that are likely to have substantial impact on patient outcomes, whose development will be overlooked by industry due to high cost-of-goods, small market size, or highly specialized handling – such as personalized MSC therapies – where a margin-driven business model is less apparent. Further headwinds to deployment of industrial MSC therapies include the $US170,000 price point of MSC platforms for GvHD in Japan and how this informs likely pricing of analogous cell pharmaceuticals in USA for pediatric GvHD. Cell drug affordability for the marginally or underinsured American patients will be a tremendous burden upon the distributive justice notion embraced by many not-for-profit academic healthcare institutions and hospital systems in the USA and Europe. Alternate means of development and deployment of promising yet marginalized cell technologies are required to meet the public’s expectation of access to effective cell therapy. The remedy may well reside within Academic Health Centers affiliated with research-intensive universities, which serve as the engines for discovery and development of cell technologies in a patient beneficent manner as espoused by the 21st Century Cures Act (H.R.34 – 21st Century Cures Act, 114th Congress, 2016).
In an effort to harness the technological benefits of this biopharmaceutical space, the Japanese FDA has put simplified regulatory requirements in place to expedite marketing approval of cell technologies and therapies developed in Japan(Hara et al., 2014). In essence, cell-based pharmaceuticals are granted conditional marketing approval with evidence of safety and presumption of efficacy, with a seven year window to demonstrate scientifically sound efficacy. Early marketing approval allows for product pricing and reimbursement that is permissive for development. In parallel, the European Medicines Agency has a “hospital exemption” clause that allows for not-for-profit public institutions to make cellular therapies available whose manufacturing costs – the lead headwind to development by academic health centers – are borne by the public health payer system. In the USA, there are 3 recent programs for accelerated approval at the end of a RDBPC Phase 2b trial that include (a) Fast Track, (b) Breakthrough Designation, and more recently (c) the 21st Century Cures Act, under which a drug is eligible for regenerative medicine advanced therapy (RMAT) designation. An argument can be made that the creation of a novel regulatory path designed to allow not-for-profit academic health centers to secure early marketing approval (akin to the Japanese regulatory model) or at least ability to secure cost recovery of cell drug manufacture costs (akin to European Hospital exemption) would serve the public good. Indeed, recent guidance from the FDA suggests such a path where multiple manufacturers, which may be individual physicians or groups of physicians, enter into a cooperative development agreement(Marks and Gottlieb, 2018). These manufacturers then produce the product at different sites according to the same protocol, which includes appropriate quality-control procedures to help ensure consistency between different lots produced at different sites. Patients are enrolled at each of the sites that are manufacturing the product in a multicenter clinical trial protocol. Once the data from the multicenter trial are analyzed to evaluate the safety and efficacy of the product, the individual physicians or groups of physicians submit a biologics licensing application that includes the manufacturing protocol used, the clinical data obtained at the individual site, and the results of the multicenter clinical trial showing safety and efficacy. This ultimately results in the issuance of a site-specific biologics license for the product made by each physician or group of physicians. These types of activities would complement the traditional industrial development and deployment of scalable MSC technologies.
Conclusion
There well may be very meaningful differences in MSC cell preparation, fitness, and functionality when comparing MSC tissue source, culture methods, and expansion levels. However, non-manufacturing variables, such as handling at point-of-care including thawing, route of delivery, and dosing, may well override any MSC functionality differences. The recent success of using MSCs for treating fistular Crohn’s disease provides a bridgehead from which technical embellishments can be implemented to increase clinical utility. Pre-clinical animal data supports the notion that the use of augmented MSCs prior to infusion either by use of pharmaceutical or cytokine pre-activation(Guess et al., 2017), genetic engineering, or reprogramming enhances their pharmaceutical potency. The recognition of MSC efferocytosis and its exploitation for immune modulation may provide novel translational strategies as well. Therein lies the path for MSC v2.0, where an understanding of potency and failures informs engineered solutions in cell manufacturing, banking, and point-of-care deployment, and importantly rational selection of subjects based on clinical and biological parameters permissive to clinical effectiveness.
Supplementary Material
Acknowledgments
JG is the current and LS the former Chair of the International Society of Cell Therapy (ISCT) MSC Committee. This work was supported in part by NIH/NIDDK R01DK109508 to JG.
Footnotes
Declaration of interests
The authors declare no competing interests.
Web Resources
- Reicin C (2012). Westlaw Journal. Published online October 2012. https://www.torys.com/Publications/Publications/AR2012-32.pdf
- Minutes of Proceedings of House of Commons (Canada) Standing Committee on Health (2016). Published online May 2016. http://www.ourcommons.ca/DocumentViewer/en/42-1/HESA/meeting-12/evidence
- JCR Pharmaceuticals Co., Ltd. Annual Report (2017). Published online March 2017. http://www.jcrpharm.co.jp/wp2/wp-content/uploads/2017/10/JCR-AR2017_English_FINAL1.pdf
- H.R.34 – 21st Century Cures Act, 114th Congress (2016). Published online December 2016. https://www.congress.gov/bill/114th-congress/house-bill/34/text
References
- Abumaree MH, Stone PR & Chamley LW 2006. The effects of apoptotic, deported human placental trophoblast on macrophages: possible consequences for pregnancy. J Reprod Immunol, 72, 33–45. [DOI] [PubMed] [Google Scholar]
- Ankrum JA, Ong JF & Karp JM 2014. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol, 32, 252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askelund KJ & Chamley LW 2011. Trophoblast deportation part I: review of the evidence demonstrating trophoblast shedding and deportation during human pregnancy. Placenta, 32, 716–23. [DOI] [PubMed] [Google Scholar]
- Bach F & Hirschhorn K 1964. Lymphocyte Interaction: A Potential Histocompatibility Test in Vitro. Science, 143, 813–4. [DOI] [PubMed] [Google Scholar]
- Ball LM, Bernardo ME, Roelofs H, Van Tol MJ, Contoli B, Zwaginga JJ, Avanzini MA, Conforti A, Bertaina A, Giorgiani G, Jol-Van Der Zijde CM, Zecca M, Le Blanc K, Frassoni F, Egeler RM, Fibbe WE, Lankester AC & Locatelli F 2013. Multiple infusions of mesenchymal stromal cells induce sustained remission in children with steroid-refractory, grade III-IV acute graft-versus-host disease. Br J Haematol, 163, 501–9. [DOI] [PubMed] [Google Scholar]
- Bartunek J, Behfar A, Dolatabadi D, Vanderheyden M, Ostojic M, Dens J, El Nakadi B, Banovic M, Beleslin B, Vrolix M, Legrand V, Vrints C, Vanoverschelde JL, Crespo-Diaz R, Homsy C, Tendera M, Waldman S, Wijns W & Terzic A 2013. Cardiopoietic stem cell therapy in heart failure: the C-CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage-specified biologics. J Am Coll Cardiol, 61, 2329–38. [DOI] [PubMed] [Google Scholar]
- Bartunek J, Davison B, Sherman W, Povsic T, Henry TD, Gersh B, Metra M, Filippatos G, Hajjar R, Behfar A, Homsy C, Cotter G, Wijns W, Tendera M & Terzic A 2016. Congestive Heart Failure Cardiopoietic Regenerative Therapy (CHART-1) trial design. Eur J Heart Fail, 18, 160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo ME & Fibbe WE 2013. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell, 13, 392–402. [DOI] [PubMed] [Google Scholar]
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ & Wang CY 2013. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med, 19, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AI 1991. Mesenchymal stem cells. J Orthop Res, 9, 641–50. [DOI] [PubMed] [Google Scholar]
- Carty F, Mahon BP & English K 2017. The influence of macrophages on mesenchymal stromal cell therapy: passive or aggressive agents? Clin Exp Immunol, 188, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai R, Rajan D, Qayed M, Arafat D, Garcia M, Liu Y, Kugathasan S, Anderson LJ, Gibson G & Galipeau J 2018. Potency Analysis of Mesenchymal Stromal Cells Using a Combinatorial Assay Matrix Approach. Cell Rep, 22, 2504–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter TM, Moore MA & Sheridan AP 1977. Maintenance of hemopoietic stem cells and production of differentiated progeny in allogeneic and semiallogeneic bone marrow chimeras in vitro. J Exp Med, 145, 1612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Koster KM & Murphy PS 2017. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J Immunol, 198, 1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fibbe WE, Dazzi F & Leblanc K 2013. MSCs: science and trials. Nat Med, 19, 812–3. [DOI] [PubMed] [Google Scholar]
- Francois M, Romieu-Mourez R, Li M & Galipeau J 2012. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther, 20, 187–95. [DOI] [PubMed] [Google Scholar]
- Franks LM, Chesterman FC & Rowlatt C 1970. The structure of tumours derived from mouse cells after “spontaneous” transformation in vitro. Br J Cancer, 24, 843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF & Keiliss-Borok IV 1974. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation, 17, 331–40. [DOI] [PubMed] [Google Scholar]
- Fung M, Yuan Y, Atkins H, Shi Q & Bubela T 2017. Responsible Translation of Stem Cell Research: An Assessment of Clinical Trial Registration and Publications. Stem Cell Reports, 8, 1190–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau J 2013. The mesenchymal stromal cells dilemma--does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy, 15, 2–8. [DOI] [PubMed] [Google Scholar]
- Galipeau J, Krampera M, Barrett J, Dazzi F, Deans RJ, Debruijn J, Dominici M, Fibbe WE, Gee AP, Gimble JM, Hematti P, Koh MB, Leblanc K, Martin I, Mcniece IK, Mendicino M, Oh S, Ortiz L, Phinney DG, Planat V, Shi Y, Stroncek DF, Viswanathan S, Weiss DJ & Sensebe L 2016. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy, 18, 151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galleu A, Riffo-Vasquez Y, Trento C, Lomas C, Dolcetti L, Cheung TS, Von Bonin M, Barbieri L, Halai K, Ward S, Weng L, Chakraverty R, Lombardi G, Watt FM, Orchard K, Marks DI, Apperley J, Bornhauser M, Walczak H, Bennett C & Dazzi F 2017. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci Transl Med, 9. [DOI] [PubMed] [Google Scholar]
- Guess AJ, Daneault B, Wang R, Bradbury H, La Perle KMD, Fitch J, Hedrick SL, Hamelberg E, Astbury C, White P, Overolt K, Rangarajan H, Abu-Arja R, Devine SM, Otsuru S, Dominici M, O’donnell L & Horwitz EM 2017. Safety Profile of Good Manufacturing Practice Manufactured Interferon gamma-Primed Mesenchymal Stem/Stromal Cells for Clinical Trials. Stem Cells Transl Med, 6, 1868–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara A, Sato D & Sahara Y 2014. New Governmental Regulatory System for Stem Cell–Based Therapies in Japan. Therapeutic Innovation & Regulatory Science, 48, 681–688. [DOI] [PubMed] [Google Scholar]
- Herreros MD, Garcia-Arranz M, Guadalajara H, De-La-Quintana P, Garcia-Olmo D & Group FC 2012. Autologous expanded adipose-derived stem cells for the treatment of complex cryptoglandular perianal fistulas: a phase III randomized clinical trial (FATT 1: fistula Advanced Therapy Trial 1) and long-term evaluation. Dis Colon Rectum, 55, 762–72. [DOI] [PubMed] [Google Scholar]
- Hess DC, Wechsler LR, Clark WM, Savitz SI, Ford GA, Chiu D, Yavagal DR, Uchino K, Liebeskind DS, Auchus AP, Sen S, Sila CA, Vest JD & Mays RW 2017. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol, 16, 360–368. [DOI] [PubMed] [Google Scholar]
- Kfoury Y & Scadden DT 2015. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell, 16, 239–53. [DOI] [PubMed] [Google Scholar]
- Koc ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI & Lazarus HM 2000. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol, 18, 307–16. [DOI] [PubMed] [Google Scholar]
- Kurtzberg J, Prasad V, Grimley M, Horn B, Carpenter P, Jacobsohn D & Prockop S 2010. Allogeneic human mesenchymal stem cell therapy (Prochymal®) as a rescue agent for severe treatment resistant GVHD in pediatric patients. Biology of Blood and Marrow Transplantation, 16, S169. [DOI] [PubMed] [Google Scholar]
- Lalu MM, Mcintyre L, Pugliese C, Fergusson D, Winston BW, Marshall JC, Granton J, Stewart DJ & Canadian Critical Care Trials, G. 2012. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS One, 7, e47559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus HM, Haynesworth SE, Gerson SL, Rosenthal NS & Caplan AI 1995. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant, 16, 557–64. [PubMed] [Google Scholar]
- Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M, Dini G, Egeler RM, Bacigalupo A, Fibbe W, Ringden O, Developmental Committee of the European Group For, B. & Marrow T 2008. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet, 371, 1579–86. [DOI] [PubMed] [Google Scholar]
- Luk F, De Witte SF, Korevaar SS, Roemeling-Van Rhijn M, Franquesa M, Strini T, Van Den Engel S, Gargesha M, Roy D, Dor FJ, Horwitz EM, De Bruin RW, Betjes MG, Baan CC & Hoogduijn MJ 2016. Inactivated Mesenchymal Stem Cells Maintain Immunomodulatory Capacity. Stem Cells Dev, 25, 1342–54. [DOI] [PubMed] [Google Scholar]
- Marks P & Gottlieb S 2018. Balancing Safety and Innovation for Cell-Based Regenerative Medicine. N Engl J Med, 378, 954–959. [DOI] [PubMed] [Google Scholar]
- Martin PJ, Uberti JP, Soiffer RJ, Klingemann H, Waller EK, Daly AS, Herrmann RP & Kebriaei P Prochymal Improves Response Rates In Patients With Steroid-Refractory Acute Graft Versus Host Disease (SR-GVHD) Involving The Liver And Gut: Results Of A Randomized, Placebo-Controlled, Multicenter Phase III Trial In GVHD. Biology of Blood and Marrow Transplantation, 16, S169–S170. [Google Scholar]
- Mendicino M, Bailey AM, Wonnacott K, Puri RK & Bauer SR 2014. MSC-based product characterization for clinical trials: an FDA perspective. Cell Stem Cell, 14, 141–5. [DOI] [PubMed] [Google Scholar]
- Moll G, Geissler S, Catar R, Ignatowicz L, Hoogduijn MJ, Strunk D, Bieback K & Ringden O 2016. Cryopreserved or Fresh Mesenchymal Stromal Cells: Only a Matter of Taste or Key to Unleash the Full Clinical Potential of MSC Therapy? Adv Exp Med Biol, 951, 77–98. [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EG, Willemze R & Fibbe WE 2006. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood, 108, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA & Mezey E 2009. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med, 15, 42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panes J, Garcia-Olmo D, Van Assche G, Colombel JF, Reinisch W, Baumgart DC, Dignass A, Nachury M, Ferrante M, Kazemi-Shirazi L, Grimaud JC, De La Portilla F, Goldin E, Richard MP, Diez MC, Tagarro I, Leselbaum A, Danese S & Collaborators, A. C. S. G. 2018. Long-term Efficacy and Safety of Stem Cell Therapy (Cx601) for Complex Perianal Fistulas in Patients With Crohn’s Disease. Gastroenterology, 154, 1334–1342 e4. [DOI] [PubMed] [Google Scholar]
- Panes J, Garcia-Olmo D, Van Assche G, Colombel JF, Reinisch W, Baumgart DC, Dignass A, Nachury M, Ferrante M, Kazemi-Shirazi L, Grimaud JC, De La Portilla F, Goldin E, Richard MP, Leselbaum A, Danese S & Collaborators, A. C. S. G. 2016. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: a phase 3 randomised, double-blind controlled trial. Lancet, 388, 1281–90. [DOI] [PubMed] [Google Scholar]
- Phinney DG, Galipeau J, Krampera M, Martin I, Shi Y & Sensebe L 2013. MSCs: science and trials. Nat Med, 19, 812. [DOI] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG & Ravichandran KS 2014. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol, 14, 166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders ME, Dreyer GJ, Bank JR, Roelofs H, Heidt S, Roelen DL, Zandvliet ML, Huurman VA, Fibbe WE, Van Kooten C, Claas FH, Rabelink TJ & De Fijter JW 2015. Safety of allogeneic bone marrow derived mesenchymal stromal cell therapy in renal transplant recipients: the neptune study. J Transl Med, 13, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensebe L & Fleury-Cappellesso S 2013. Biodistribution of mesenchymal stem/stromal cells in a preclinical setting. Stem Cells Int, 2013, 678063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensebe L, Tarte K, Galipeau J, Krampera M, Martin I, Phinney DG, Shi Y & Therapy M. S. C. C. O. T. I. S. F. C. 2012. Limited acquisition of chromosomal aberrations in human adult mesenchymal stromal cells. Cell Stem Cell, 10, 9–10; [DOI] [PubMed] [Google Scholar]
- Shanks N, Greek R & Greek J 2009. Are animal models predictive for humans? Philos Ethics Humanit Med, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipp D 2015. Conditional approval: Japan lowers the bar for regenerative medicine products. Cell Stem Cell, 16, 353–6. [DOI] [PubMed] [Google Scholar]
- Teerlink JR, Metra M, Filippatos GS, Davison BA, Bartunek J, Terzic A, Gersh BJ, Povsic TJ, Henry TD, Alexandre B, Homsy C, Edwards C, Seron A, Wijns W, Cotter G & Investigators C 2017. Benefit of cardiopoietic mesenchymal stem cell therapy on left ventricular remodelling: results from the Congestive Heart Failure Cardiopoietic Regenerative Therapy (CHART-1) study. Eur J Heart Fail, 19, 1520–1529. [DOI] [PubMed] [Google Scholar]
- Turner L & Knoepfler P 2016. Selling Stem Cells in the USA: Assessing the Direct-to-Consumer Industry. Cell Stem Cell, 19, 154–157. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen X, Cao W & Shi Y 2014. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol, 15, 1009–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.