

Abstract
The standard proteinogenic amino acids grant access to a myriad of chemistries that harmonize to create life. Outside of these twenty canonical protein building blocks are countless noncanonical amino acids (ncAAs), either found in nature or created by man. Interest in ncAAs has grown as research has unveiled their importance as precursors to natural products and pharmaceuticals, biological probes, and more. Despite their broad applications, synthesis of ncAAs remains a challenge, as poor stereoselectivity and low functional-group compatibility stymie effective preparative routes. The use of enzymes has emerged as a versatile approach to prepare ncAAs, and nature’s enzymes can be engineered to synthesize ncAAs more efficiently and expand the amino acid alphabet. In this tutorial review, we briefly outline different enzyme engineering strategies and then discuss examples where engineering has generated new ‘ncAA synthases’ for efficient, environmentally benign production of a wide and growing collection of valuable ncAAs.
Key Learning Points:
Important applications of ncAAs in medicine, chemistry, and biology
Advantages and disadvantages of current approaches for synthesizing ncAAs
Strategies for enzyme engineering
Cases where ncAA synthases have been created or optimized with enzyme engineering
Opportunities for further exploration and progress in biocatalytic ncAA synthesis
1. Introduction
The twenty canonical L-α-amino acids (Scheme 1a) that serve as the primary basis of protein structure and function comprise only a small fraction of biologically and technologically important amino acids. Noncanonical amino acids (ncAAs), which are not naturally incorporated into proteins during translation, contain unusual side chains, D stereochemistry, or atypical backbone connectivity (Scheme 1b). These features in turn impart distinct chemical and biological properties, such as greater stability in vivo. These properties have elicited considerable interest, and ncAAs are used as therapeutics1 and synthetic intermediates,2 and are even encoded directly into proteins to confer useful new features.3
Scheme 1.

(a) Structure of L-a-amino acids and (b) examples of noncanonical amino acids (ncAAs).
Noncanonical amino acids are challenging to synthesize because they often contain a stereocenter at the α-carbon, which must be set in a precise configuration. In addition, the amine and carboxylic acid groups are reactive and often have to be protected. These problems are compounded when ncAAs have complex side chains that contain additional stereocenters or reactive functional groups. Nature circumvents such challenges by using enzymes, which bind and position substrates to accelerate a specific reaction, making enantiopure amino acids in aqueous media without the need for protecting groups. However, many enzymes that produce ncAAs in nature are not suitable for preparative ncAA synthesis due to low activity, poor endogenous expression and stability, need for allosteric activation, or limited substrate scope. Furthermore, many ncAAs are naturally synthesized via complicated multi-enzyme cascades, which may be difficult to identify and use for synthesis at scale. New strategies are needed for ncAA synthesis, and engineering new or improved enzymes offers some promising leads.
A number of enzymes have activities that could be used to make ncAAs, and protein engineering has been an indispensable tool for expanding this latent potential. Researchers have been able to requisition existing enzymes and engineer them to create high-yielding biocatalytic platforms that generate enantiomerically pure ncAAs. We will refer to these enzymes as ‘ncAA synthases’, since they are used to couple two molecules without requiring additional energetic input (e.g., ATP). Well-designed mutagenesis and screening strategies facilitate the engineering process, and iterative rounds of mutagenesis and screening (directed evolution) can produce greatly improved ncAA synthases; expanded substrate scopes, enhanced stability and heterologous expression, and increased yields are all achievable with the appropriate experimental design. In this tutorial review, we will illustrate principles of protein engineering and evolution in the context of ncAA synthases. To focus the scope of this review, we will primarily explore the synthesis and applications of α-amino acids, and direct the readers interested in different configurations to other excellent works4,5. We will begin by introducing the importance of ncAAs in chemistry and medicine and comparing current methods for producing ncAAs. We will then cover principles of protein engineering and evolution before exploring cases where researchers have applied these principles to address fundamental shortcomings in the synthesis of valuable ncAAs. Finally, we will discuss how directed evolution could provide future biocatalyst improvements.
2. Applications of ncAAs in chemistry, medicine, and biology
Even with advances like photo-redox chemistry, metal-catalyzed cross-coupling, and asymmetric catalysis, the bottleneck for drug discovery is chemical synthesis.2 Important pharmaceutical functionalities such as chiral amines, N-heterocycles, and unprotected polar groups are challenging to work with in synthesis, but the incorporation of ncAAs directly into synthetic pipelines can bypass many of these difficulties. For example, the diabetes medication saxagliptin (Scheme 2) contains a chiral amine as well as a challenging β-quaternary center that is important for the drug’s activity.1 Improved methods to synthesize this drug have focused on producing the ncAA L-α−3-hydroxy-1-adamantyl-glycine enzymatically for use as a building block in the synthesis of saxagliptin.6 Other medicines contain alkaloids, pharmaceutically important natural products derived from amino acids. Alkaloids and similar compounds make up many essential medicines such as dopamine (heart failure), codeine and morphine (analgesic), vincristine and irinotecan (cancer), and quinine (antimalarial).7 As biologically active and synthetically useful molecules, it is unsurprising that ncAAs are present in 12% of the 200 top-grossing drugs.8 Incorporating ncAAs and ncAA-derived products into synthetic pipelines allows important pharmaceuticals to be synthesized more easily than ever before. However, few ncAAs are readily available, and improved synthetic and biocatalytic methods are needed to realize their full potential.
Scheme 2.

The ncAA L-α−3-hydroxy-1-adamantyl-glycine is a building block of the diabetes drug saxagliptin.
Protein therapeutics including peptides and antibodies also make use of ncAAs. Therapeutic peptides have been used since the 1920s, when insulin was extracted from animal pancreases for diabetes treatment. Peptide drugs remain important to this day, with more than 60 approved for use.1 However, most natural peptides are not suitable therapeutics because they are present only in low concentrations and are susceptible to proteolysis, limiting bioavailability. Incorporation of ncAAs with the D-configuration, unnatural backbones, or bulky side chains can reduce proteolysis, and modified side chains can tune biological specificity and pharmacokinetics.1 For example, cyclic antimicrobial peptides such as daptomycin disrupt the membranes of infectious microorganisms. Daptomycin incorporates the ncAAs kynurenine, ornithine, and (2S,3R)-methylglutamate, as well as three D-amino acids (Scheme 3).
Scheme 3.

Daptomycin, a cyclic peptide antibiotic, includes the ncAAs kynurenine (pink), ornithine (purple), (2S,3R)-methylglutamate (blue), as well as D-amino acids (green).
Antibodies and antibody-drug conjugates (ADCs) are another class of protein therapeutics that benefit from access to ncAAs. ADCs are versatile therapeutics composed of a chemotoxic agent coupled via an amino acid linker to an antibody that specifically targets a cellular component with limited side effects.9 A common linker is a dipeptide composed of valine and the ncAA citrulline, which is cleaved in the lysosome to release the toxic ‘payload’ (Fig 1). The linker and payload are typically attached to the antibody via non-specific modifications of surface-exposed cysteine and lysine residues. With this non-specific conjugation, the payload may be attached at different positions and in different concentrations, resulting in a heterogeneous drug whose pharmacokinetics, safety, and efficiency are not well-defined. Incorporation of ncAAs into ADCs can provide site-specific, bio-orthogonal attachment points for the linker, affording tunable and reproducible control over the payload concentration.9
Fig 1.

Antibody-drug conjugates (ADCs) target drugs to specific locations through the high-affinity interactions of the antibody to its antigen. In this simple example, the drug is attached to an antibody by site-specific incorporation of the ncAA selenocysteine (which can react with maleimides under different conditions than cysteine) and a cleavable valinecitrulline linker (blue). The drug is released following cleavage of the linker (indicated with a red dashed line).
Amino acid sequence dictates protein tertiary structure and affects protein function, localization, recognition, and post-translational modification. Consequently, incorporation of ncAAs at certain positions within a protein sequence can be used to modulate the physical and chemical properties of that protein. For example, global replacement of methionine with selenomethionine provides heavy atoms for X-ray crystallography,10 while replacement of amino acids with their fluorinated analogues can influence the substrate specificity and stability of enzymes.11 Furthermore, genetic code expansion permits the site-specific incorporation of ncAAs into proteins where promiscuous global replacement might be undesirable or impossible.3,10 For example, ncAAs with side chains such as the environmentally sensitive fluorophore 7-hydroxycoumarin, the metal chelator 2,2’-bipyridine, and the metal-binding fluorophore 8-hydroxyquinoline have unique properties that can be used to probe biomolecular interactions or induce metal-dependent assembly or fluorescence.12,13 Additionally, ncAAs with reactive unsaturated aliphatic, azido, and carbonyl side chains can be used as site-specific bio-orthogonal handles for chemical modification.14 The ability to selectively manipulate proteins through the incorporation of ncAAs promotes the understanding and engineering of protein stability, activity, and mechanism.
3. Methods for ncAA production
Although ncAAs are valuable chemical and biological tools, their applications are limited by inefficient routes of production. The most popular approaches, such as extraction from protein hydrolysate, fermentation, chemical synthesis, and biocatalysis fall short in terms of cost, yield, or scope.15–17 Extracting amino acids from hydrolyzed proteins is excellent for large-scale production, especially when sourced from inexpensive industrial byproducts such as hair, meat, or plants. However, this is only suitable for naturally occurring ncAAs with unique physicochemical properties that enable purification, such as reactive side chains or extreme pH stability.
Amino acids are also produced on a large scale by microorganisms that convert sugars and other feedstocks into the desired products.15 Bacterial strains of Escherichia coli or Corynebacterium glutamicum have been extensively engineered for efficient metabolism and mitigated stress response to enhance yields.18 Production by fermentation, however, requires an organism with the capacity to synthesize the ncAA. This is a major complication, since biosynthetic pathways for many ncAAs either give poor yields or are simply unknown.
Chemical synthesis can access numerous ncAAs by employing intermediates such as serine-derived lactones, hydantoins, or aziridines (Scheme 4, blue).19,20 A significant benefit of chemical synthesis approaches is their broad applicability, allowing a variety of ncAAs to be produced from a single synthetic pipeline. Limitations are also apparent: chemical synthesis can be labor-intensive, utilize hazardous reagents and produce significant waste products, or generate racemic products that require further purification.
Scheme 4.

Selected representative methods for synthesizing ncAAs using chemical synthesis (blue) or biocatalysis (red).
An increasingly useful approach to preparing ncAAs is biocatalysis, which can either replace or supplement chemical synthesis and fermentation with enzymes (Scheme 4, red).4,16 Enzyme-catalyzed reactions benefit from mild reaction conditions and a broad range of biocatalysts that can be used in derivatization or bond-forming reactions. For example, aminotransferases are used to form chiral amines by transferring the amino group of one amino acid to a prochiral α-keto acid while setting the stereochemistry at the α-carbon. The process for manufacturing Januvia, a diabetes drug, incorporates an engineered aminotransferase that replaces two steps of the chemical synthesis route.21 Other enzymes such as lyases capitalize on accessible, non-hazardous starting materials to synthesize diverse optically pure ncAAs. For example, ammonia lyases can catalyze the asymmetric amination of inexpensive, prochiral substrates such as fumarate or cinnamic acid derivatives to make optically pure ncAAs (discussed in Section 5.1).22,23 Other enzymes, such as tyrosine phenol lyase (TPL)24 and tryptophan synthase (TrpS),25,26 can generate more complexity from even simpler substrates by coupling a nucleophilic side chain to an amino acid backbone (discussed in Section 5.2).
Although biocatalysts perform impressive chemical transformations, applications are limited by reaction and substrate scope as well as enzyme stability and compatibility with process conditions. Improved biocatalysts are needed for simple, green, and cost-effective access to a variety of ncAAs. Protein engineering can improve catalyst performance, expand the scope of substrates the enzymes can accept, and even generate new activities. Methods of directed enzyme evolution are especially powerful for rapid engineering. In the following sections of this review, we will focus on opportunities for engineering ncAA synthases that generate complex products from simple (e.g., prochiral or inexpensive) starting materials. We believe that these biocatalysts offer special advantages that will significantly improve the synthesis of ncAAs.
4. Enzyme engineering strategies
This review will illustrate several successful strategies for engineering improved ncAA synthases. Because the relationship between enzyme sequence and function is still largely unknown, we will focus primarily on methods in which collections of mutant enzymes—called ‘libraries’—are generated and screened for desired properties. This process provides the basis for discovering beneficial mutations. A single round of mutation and screening is sufficient to engineer an improved enzyme as long as the library contains the improved variant and screening can accurately identify it. Indeed, several examples described in Section 5 describe enzymes that have shown substantial improvements in a single round of mutation and screening. However, accumulating mutations in iterations of mutagenesis and screening, an approach known as directed evolution, can produce even better enzymes. The mutagenesis and screening strategies are the foundation of enzyme engineering, and success in an engineering project rests on the appropriate selection of complementary methods.
The first step is to select an enzyme as the starting point, known as the ‘parent’ enzyme. Even poor baseline activity for the reaction of interest can be the foundation of a good evolved enzyme, provided improvements in activity can be measured reliably, as discussed below. If the starting activity is too low, it can be worthwhile to explore homologous enzymes as parents, as they can differ drastically in substrate scope, activity, and stability.27 Enzymes with high stability are often preferred parents, especially for directed evolution, as they can support the accumulation of activating but often destabilizing mutations.28
Starting from the parent gene, diversity is introduced to produce a collection of mutant genes, which are used to transform a suitable host organism. Standard microbiology techniques are typically used to array individual clones—containing individual mutant genes—into physically separated compartments, such as the wells of microtiter plates, where gene expression and protein production take place. The resulting library of enzyme variants is then assayed for the desired activity with an appropriate screen. (It should be noted that some screening techniques, such as those that use cell sorting instruments, as well as most types of selections, do not require that the cells containing the protein variants be physically separated prior to screening or selection, but instead rely on the technique itself to accomplish separation. These methods, however, provide enrichment rather than full separation of individual clones.) The sensitivity and reproducibility of the screen determines the improvement that can be measured reliably and therefore discovered in these experiments, while the mutagenesis method determines the sequence diversity that is searched. Mutagenesis and screening methods should be evaluated together, as the combination is critical to the success of any enzyme engineering project. The goal is to generate libraries that are sufficiently rich in improved enzyme variants that the screening method can find them efficiently.
The first rule to remember when developing a protein engineering screen is: you get what you screen for. In other words, the screen needs to report faithfully on the desired property or set of properties. Developing a very high throughput screen for synthesis of a ncAA can be challenging, especially when the starting yield is low, and/or the product amino acid does not have a chromophore or fluorophore that make it readily visible in a complex medium with high concentrations of substrates and other species. Indirect assays, such as surrogate screens (see Section 5.1.2) or selections (a method of associating the fitness of an organism with the activity of an enzyme) can be used, but this can and often does result in variants that perform well in the surrogate screen or selection but not in the desired task; there are of course many notable successes. Additionally, if the assay requires purification steps or additional reactions, the throughput may be limited to a few hundred, rather than thousands, of samples per day. The sample throughput capacity and cost determine whether one has to take a more ‘designed’ approach to making variants or whether one can use more agnostic mutagenesis methods such as random mutagenesis of the whole sequence. Because new mutagenesis and library construction protocols appear regularly, we will not go into any detail on specific methodologies. Rather, we will discuss four general classes of library construction and the screening techniques commonly associated with them. Readers may be interested in recent reviews from the Hilvert29 and Liu30 groups for further details on selections and library construction methods.
The mutagenesis approach requiring the highest level of design is one in which the protein engineer has identified one or more specific residues that influence enzyme behavior and then decides which mutation to make at those residues using site-directed mutagenesis methods. Making informed decisions, however, usually requires detailed structural information, and success depends on having an accurate assessment of how a given mutation will affect biocatalyst structure and function. An example of site-directed mutagenesis is targeted modification of the active site to improve substrate access. Bulky residues (e.g., leucine or phenylalanine) in the active site may be mutated to smaller ones (e.g., alanine) to improve binding of larger substrates. Such ‘designed’ mutations can provide a starting point for further improvement by directed evolution or may even be sufficient to generate an efficient biocatalyst. Since this approach explores a very limited sequence space, success depends entirely on the validity of the hypothesis. It is important to keep in mind that failed attempts are rarely published.
A somewhat more agnostic approach is to use site-saturation mutagenesis to sample most or all possible mutations at a given residue or set of residues. Beneficial mutations found by screening saturation mutagenesis libraries can be accumulated in sequential rounds (see ref. 31 for further reading), by recombination (see below), or by screening combinatorially saturated mutant libraries. If positions are sampled one or two at a time, these libraries are small enough to be analyzed by techniques such as high-performance liquid chromatography (HPLC), gas chromatography (GC), mass spectrometry (MS), and even thin layer chromatograph (TLC) or nuclear magnetic resonance (NMR). Of course, success with site-saturation mutagenesis depends on whether the engineer has chosen the right residue(s) to target. For many enzymes and properties this is not easy, and libraries at many positions may have to be screened in order to find beneficial mutations.
In directed evolution, random mutagenesis is often used to introduce mutations at a low frequency throughout the gene of interest. A great advantage of random mutagenesis is that no prior knowledge of the enzyme structure or mechanism is required: the experiment tells you what is important. For example, random mutagenesis and screening can identify mutations that mimic allosteric activation, which may be distributed throughout the protein.32 Random mutagenesis and screening often finds activating mutations that are distant from the active site or substrate binding pocket, at residues that would likely not be chosen by any ‘design’ methods. (Examples of this can be seen in Section 5.2.2.) However, there are costs: many randomly-mutated variants are parent-like, or unaltered in the property screened, and therefore significant screening effort is required to identify the rare beneficial mutations. Often this means screening hundreds to thousands of samples. Furthermore, random mutagenesis by the most convenient method, error-prone PCR, only accesses a fraction of all the possible single amino acid substitutions (roughly 6 out of 19) as it traverses the codon table via single nucleotide mutations; at the low mutation rates used for directed evolution, there is an underwhelming probability of making two mutations within the same codon.
Recombination, performed either in vitro or in vivo, parallels the natural events of homologous recombination, such as the shuffling of chromosomal DNA during meiosis or diversity generation during V(D)J antibody recombination.33,34 Rather than generating new mutations, recombination navigates the evolutionary landscape by leveraging genetic diversity that already exists. One can recombine homologous protein sequences to make chimeric proteins or recombine previously identified mutations to create new combinations in a single sequence. The latter is useful when several beneficial mutations are found in one generation of random mutagenesis. Stemmer’s ‘DNA shuffling’ method35 and Zhao’s ‘staggered extension’ method36 perform random mutagenesis and recombination in one operation. Sequence information is all that is necessary for most recombination methods.
In choosing a mutagenesis strategy, the goal is to find and accumulate beneficial mutations with the least combined mutagenesis and screening effort. Site-directed and site-saturation mutagenesis are highly focused approaches and produce small libraries that can be screened with relatively low-throughput techniques. The downside is that the targeted positions are limited and may not yield beneficial mutations. Random mutagenesis samples a far greater sequence space, but the frequency of beneficial mutations can be low, and a higher-throughput screen is generally needed to find improvements. As any library is likely to contain variants exhibiting a range of performance, it is crucial to establish a robust screening system that discriminates improved enzyme variants from the parent.37 Improved variants can always be used for subsequent rounds of engineering in a directed evolution approach. Making incremental improvements is an effective way to navigate the enzyme’s fitness landscape to create an exceptional enzyme for a given task; this evolutionary search process can even create entire lineages of enzymes that excel at different tasks (see Fig 9 in Section 5.2.2).
Fig 9.

Evolutionary lineage of TrpB-based ncAA synthases. Starting with the isolated TrpB enzyme subunit from Pyrococcus furiosus (PfTrpB), directed evolution has been used to improve standalone Trp production (red),26 as well as activity with L-threonine (orange),46 substituted indoles (green and blue),49 and bulkier β-branched L-serine analogues (pink).48 It has also been engineered to produce Trp analogues such as 4-cyanoTrp at lower temperatures (purple).50 Intermediate variants in the evolution are shown as nodes, with representative variants shown with their structural models. Nodes are connected by lines that represent the mutagenesis approach. Transfer of activating mutations to a homologous TrpB from Thermotoga maritima (TmTrpB) resulted in variants that exhibited different substrate preferences than PfTrpB variants; where PfTrpB was adept at 6- and 7-substituted Trp production, TmTrpB was adept at 4- and 5-substituted Trp production. (Nomenclature notes: Pf2B9‡ is Pf2B9 with three mutations: L161V, M139L, L212P; Tm2F3-I184F is shortened to Tm2F3* for consistency.)
Although not discussed here, computational models are also showing promise for enzyme engineering, with small in silico libraries yielding high frequencies of activating mutations that reduce screening efforts. This typically requires identifying a parent enzyme with an appropriate and well-understood mechanism and then computationally redesigning that enzyme based on the desired mechanism. When used correctly, this approach can produce a slightly active enzyme for further evolution or can even create highly proficient biocatalysts, as demonstrated in a recent report from the Janssen and Wu groups on the development of β-ncAA synthases.5 Importantly, it is now possible to order entire libraries of mutant genes made to individual specifications from various DNA suppliers. While expensive, synthetic libraries of individual genes reduce labor associated with library construction and validation and also reduce screening requirements compared to libraries made by various randomization methods.
5. Engineering improved ncAA synthases
In this section, we discuss examples where enzyme engineering principles described above have been applied to improve biocatalytic ncAA synthesis. These examples highlight key aspects of engineering proteins through mutation and screening to quickly achieve functional goals and cover different parent enzymes, mutation strategies, screening approaches, and reaction conditions.
5.1. Asymmetric carbon-nitrogen bond formation by ammonia lyases
Ammonia lyases catalyze reversible carbon-nitrogen bond cleavage to produce a trans-α,β-unsaturated carboxylic acid and ammonia (Scheme 5).38 Of interest in this review are ammonia lyases that competently catalyze the reverse reaction, resulting in the asymmetric addition of ammonia to synthesize enantiopure α-amino acids. This occurs with readily available, prochiral substrates such as fumarate or cinnamic acid analogues. Ammonia is activated within the enzyme, either by internal residues or by a special electrophilic group, which facilitates nucleophilic attack at the electrophilic alkene followed by proton donation to the β-carbon by a catalytic base (Scheme 5). Ammonia lyases act on a diverse set of substrates, such as aspartate (aspartate ammonia lyases, or DALs), β-methylaspartate (MALs), and the aromatic amino acids (PALs, TALs, and HALs for phenylalanine, tyrosine, and histidine, respectively). This class of enzymes has the potential to access diverse types of functional ncAA products, from β-substituted aspartate analogues to aromatic ncAAs (also called arylalanines).
Scheme 5.

Generalized mechanism of asymmetric amination by ammonia lyases.
5.1.1. MAL: Accessing bulky β-substituted aspartate analogues.
Of all the ammonia lyases, those that act on aspartate and its analogues are among the most specific.38 The MAL catalytic cycle requires the presence of a β-carboxylate (the aspartate side chain, labeled blue in Scheme 6), which renders the α-carbon electrophilic and provides necessary acidity at the β-carbon (labeled red in Scheme 6). In MALs, the species with the electrophilic α-carbon (Scheme 6, rightmost resonance species) is further stabilized by a bound magnesium ion that coordinates the negatively charged β-carboxylate. Because of this, substitutions are better tolerated at the α-carboxylate group by MALs, enabling production of certain β-ncAAs rather than the corresponding α-ncAA that lacks the aspartate side chain (Scheme 7). This approach has recently been used with great success to computationally engineer an aspartate ammonia lyase for β-amino acid production.5 Therefore, if used to synthesize α-ncAAs, MALs are best suited to synthesize aspartate analogues as they contain the necessary β-carboxylate.
Scheme 6.

Important species and features in the reaction of β-methylaspartate ammonia lyase (MAL).
Scheme 7.

MAL adds ammonia at the β-carbon of a carboxylate.
Wild-type MALs can produce β-methylaspartate—their eponymous natural substrate—from the prochiral substrate 2-methylfumarate (Scheme 6, R = methyl). MALs show moderate activity on substrates with slightly larger β-substituents such as ethyl, propyl, and ethoxy groups.23 Unfortunately, some of the more valuable aspartate analogues, such as the excitatory neural ligand transporter inhibitor threo-3-benzyloxyaspartate, fail to show even trace activity with MAL, which is attributed to the large size of the β-substituent.
To improve the biocatalytic synthesis of bulkier β-branched aspartate analogues, Raj and colleagues engineered MAL from the bacterium Clostridium tetanomorphum (CtMAL).23 A crystal structure of β-methylaspartate in complex with CtMAL suggested that three residues, F170, Y356, and L384, were likely to sterically occlude the binding of substrates with larger β-substituents (Fig 3). The authors prepared three separate site-saturation mutagenesis libraries targeting these residues, hypothesizing that the active site could be modified to enable access by bulkier substrates. After expressing the corresponding CtMAL variants in E. coli, they screened for amination of the model bulky substrate 2-hexylfumarate, toward which the wild-type enzyme showed no detectible activity. The authors used an absorbance assay to screen for 2-hexylfumarate amination by monitoring the change in 270-nm absorbance in the reaction mixture over time. Due to the conjugation between the terminal carboxylate groups in fumarate analogues, the substrate absorbs strongly at 270 nm (shown in Scheme 6). Following substrate amination to produce an α-amino acid, this conjugation is broken and the absorbance at 270 nm is significantly decreased. Using this approach, the authors identified L384A as a beneficial mutation that increased the yield of β-hexylaspartate from 0% to 85% (Table 1). The CtMAL L384A variant was also able to react with fumarate, 2-hexylfumarate, and 2-benzyloxyfumarate, giving nearly complete conversion within an hour at room temperature.
Fig 3.

Engineering the active site of MAL from Clostridium tetanomorphum (CtMAL) to tolerate larger substrates. A crystal structure of CtMAL (PDB ID: 1KKR) in complex with its native ncAA substrate β-methylaspartate (white) suggested that three bulky residues (orange) might restrict the size of the β-substituent that could productively bind in the active site. The β-substituent and targeted residues are shown with space-filling spheres to illustrate the steric requirements of the active site, and the magnesium ion (purple) and catalytic base (K331) are also shown. Site-saturation mutagenesis of these residues identified one mutation—L384A—that greatly enhanced the ncAA activity of CtMAL.23
Table 1.
Yields and stereoselectivities for the amination of fumarate analogues by wild-type and engineered CtMAL variants are shown for four representative products under multiple conditions.23
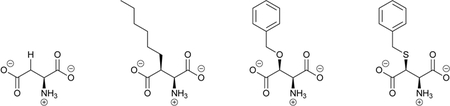 |
||||
|---|---|---|---|---|
| L-aspartate | threo-L-2-hexylaspartate | threo-L-2-benzyloxyaspartate | threo-L-2-benzylthioaspartate | |
| Wild-type CtMAL yielda | 100% | 0% | 0% | 0% |
| CtMAL L384A yielda | 99% | 53% | 60% | 42% |
| CtMAL L384A yieldb | ~100% | ~85% | ~90% | ND |
| ee (de) | >99% l ( - ) | ND (>95% threo) | >99% l (>95% threo) | ND (20% threo) |
0.01 mol% catalyst, 10-fold molar excess ammonia.
0.05 mol% catalyst, 5 M (167-fold molar excess) ammonia.
Chemical synthesis of β-branched aspartate analogues is challenging because, in addition to containing two carboxylic acid groups and a reactive amine, they are diastereomeric, having stereocenters at both the α- and β-carbon positions. CtMAL L384A preferentially produces the threo isomer from the prochiral substrate (Scheme 6), mirroring the native enzyme. This specificity enabled synthesis of the valuable therapeutic compound threo-3-benzyloxyaspartate in >95% excess over the erythro isomer. However, varying degrees of diastereomeric excess (de) were seen for other substrates, especially for those that contained thioether bonds, such as 2-benzylthioaspartate (Table 1). Nonetheless, this rapid engineering of CtMAL to access therapeutically relevant, optically pure, diastereomeric aspartate analogues from prochiral starting materials demonstrates the utility of protein engineering in ncAA synthesis.
5.1.2. PAL: Efforts to evolve a D-arylalanine synthase.
Aromatic ncAAs have diverse applications.1,3 This has made ammonia lyases with expansive aromatic substrate scopes, such as PALs, a prime target for application and for engineering.38 PALs natively catalyze the reversible decomposition of L-phenylalanine to trans-cinnamic acid and ammonia (Scheme 8). This is accomplished with the aid of the highly electrophilic 4-methylidine-5-imidazolone (MIO) group, which is unique in that it is not formally a cofactor. Rather, it is formed intramolecularly from a short three-amino acid sequence within the active site, in a manner reminiscent of GFP chromophore maturation.38 In the synthetic direction, the electrophilic MIO group enables amination by activating ammonia for nucleophilic attack, forming a stereospecific carbon-nitrogen bond (Scheme 8). The catalytic base of PALs must be even stronger than that of the aspartate-specific ammonia lyases, as aryl side chains do not render the protons of the β-carbon as labile as carboxylate groups do. PALs are an example of productive wild-type ncAA synthases with broad arylalanine-synthase activity that can be used to synthesize ncAAs in vitro. For example, wild-type PALs readily aminate halogenated cinnamic acids to generate the corresponding halogenated phenylalanine analogues.22
Scheme 8.

Generalized 4-methylidine-5-imidazolone (MIO)-dependent reaction for L-arylalanine synthesis by phenylalanine ammonia lyase (PAL).
The Turner group has carried out extensive engineering of PAL from the cyanobacterium Anabaena variabilis (AvPAL) for straightforward arylalanine synthesis. AvPAL has a broad substrate scope and is able to synthesize many different types of L-arylalanines with high activity and enantioselectivity. However, it was noted that the enantioselectivity of the reaction is compromised with electron-deficient substrates, such as nitrocinnamic acid.39 Further analysis suggested that these substrates go through a MIO-independent pathway. In this case, the enzyme is still required for proton abstraction at the β-carbon, but ammonia does not need to be activated by MIO for nucleophilic attack, due to the enhanced electrophilicity of the α-carbon (Scheme 9). The effect is strong enough to produce a racemic mixture whenever electron density is directed away from the electrophilic alkene, i.e., with para or ortho substitutions; meta-substituted substrates are less affected.
Scheme 9.

The MIO-independent pathway of PAL from Anabaena variabilis (AvPAL).
While this is an unfortunate side reaction of AvPAL, the L-α-amino acids have a privileged position in nature, and enzymes that interact with them are abundant. A biocatalytic toolbox is lacking for the D-configured amino acids, which are particularly useful in peptide-based therapeutics as they can reduce degradation in vivo (see Section 2). For example, the gonadotropin-releasing hormone antagonist cetrorelix contains three D-arylalanine ncAAs (Scheme 10). Therefore, this effect could potentially be leveraged as starting activity for the evolution of a D-arylalanine synthase.
Scheme 10.

The synthetic peptide cetrorelix contains three D-arylalanine ncAAs (blue).
To improve the practicality and efficiency of synthesizing D-arylalanine products, Parmeggiani and colleagues engineered AvPAL to increase its potential as a D-arylalanine synthase.40 Site-saturation mutagenesis libraries were prepared by targeting 48 residues near the active site that they hypothesized might influence the enantioselectivity of the reaction. To design a screen, the authors considered an approach similar to the MAL reaction discussed in Section 5.1.1, as the PAL reaction results in a wavelength change at 290 nm during the reaction. However, this approach does not report on the stereochemistry. Parmeggiani and colleagues addressed this by implementing an enzyme cascade that specifically interacted with D-amino acids, producing an intense color change. This would only occur within E. coli colonies expressing an AvPAL variant with improved D-arylalanine synthase activity (Fig 4). This high-throughput technique afforded rapid sampling of ~5,000 colonies, with numerous AvPAL variants showing significantly faster rates of D-arylalanine production.
Fig 4.

Enhanced enantiomer detection via an enzyme cascade. A D-amino acid oxidase (DAAO) can be used to generate H2O2 from the D-arylalanine product. Horseradish peroxidase (HRP) uses H2O2 to oxidize colorless 3,3‘-diaminobenzidine (DAB), forming an orange-brown dye. While this screen successfully identified AvPAL variants that produced D-arylalanines at an increased rate, it failed to find variants that could produce enriched mixtures of D-arylalanines over L-arylalanines.40
However, the screen had an important drawback: it only reported on D-amino acid production and provided no information on the corresponding production of an L-amino acids that might be expected from the less enantioselective MIO-independent mechanism. Indeed, further examination demonstrated that the variants identified in the screen did not change the distribution of enantiomers; they simply produced a racemic mixture more rapidly. It is not clear that mutations at the 48 targeted residues actually failed to enrich D-arylalanine production, because the screen only reported on an increase in D-arylalanine production, exactly as designed. For example, it could be the case that some mutants enriched for the D-isomer, but did so more slowly and were therefore not apparent in the screen. Additional controls to differentiate racemic from enantioenriched product formation could be implemented in future studies, but this would make an already complex screen even more complicated. A recent report by Zhu and colleagues demonstrated that a single active-site mutation to AvPAL (N347A), introduced by site-directed mutagenesis, resulted in a 2.3-fold enrichment in production of D-p-nitrophenylalanine over L-pnitrophenylalanine by influencing the stereoselectivity of the reaction within the enzyme.41 AvPAL-N347A may provide valuable starting activity for future directed evolution of an enantioselective D-arylalanine synthase.
5.2. Synthesis of designer ncAAs through direct side chain addition to amino-acrylate intermediates
Ideally, a ncAA synthase would be modular, in that it would attach desired side chains to an amino acid backbone with perfect stereoselectivity. An advantage typically associated with chemical synthesis, modularity allows different pieces to be incorporated into a diverse array of products with the same technique (see Section 3, Scheme 4). The pyridoxal 5’-phosphate (PLP)-dependent enzymes tyrosine phenol lyase (TPL) and tryptophan synthase (TrpS) have this attractive feature. These enzymes catalyze the β-elimination of an L-amino acid substrate to form an electrophilic amino-acrylate intermediate (Fig 5a). The amino-acrylate is a versatile electrophile that allows diverse nucleophilic substrates to be incorporated as amino acid side chains to form new L-α-amino acids. Due to the range of acceptable nucleophiles, these enzymes are capable of C–C bond formation as well as C–N and C–S bond formation. These enzymes also act with perfect enantioselectivity, as the stereochemistry at the α-carbon is retained through proton abstraction and donation on the same face of the amino-acrylate by the active-site lysine.25
Fig 5.

(a) Generalized reaction for pyridoxal 5’-phosphate (PLP)-dependent ncAA synthesis. Simplified reactions that follow the overall scheme in (a) are shown for the PLP-dependent enzymes (b) tyrosine phenol lyase (TPL) and (c) the β-subunit of tryptophan synthase (TrpB).
5.2.1. TPL: Active-site remodeling of tyrosine-analogue synthases.
Tyrosine phenol lyase (TPL) catalyzes the degradation of L-tyrosine (Tyr) to phenol, pyruvate, and ammonia through a β-elimination reaction (Figs 5b and 6). The reaction is readily reversible, and the addition of excess of ammonia and pyruvate shifts the equilibrium to favor Tyr production. This occurs by promoting the formation of the electrophilic amino-acrylate intermediate, which then reacts with phenol to form a C–C bond via an electrophilic aromatic substitution mechanism.42 Phenol is nucleophilic at positions para and ortho to the electron-donating hydroxyl group, and the enzyme positions this substrate such that the C–C bond is formed exclusively at the para position.
Fig 6.

Expanding the substrate scope of TPL from Citrobacter freundii (CfTPL) through enzyme engineering. The wild-type enzyme shows activity only with phenol, catechol (3-hydroxyphenol), and various fluorophenols to synthesize the corresponding tyrosine (Tyr) analogue. A crystal structure of CfTPL (PDB: 2YCN) in complex with the ncAA 3-fluoroTyr (wild-type activity, R1 = F, R2 = H) suggests that the active site is packed with hydrophobic residues that might sterically occlude larger substituents on the phenolic substrate. Based on this hypothesis, Seisser et al. in 2010 targeted five residues in the active site to replace with valine (F36V, M288V, M379V, F448V) or serine (T125S) and identified the variant CfTPL-M379V, which could produce 3-methylTyr, 3-methoxyTyr, and 3-chloroTyr.24 Two 2013 studies from the Wang group targeted three of these residues (F36, M288, and F448) with site-saturation mutagenesis to identify variants that could synthesize the ncAAs 3-(methylthio)-L-tyrosine (MtTyr, a Tyr-Cys cofactor mimic, provided by F36L mutation; blue)44 and 2-amino-3-(8-hydroxy-5-quinolinyl)-L-alanine (HqAla, a metal-chelating ncAA, provided by double mutant M288S/F448C; purple).12
TPL has been used for industrial-scale Tyr production as well as for the preparation of important ncAAs.18 As early as the 1970s, TPL was found to synthesize the therapeutic Tyr analogue L-DOPA directly from catechol, ammonia, and pyruvate (Fig 6, R1 = OH, R2 = H). Fluorotyrosine, used to study redox-active tyrosine residues, is also synthesized by TPL via addition of the corresponding fluorinated phenol (Fig 6).43 Unfortunately, wild-type TPL variants fail to produce Tyr analogues with substituents larger than fluorine; catechol seems to be the exception.
TPL variants have been engineered to expand the nucleophiles accepted in the reaction. For example, 3-methyltyrosine (3-MeTyr), an anticancer drug precursor, is synthesized through a three-step, low-yielding, racemic chemical synthesis with many protecting and deprotecting steps, followed by biocatalytic kinetic resolution.24 To improve synthesis of 3-MeTyr, Seisser and colleagues engineered TPL from the bacterium Citrobacter freundii (CfTPL), one of the most extensively studied TPL variants.24 Using site-directed mutagenesis, the authors targeted residues in the active site (F36, T125, M288, M379, and F448) that were hypothesized to interact unfavorably with substituted phenols (Fig 6). To reduce the steric restrictions, the hydrophobic residues were individually mutated to valine, while T125 was mutated to serine, and these variants were then screened by HPLC. While many of the mutations increased the production of 3-substituted phenols, the M379V variant was found to produce 3-MeTyr, 3-methoxyTyr (another anticancer precursor), and 3-chloroTyr (an atherosclerosis marker) with good yields (Fig 6, red).
Engineering efforts by the Wang group have expanded the capacity of CfTPL to synthesize designer ncAAs that can be incorporated into a protein and exhibit a specialized function. In two studies they used site-saturation mutagenesis and a thin-layer chromatography (TLC)-based screen to identify CfTPL variants that could synthesize the desired ncAA. The first study focused on making 3-(methylthio)-L-tyrosine (MtTyr; Fig 6, purple), which is a Tyr-Cys cofactor mimic.44 The Tyr-Cys cofactor is known to modulate enzyme kinetics and is common in metalloenzymes. However, Tyr-Cys cross-linking can only occur when the cysteine residue is positioned in a particular orientation relative to the tyrosine residue, which is difficult or impossible to engineer in many proteins. The authors reasoned that MtTyr might offer the properties of the Tyr-Cys cofactor in a single residue and that a TPL variant could be used to synthesize it in the laboratory. Wild-type CfTPL was selected as the parent for engineering a MtTyr synthase due to its well-characterized ability to generate other Tyr analogues.44 However, CfTPL had no activity toward the o-(methylthio)phenol nucleophile that forms the side chain of MtTyr. To accommodate MtTyr, Wang and colleagues individually targeted three active-site residues (F36, M228, and F448) for site-saturation mutagenesis. The authors chose only 96 E. coli clones from these libraries for analysis using the TLC-based screen, which identified CfTPL F36L as having improved activity. This variant synthesized MtTyr with 40% yield at preparative scale, which could then be purified and incorporated into proteins of interest using an evolved orthogonal amino-acyl tRNA synthetase and amber stop codon suppression technology (as described in ref. 10).
In a following study, the Wang group engineered CfTPL to synthesize the Tyr analogue 2-amino-3-(8-hydroxy-5-quinolinyl)-L-alanine (HqAla; Fig 6, purple).12 HqAla contains the bidentate metal chelator 8-hydroxyquinoline (8-HQ) as its side chain. 8-HQ is a common organic ligand of metal complexes noted for its high quantum yield of fluorescence, particularly when bound to zinc(II). Wild-type CfTPL again had no activity with the desired nucleophile (8-HQ), so the authors repeated site-saturation mutagenesis targeting active site residues F36, M228, and P448. This study targeted all three sites simultaneously, generating a library of 4,096 possible variants. CfTPL variants were again analyzed by TLC, and 1024 clones were screened to identify the double active-site mutant M228S/F448C that produced HqAla with 40% yield. Again, the synthesis of the ncAA was sufficient for downstream experiments and could be used to create proteins that exhibited zinc-dependent fluorescence, highlighting the capacity of enzyme engineering to access new chemical and biological space rapidly and effectively.
One mechanistic limitation of TPL is that the reaction is under thermodynamic control. The forward and reverse reaction rates of the net reaction depend strongly on the concentrations of products and reactants, and excess reactants are needed to drive product formation. Ammonia lyases (discussed in Section 5.1) also have this limitation, as do aminotransferases. The need for excess reagents is not a major issue when using TPL and ammonia lyases for preparative-scale synthesis, since ammonia and pyruvate are inexpensive and easy to exclude during purification. Nonetheless, it would be preferable to have the reaction under kinetic control such that product formation is effectively irreversible, improving atom economy and making in vivo applications more accessible. This type of reaction is possible when using CfTPL with specialized substrates. For example, S-(o-nitrophenyl)-L-cysteine can undergo rapid β-elimination in the presence of CfTPL, as the nitrothiophenol side chain acts as a good leaving group (Fig 7).42 This subsequently forms the reactive amino-acrylate intermediate, which is attacked by phenol to produce Tyr. Furthermore, because CfTPL binds S-(o-nitrophenyl)-L-cysteine more tightly than Tyr, as long as S-(o-nitrophenyl)-L-cysteine is present in the reaction it preferentially undergoes β-elimination and inhibits Tyr degradation. This approach gives yields of ~70%.
Fig 7.

The use of a specialized substrate, S-(o-nitrophenyl)-L-cysteine, affords kinetic control over Tyr production with CfTPL. This substrate binds in the active site (1) and rapidly forms the amino-acrylate (2), due to the good leaving group at the β-carbon (blue). The amino-acrylate (red) can either undergo nucleophilic attack by phenol (3, green) or deaminate to pyruvate and ammonia (4). Since TPL has a higher affinity for the substrate, S-(o-nitrophenyl)-L-cysteine, than for the Tyr product (5), TPL selectively binds the substrate, thereby reducing degradation of the Tyr product and providing higher yields.
5.2.2. TrpB: Evolution of stand-alone TrpB function from an allosteric TrpS complex.
Due to the reversible nature of enzymatic reactions under thermodynamic control, the ncAA synthases discussed thus far have suffered from inherently low substrate coupling efficiencies, with a high concentration of one or more substrates remaining upon reaching equilibrium. Although there are ways to circumvent this by using specialized substrates, an ideal biocatalyst would couple stoichiometric proportions of simple substrates at high rates and with quantitative yields. Additionally, these biocatalysts could have applications in vivo, as physiological concentrations of reactants could be sufficient to form products. To accomplish this, the enzymatic reaction should be under kinetic control. This is the case for tryptophan synthase (TrpS), which catalyzes the final steps of L-tryptophan (Trp) biosynthesis.
TrpS a heterodimeric complex composed of an α-subunit (TrpA) that allosterically regulates the β-subunit (TrpB).25 In the native reaction, indole glycerol-3-phosphate undergoes a retro-aldol reaction in TrpA to release indole. This induces TrpB to catalyze the β-elimination of L-serine (Ser), which generates the amino-acrylate intermediate (Fig 8a). Indole then diffuses through a hydrophobic tunnel connecting the subunits and attacks the amino-acrylate to form Trp (Fig 8b). The wild-type TrpS enzyme can perform this C–C bond-forming reaction with an array of indole analogues in vitro, synthesizing substituted Trp analogues in a single step.25 Numerous Trp derivatives have been made using this strategy. For example, the Goss group demonstrated that Salmonella enterica TrpS can use 7-chloroindole and Ser to form 7-chloroTrp, part of the antibiotic rebeccamycin (Scheme 11a).45 This reaction occurs in a single step, whereas nature would require an additional Trp halogenase to add the chloro substituent to Trp. Additionally, nonindole nucleophiles have been used to form C–S and C–N bonds, demonstrating that TrpS can also be a platform for the production L-cysteine and L-β-aminoalanine ncAAs.25
Fig 8.

The reaction of TrpB is under kinetic control, and parallels that of TPL with specialized substrates (as shown in Fig 7). (a) Formation of the reactive amino-acrylate intermediate (red) is favored by the β-elimination of L-serine and exclusion of water (blue) from the hydrophobic active site. (b) Indole (green) is activated for nucleophilic attack within the active site to form tryptophan (Trp); little to no β-elimination of Trp occurs when the enzyme is provided with Trp as its only substrate,32 suggesting that this step is effectively irreversible. (c) Nonproductive deamination of the amino-acrylate produces pyruvate and ammonia, a step that has been disfavored throughout the directed evolution of TrpB variants.
Scheme 11.

(a) Chlorotryptophan is found in the antibiotic natural product rebeccamycin; (b) β-methyltryptophan is found in natural products indolmycin and streptonigrin.
Although TrpS is an impressive biocatalyst, there are roadblocks for its application. Expression of the TrpS complex is metabolically challenging for the host cell, and the need for both the TrpA and TrpB subunits complicates expression and engineering. TrpB performs the synthetically interesting β-substitution reaction between indole and Ser to generate Trp, while TrpA generates indole in situ so that this toxic metabolite is not released into the cytosol. If the indole analogues are added exogenously, then TrpA is superfluous, but removing TrpA significantly decreases the activity of TrpB, due to the allosteric interactions between the subunits of TrpS.26
Buller and colleagues engineered a stand-alone TrpB ncAA synthase by directed evolution of TrpB from the hyperthermophilic archaeon Pyrococcus furiosus (PfTrpB; evolution shown in Fig 9, red).26 Because it was unknown whether directed evolution could recover the activity lost by removal of TrpA and, if so, what mutations would be beneficial, Buller used random mutagenesis to evolve the stand-alone PfTrpB. Variants were screened for Trp formation by monitoring an increase in 290-nm absorption, caused by a slight red-shift in the absorption of indole as it is converted to Trp. Impressively, nearly 4% of the 528 first-generation variants of PfTrpB screened displayed an increase in Trp formation. This is a higher rate of beneficial mutations than is usually seen in a random mutagenesis experiment and shows that there are many possible ways to reactivate TrpB. The greatest single improvement came from a T292S mutation that restored the kcat to that of wild-type PfTrpS (PfTrpB2G9, which we will simplify to Pf2G9). An additional five mutations (P12L, E17G, I68V, F274S, and T321A) resulted in a PfTrpB variant whose kcat exceeded that of the wild-type TrpB threefold (Pf0B2). Interestingly, none of these six beneficial mutations were in the active site, but rather were distributed throughout the TrpB structure. Further analysis showed that these mutations recapitulate the action of TrpA and stabilize the enzyme’s ‘closed’ conformation, a state that promotes formation of the reactive amino-acrylate intermediate.32
After establishing a stand-alone PfTrpB platform, Buller’s team engineered catalysts for making β-methyltryptophan (β-MeTrp) analogues.46 The ncAA β-MeTrp is a component of biologically important molecules such as indolmyin and streptonigrin (Scheme 11b). The presence of two adjacent stereocenters makes chemical synthesis of β-MeTrp challenging, but biocatalysts exhibit powerful spatial control over the diastereomeric configuration (see Section 5.1.1). Thus, it was reasoned that using the β-branched substrate L-threonine (Thr) might enable production β-MeTrp in a single step. This would be especially useful if the chirality—which is lost at both the α and β positions upon amino-acrylate formation—could be retained in the product (Scheme 12). (For clarity, we will refer to the β-substituted amino-acrylate intermediates simply as amino-acrylates.) Furthermore, it would be a significant simplification over the natural β-MeTrp biosynthetic pathway, which starts with Trp and recruits an additional three enzymes for methylation at the β-position. Although wild-type PfTrpS has been shown to use Thr as a substrate, this process is highly inefficient. When indole glycerol-3-phosphate is used as the source of indole, the enzyme shows a >82,000-fold preference for Ser over Thr.47
Scheme 12.

Reaction of TrpB with threonine, showing the loss of chirality in the β-methylamino-acrylate (amino-crotylate) intermediate.
To encourage activity with Thr, Herger et al. applied random mutagenesis and recombination to the stand-alone variant Pf4D11.46 Following two rounds of mutagenesis and screening for (2S,3S)-β-methyltryptophan (β-MeTrp) formation at 290 nm from a mixture with Thr and indole, a new variant (Pf2B9) was identified that incorporated three mutations (Fig 9, orange). Pf2B9 boosted production of β-MeTrp >6,000-fold relative to wild-type PfTrpB. Furthermore, Pf2B9 accepted an array of indole substrates: seven (2S,3S)-β-MeTrp analogues were synthesized on preparative scale with >99% de. While the formation of Trp analogues is irreversible, PfTrpB was found to be limited by the unproductive abortive deamination of the amino-acrylate intermediate (Fig 8c), which consumes an equivalent of amino acid substrate. However, adding excess Ser or Thr to the reaction can reduce this effect and improve Trp production, albeit with lower coupling efficiency.
Next, Boville and colleagues sought to expand the substrate scope of PfTrpB to synthesize more complex β-branched ncAAs, such as (2S,3S)-β-ethyltryptophan (β-EtTrp) and (2S,3S)-β-propyltryptophan (β-PrTrp) (Fig 9, pink).48 Pf2B9 was selected as the parent enzyme due to its activity with Thr, but even this evolved variant had poor baseline activity with (2S,3R)-β-ethylserine (β-EtSer) and (2S,3R)-β-propylserine (β-PrSer). Modeling β-EtSer into the Pf2B9 active site suggested that residue L161 clashed with the bulkier β-ethyl substituent. Based on the hypothesis that a smaller side chain would reduce steric hinderance, L161 was mutated to alanine (Pf2B9 L161A), which proved to increase activity with indole and β-ethylserine. Two rounds of random mutagenesis and screening for increased absorption at 290 nm generated a variant with two additional beneficial mutations (Pf8C8), which was then used as the parent for recombination with eight previously identified mutations. This final variant, Pf7E6, was found to be a robust biocatalyst for four amino acid substrates (Ser, Thr, β-EtSer, β-PrSer) and ten indole analogues, which enabled Boville and coworkers to generate dozens of β-branched ncAAs. Mechanistic studies showed that the versatility of Pf7E6 arose from a more stable amino-acrylate than found in Pf2B9. This further reduced the frequency of the unproductive deamination reaction, and single equivalents of substrate could be used to achieve high yields.48
Many desirable ncAAs still remained inaccessible using TrpS and the evolved TrpBs. In particular, bulky or electron-deficient indoles, particularly 4-substituted indoles, remained challenging substrates; bulky groups introduce additional steric factors that interfere with substrate positioning, while electron-withdrawing groups attenuate nucleophilicity (Scheme 13). To address this, Romney and colleagues targeted production of 4-nitroTrp,49 one of the most difficult products for PfTrpB and a valuable precursor in the synthesis of tumor-promoting indolactam V and the natural herbicide thaxtomin A (Scheme 14). Multiple rounds of random mutagenesis and recombination identified the variant Pf2A6, which increased the yield of 4-nitroTrp to 95% (Fig 9, green). To improve the sensitivity of the screen during the evolution, Romney and colleagues implemented an organic extraction step to quickly and reliably remove excess nitroindole from the aqueous media, reducing the background signal of the colorimetric screen. While Pf2A6 was most active with 4-nitroTrp, it was observed that the early mutations M139L and N166D were broadly activating for 4-, 5-, 6-, and 7-nitroTrp, while the later mutations were specific to 4-nitroTrp. Results of assaying for pyruvate formation suggested that this early general improvement was due to further stabilization of the amino-acrylate intermediate, which was not as readily hydrolyzed and thus more available for attack by weakly nucleophilic substrates.
Scheme 13.

The 4-nitro groups disfavors nucleophilic attack (black arrows) by electron withdrawal (red arrows) and steric repulsion (blue dashed line).
Scheme 14.

Tumor-promoting indolactam V and the herbicide thaxtomin A are synthesized from 4-nitrotryptophan.
Through additional directed evolution, Romney et al. engineered PfTrpB for high activity toward 4-, 6-, and 7-nitroindole.49 However, the available variants were still poorly active with 5-nitroindole, as well as many other 4-, and 5-substituted indoles. Murciano-Calles et al. had previously shown that the activating mutations found in PfTrpB could be transferred to TrpBs isolated from other species, such as the hyperthermophilic bacterium Thermotoga maritima (TmTrpB).27 Despite being from different orders of life and having only 64% sequence identity, many of the activating mutations found in PfTrpB were also beneficial when made at the corresponding residues in TmTrpB, suggesting that both of these TrpS complexes were regulated using similar allosteric mechanisms. However, these enzymes displayed varied activity with different substituted indoles: PfTrpB was the best for many 6- and 7-substituted Trp analogues, while TmTrpB was superior for many 4- and 5-substitutions (Fig 9, blue). Romney and colleagues capitalized on this discovery and used both PfTrpB and TmTrpB variants to synthesize challenging nitroTrps, cyanoTrps, haloTrps, and di-substituted Trps.49 This work culminated in a panel of biocatalysts tuned to the substrates of interest. More than 20 Trp analogues could be prepared with good yields (ten exceeded 90% yield) from equimolar amounts of substrates and with low biocatalyst loadings.
Certain valuable Trp analogues still proved difficult to synthesize with the expanded TrpB platform, including 4-cyanoTrp, a blue-fluorescent ncAA useful for in vitro and in vivo imaging. In a single round of random mutagenesis followed by recombination, Boville et al. identified variant Tm9D8*, which improved yields of 4-cyanoTrp from 24% to 78% (Fig 9, purple).50 Intriguingly, the mutations in Tm9D8* shifted the temperature profile of the biocatalyst to lower temperature. Previously, the thermophilic TrpB variants showed peak performance at 75 °C. However, Tm9D8* achieves 76% yield at 37 °C, while at 75 °C only 46% yield is achieved. Furthermore, this temperature shift occurs without decreasing the thermostability of the enzyme, which is still active after heat-treatment above 90 °C. The high stability facilitates purification from mesophilic hosts like E. coli, as demonstrated by the gram-scale production of 4-cyanoTrp using heat-treated lysate. The high activity at lower temperature also creates potential in vivo applications.
6. Conclusions and Future Directions
Noncanonical amino acids are valuable as final products and intermediates, and enzymes provide a straightforward, cost-effective, and green method for their production. These enzymes can be optimized for activity, selectivity, stability and other important properties. Enzymes such as ammonia lyases, tyrosine phenol lyase, and tryptophan synthase can perform reactions on substrates well beyond their natural activities and produce optically pure ncAAs from simple and inexpensive starting materials. However, only a few select classes of enzymes have been engineered to date; there is still a need for new classes of ncAA synthases that access an even wider range of amino acids.
Engineering useful new ncAA synthases requires suitable starting enzymes. Well-characterized enzymes often serve as the basis for engineering new reactions, but homologous and even previously uncharacterized enzymes allow protein engineers to access a wealth of untapped functional and sequence diversity. Information gained from engineering one enzyme can be transferred to other family members. Beneficial mutations found in PfTrpB, as discussed in Section 5.2.2, were often beneficial when transferred to homologous TrpB enzyme subunits from other, even distantly related species. These variants had notable and useful differences in their substrate scopes, which facilitated further evolution.49 Future work can identify new enzymes in natural biosynthetic pathways that can be exploited for ncAA synthesis and serve as a launchpad for further evolution.
In addition to identifying new enzymes, it is important to explore previously studied variants and report on more complete substrate scopes rather than just a few key substrates. This can help inform the enzyme’s mechanism and function and facilitates the selection of even better parent enzymes in future engineering. For example, Wang’s group could have used Seisser’s CfTPL-M379V variant24 as the parent for engineering activity for MtTyr production,44 as 3-methoxyTyr and MtTyr are structurally similar. Alternatively, Seisser and coworkers could have reported MtTyr and other Tyr analogue activity with their CfTPL variant, which would have provided additional information on how small alterations influenced the reactivity of the enzyme, if at all. Such information allows researchers to better traverse the vast and diverse landscape of protein function; stopping at a single beneficial mutation or product is shortsighted and leaves behind important opportunities.
With new discoveries and unbridled engineering, nature’s remarkable biosynthetic machinery has the potential to change how we approach chemistry, biology, and medicine. Enzymes such as ncAA synthases have proven to be potent and malleable biocatalysts, and protein engineering is sure to bring more versatile tools into the fold.
Fig 2.

Protein engineering by mutagenesis and screening. A successful engineering strategy depends on the choice of starting enzyme (the ‘parent’ enzyme), the mutation strategy, and the screening approach. A good choice of parent enzyme is typically one that already exhibits that desired activity (but poorly), or an enzyme that performs a mechanism that is compatible with that activity. Homologous enzyme variants can also be worthwhile to test, as are thermophilic enzymes. After parent selection, an appropriate mutagenesis technique is applied to the parent gene. Depending on the amount of information known and the functional goals, site-directed or site-saturation mutagenesis can target a single position of the enzyme, while random mutagenesis can blindly explore the entire sequence, and recombination can combine known activating mutations or swap genetic diversity between different variants. Following this, the choice of mutation strategy will largely influence the choice of screening technique. Colorimetric screens (illustrated here) are wise choices for large libraries, while smaller libraries can be screened with lower-throughput techniques, such as HPLC. Once an improved variant is—or multiple improved variants are—identified, the cycle can be repeated, which is called directed evolution. Depending on the results, the same mutation strategy and screen can be used, or an entirely new approach can be adopted if appropriate.
Acknowledgements
The authors would like to thank Dr. Sabine Brinkmann-Chen, Dr. David Romney, Prof. Andrew Buller, and Bradley Boville for helpful discussion and comments on the manuscript. C.E.B. was supported by a postdoctoral fellowship from the Resnick Sustainability Institute. This work was funded by the Jacobs Institute for Molecular Engineering for Medicine (Caltech) and the Rothenberg Innovation Initiative (formerly CI2).
Footnotes
Conflicts of Interest
There are no conflicts to declare.
References
- 1.Blaskovich MAT, J. Med. Chem, 2016, 59, 10807–10836. [DOI] [PubMed] [Google Scholar]
- 2.Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM and Wood A, Nat. Chem, 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- 3.Young DD and Schultz PG, ACS Chem. Biol, 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue Y-P, Cao C-H and Zheng Y-G, Chem. Soc. Rev, 2018, 47, 1516–1561. [DOI] [PubMed] [Google Scholar]
- 5.Li R, Wijma HJ, Song L, Cui Y, Otzen M, Tian Y, Du J, Li T, Niu D, Chen Y, Feng J, Han J, Chen H, Tao Y, Janssen DB and Wu B, Nat. Chem. Biol, 2018, 14, 664–670. [DOI] [PubMed] [Google Scholar]
- 6.Park ES and Shin JS, J. Mol. Catal. B Enzym, 2015, 121, 9–14. [Google Scholar]
- 7.Ehrenworth AM and Peralta-Yahya P, Nat. Chem. Biol, 2017, 13, 249–258. [DOI] [PubMed] [Google Scholar]
- 8.McGrath NA, Brichacek M and Njardarson JT, J. Chem. Educ, 2010, 87, 1348–1349. [Google Scholar]
- 9.Thu T, Nguyen H, Bai X and Shim H, Biodesign, 2015, 3, 154–161. [Google Scholar]
- 10.Liu CC and Schultz PG, Annu. Rev. Biochem, 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 11.Odar C, Winkler M and Wiltschi B, Biotechnol. J, 2015, 10, 427–446. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Li J, Hu C, Zhou Q, Zhang W, Hu M, Zhou J and Wang J, Angew. Chemie Int. Ed, 2013, 52, 4805–4809. [DOI] [PubMed] [Google Scholar]
- 13.Almhjell PJ and Mills JH, Curr. Opin. Struct. Biol, 2018, 51, 170–176. [DOI] [PubMed] [Google Scholar]
- 14.Boutureira O and Bernardes GJL, Chem. Rev, 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- 15.D’Este M, Alvarado-Morales M and Angelidaki I, Biotechnol. Adv, 2018, 36, 14–25. [DOI] [PubMed] [Google Scholar]
- 16.Rudroff F, Mihovilovic MD, Gröger H, Snajdrova R, Iding H and Bornscheuer UT, Nat. Catal, 2018, 1, 12–22. [Google Scholar]
- 17.Völler JS and Budisa N, Curr. Opin. Biotechnol, 2017, 48, 1–7. [DOI] [PubMed] [Google Scholar]
- 18.Lütke-Eversloh T, Santos CNS and Stephanopoulos G, Appl. Microbiol. Biotechnol, 2007, 77, 751–762. [DOI] [PubMed] [Google Scholar]
- 19.Ager DJ, in Amino Acids, Peptides, and Proteins in Organic Chemistry, Vol. 1 - Origins and Synthesis of Amino Acids, ed. Hughes AB, WILEY-VCH, 2009, pp. 495–526. [Google Scholar]
- 20.Brittain WDG and Cobb SL, Org. Biomol. Chem, 2018, 16, 10–20. [DOI] [PubMed] [Google Scholar]
- 21.Slabu I, Galman JL, Lloyd RC and Turner NJ, ACS Catal, 2017, 7, 8263–8284. [Google Scholar]
- 22.Gloge A, Zon J, Kovari A, Poppe L and Rétey J, Chem. Eur. J, 2000, 6, 3386–3390. [DOI] [PubMed] [Google Scholar]
- 23.Raj H, Szymański W, De Villiers J, Rozeboom HJ, Veetil VP, Reis CR, De Villiers M, Dekker FJ, De Wildeman S, Quax WJ, Thunnissen AMWH, Feringa BL, Janssen DB and Poelarends GJ, Nat. Chem, 2012, 4, 478–484. [DOI] [PubMed] [Google Scholar]
- 24.Seisser B, Zinkl R, Gruber K, Kaufmann F, Hafner A and Kroutil W, Adv. Synth. Catal, 2010, 352, 731–736. [Google Scholar]
- 25.Phillips RS, Tetrahedron Asymmetry, 2004, 15, 2787–2792. [Google Scholar]
- 26.Buller AR, Brinkmann-Chen S, Romney DK, Herger M, Murciano-Calles J and Arnold FH, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 14599–14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murciano-Calles J, Romney DK, Brinkmann-Chen S, Buller AR and Arnold FH, Angew. Chemie Int. Ed, 2016, 55, 11577–11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom JD, Labthavikul ST, Otey CR and Arnold FH, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeymer C and Hilvert D, Annu. Rev. Biochem, 2018, 87, 131–157. [DOI] [PubMed] [Google Scholar]
- 30.Packer MS and Liu DR, Nat. Rev. Genet, 2015, 16, 379–394. [DOI] [PubMed] [Google Scholar]
- 31.Reetz MT and Carballeira JD, Nat. Protoc, 2007, 2, 891–903. [DOI] [PubMed] [Google Scholar]
- 32.Buller AR, van Roye P, Cahn JKB, Scheele RA, Herger M and Arnold FH, J. Am. Chem. Soc, 2018, 140, 7256–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heyer WD, Cold Spring Harb. Perspect. Med, 2015, 5, 1–24. [Google Scholar]
- 34.Briney BS and Crowe JE, Front. Immunol, 2013, 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stemmer WP, Proc. Natl. Acad. Sci, 1994, 91, 10747–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao H, Giver L, Shao Z, Affholter JA and Arnold FH, Nat. Biotechnol, 1998, 16, 258–261. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J-H, Chung TDY and Oldenburg KR, J. Biomol. Screen, 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 38.Parmeggiani F, Weise NJ, Ahmed ST and Turner NJ, Chem. Rev, 2018, 118, 73–118. [DOI] [PubMed] [Google Scholar]
- 39.Lovelock SL, Lloyd RC and Turner NJ, Angew. Chemie Int. Ed, 2014, 53, 4652–4656. [DOI] [PubMed] [Google Scholar]
- 40.Parmeggiani F, Lovelock SL, Weise NJ, Ahmed ST and Turner NJ, Angew. Chemie Int. Ed, 2015, 54, 4608–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu L, Feng G and Ge F, Appl. Biochem. Biotechnol, DOI: 10.1007/s12010-018-2794-3. [DOI] [PubMed] [Google Scholar]
- 42.Phillips RS, Chen HY and Faleev NG, Biochemistry, 2006, 45, 9575–9583. [DOI] [PubMed] [Google Scholar]
- 43.Seyedsayamdost MR, Yee CS and Stubbe J, Nat. Protoc, 2007, 2, 1225–1235. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Q, Hu M, Zhang W, Jiang L, Perrett S, Zhou J and Wang J, Angew. Chemie Int. Ed, 2013, 52, 1203–1207. [DOI] [PubMed] [Google Scholar]
- 45.Smith DRM, Willemse T, Gkotsi DS, Schepens W, Maes BUW, Ballet S and Goss RJM, Org. Lett, 2014, 16, 2622–2625. [DOI] [PubMed] [Google Scholar]
- 46.Herger M, Van Roye P, Romney DK, Brinkmann-Chen S, Buller AR and Arnold FH, J. Am. Chem. Soc, 2016, 138, 8388–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buller AR, Van Roye P, Murciano-Calles J and Arnold FH, Biochemistry, 2016, 55, 7043–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boville CE, Scheele RA, Koch P, Brinkmann-Chen S, Buller AR and Arnold FH, Angew. Chemie Int. Ed, DOI: 10.1002/anie.201807998. [DOI] [PubMed] [Google Scholar]
- 49.Romney DK, Murciano-Calles J, Wehrmüller J and Arnold FH, J. Am. Chem. Soc, 2017, 139,10769–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boville CE, Romney DK, Almhjell PJ, Sieben M and Arnold FH, J. Org. Chem, 2018, 83, 7447–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]