

Abstract
GTP-binding proteins play important roles in many essential biological processes, including cell signaling, trafficking, and protein synthesis. To assess quantitatively these proteins at the whole proteome level, we developed a high-throughput targeted proteomic method based on the use of isotope-coded GTP probes and multiple-reaction monitoring (MRM) analysis. Targeted proteins were labeled with desthiobiotin-GTP probes, digested with trypsin, and the ensuing desthiobiotin-conjugated peptides were enriched with streptavidin beads for LC-MS/MS analysis. We also established a Skyline MRM library based on shotgun proteomic data acquired for 12 different human cell lines. The library contained 605 tryptic peptides derived from 217 GTP-binding proteins, representing approximately 60% of the annotated human GTP-binding proteome. By using this library, in conjunction with isotope-coded GTP probes and scheduled LC-MRM analysis, we investigated the differential expression of GTP-binding proteins in a pair of primary/metastatic colon cancer cell lines (SW480 and SW620). We were able to quantify 97 GTP-binding proteins, and we further validated the differential expression of several GTP-binding proteins by Western blot analysis. Together, we developed a facile targeted quantitative proteomic method for the high-throughput analysis of GTP-binding proteins and applied the method for probing the altered expression of these proteins involved in colon cancer metastasis.
Graphical Abstract
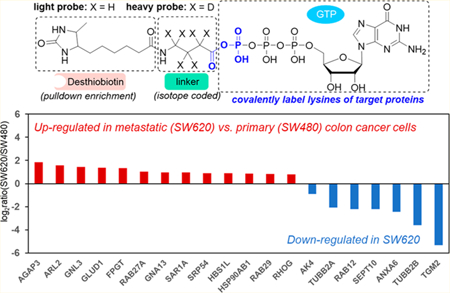
GTP-binding proteins are involved in a series of essential biological processes, including cell signaling,1 intracellular trafficking,2 proliferation,3 differentiation,4 and apoptosis.5 For example, many GTPases act as switches in signal transduction pathways that toggle between the GTP-bound active, and the GDP-bound inactive states.6 Guanine nucleotide exchange factors (GEFs) catalyze the exchange of the bound GDP with a new GTP, which activates downstream signaling pathways. On the other hand, the active forms of GTPases can be switched off through intrinsic hydrolysis of the bound GTP to GDP catalyzed by GTPase-activating proteins (GAPs). In addition, aberrant GTPase signaling has been observed in human diseases, including many different types of cancer.7 Considering the biological importance of GTP-binding proteins and their implications in human diseases, high-throughput analytical tools for assessing quantitatively GTP-binding proteins at the whole proteome scale will facilitate the exploration of the related signaling network.
Most GTPases contain a highly conserved G domain, which is responsible for GTP binding and hydrolysis.8 In this regard, developing chemical probes that target active sites of GTPases could help understand their enzymatic functions and provide important knowledge for inhibitor discovery.9 Activity-based protein profiling (ABPP) has been widely applied in probing particular families of enzymes in complex proteomes.10 Compared with other proteomic methods that measure protein expression levels, the ABPP approach takes advantage of active site labeling and thus provides direct information about the binding affinity of a protein.
Several structurally distinct ABPP probes have been reported for profiling GTP-binding proteins, though only a few have been applied in mass spectrometry-based proteomic studies.11,12 Cisar et al.13 reported a photoreactive GTP affinity probe, which could covalently photo-cross-link with target proteins with an alkyne handle for subsequent conjugation through click chemistry. Using this GTP-BP-yne probe, more than 30 annotated GTP-binding proteins were identified with shotgun proteomic method. In addition, Patricelli et al.14 and Qiu et al.15 reported a lysine-reactive nucleotide acyl phosphate probe for the proteome-wide profiling of nucleotide-binding proteins such as kinases. With this strategy, Xiao et al.16 identified 349 ATP-binding and 66 GTP-binding proteins in HL-60 cell lysates using the biotin-conjugated ATP and GTP acyl-phosphate probes, respectively. A competitive binding experiment between the ATP and the GTP probes, facilitated by stable isotope labeling by amino acids in cell culture (SILAC), revealed clear differences in proteins being labeled with the two probes, which confirmed their good selectivity based on binding affinity. Later, Xiao et al.17 applied a similar quantification strategy for profiling 6-thioguanosine triphosphate (SGTP)-binding proteins compared with GTP-binding proteins. More than 30 GTP-binding proteins were quantified and several exhibited higher binding affinities toward SGTP than GTP. Commercialized desthiobiotin-ATP and GTP probes (ActivX Biosciences) have also been employed for profiling kinome and GTPase proteome.14 In this vein, Hunter et al.18 applied this lysine-reactive biotin-GTP probe for profiling an active site inhibitor of oncogenic K-Ras G12C. They were able to detect over 100 GTP-binding proteins from the lysate of MIA PaCa cells. Very recently, Huang et al.19 developed a targeted quantitative proteomic approach based on gel fractionation coupled with multiple-reaction monitoring (MRM) analysis for the assessment of the reprogramming of small GTPase proteome during melanoma metastasis. This method is highly efficient, though the major targets are limited to small GTPases.
We report here an MRM-based quantitative proteomic method for profiling GTP-binding proteins using desthiobiotin-GTP acyl phosphate probes. We also introduced an isotope-coded linker into the GTP probe, which not only improved the coverage of GTP-binding proteins, but also facilitated quantitative analysis. Furthermore, we applied successfully this method for the quantification of GTP-binding proteins in a pair of matched primary/metastatic colon cancer cell lines (SW480/SW620) initially derived from the same patient, which enabled the identification of potential drivers and suppressors for colon cancer metastasis.
MATERIALS AND METHODS
Synthesis and Purification of Desthiobiotin-GTP Affinity Probes.
All chemicals were purchased from Sigma-Aldrich unless specifically noted. The desthiobiotin-GTP affinity probes were prepared following previously published procedures with some modifications (see Supporting Information for details).15 Briefly, the probes were synthesized by conjugating GTP with desthiobiotin or desthiobiotin linked with isotope-coded γ-aminobutyric acid (GABA). The identities of probes were confirmed by ESI-MS analyses and the probe concentrations were determined from the UV absorption of its aqueous solution at 252 nm (extinction coefficient ε = 13700 M−1 cm−1). Once dissolved in water, the GTP probes were aliquoted and stored at −80 °C until use.
Cell Lysate Preparation and Labeling with the GTP Probe.
HCT-116, HEK293T, HeLa, SW480, SW620, U2OS, WM-115, and WM-266-4 cells were obtained from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen-Gibco). Jurkat T, K562, and PC-3 cells were from ATCC and cultured in RPMI 1640 medium (Invitrogen-Gibco). GM04429 cells were kindly provided by Prof. Gerd P. Pfeifer and cultured in DMEM. All culture media were supplemented with 10% fetal bovine serum (Invitrogen-Gibco) and penicillin/streptomycin (100 IU/mL). The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2, and the culture medium was changed every 2–3 days as needed.
Approximately 2 × 107 cells were harvested, washed with cold PBS for three times, and lysed by incubating on ice for 30 min in a 1 mL lysis buffer, which contained 0.1% Triton X-100, 50 mM HEPES (pH 7.4), 100 mM NaCl, and 10 μL of protease inhibitor cocktail (100×). The cell lysates were centrifuged at 15,000g for 30 min at 4 °C, and the supernatants collected. The labeling conditions were similar as previously described.15 Briefly, after removal of endogenous nucleotides using NAP-5 columns (Amersham Biosciences), approximately 1 mg lysate was treated with 5 mM EDTA for 5 min. The GTP probes and MgCl2 were then added to the lysate until their final concentrations reached 20–100 μM and 20 mM, respectively, and the mixture was incubated at room temperature for 2 h. After the reaction, the unreacted probes were quenched with excess glycine (100 mM) for 0.5 h and removed by buffer exchange with 50 mM NH4HCO3 solution using an Amicon Ultra-4 filter (10 kDa, Millipore).
Data-Dependent LC-MS/MS Analysis.
Preparation of labeled peptide samples was adapted from a previous report with minor modifications (see Supporting Information for details).16 After labeling with 100 μM GTP probes, the lysates were subjected to filter-aided sample preparation (FASP), where trypsin was the digestion enzyme.20 The desthiobiotin-conjugated peptides were enriched from the resulting mixture using high-capacity streptavidin beads (Sigma-Aldrich) and eluted with 1% TFA in 70% CH3CN at room temperature. The samples were analyzed using online 2D-LC-MS/MS on a Q Exactive Plus quadrupole-Orbitrap mass spectrometer (Thermo Fisher) operated in data-dependent acquisition mode. The raw data from 12 cell lines were combined and processed by Maxquant (version 1.5.3.8) against Uniprot human proteome database (reviewed by Swiss-Prot, 20386 entries, downloaded on 10/17/2017). Gene ontology (GO) analyses were conducted using a web-based database DAVID Bioinformatics Resources (version 6.8, https://david.ncifcrf.gov/).
Scheduled LC-MRM Analysis.
The above-mentioned LC-MS/MS data acquired in the DDA mode were processed using Skyline (version 3.7)21 to generate an MRM library for GTP-binding proteins and calibrate iRT22 for the scheduled MRM analysis (see details in Supporting Information). For the quantitative profiling of GTP-binding proteins in colon cancer cell lines, SW480 (primary), and SW620 (metastatic) cells were labeled with light and heavy desthiobiotin-GTP probes, respectively (in the forward experiment), and combined for the subsequent steps of sample preparation. The samples were analyzed on a TSQ Vantage triple-quadrupole mass spectrometer (Thermo Fisher) coupled with a nanoelectrospray ionization source and an EASY n-LC-II HPLC system (Thermo Fisher). The data were processed using Skyline for quantitative analysis.23 All the targeted peptides were manually checked to ensure that the intensity distribution of selected transitions is consistent with the theoretical distributions in the spectral library, with dotp24 being greater than 0.8. The Skyline library and the raw files for LC-MRM analyses for the paired colon cancer cells were deposited into PeptideAtlas with the identifier number of PASS01239 (http://www.peptideatlas.org/PASS/PASS01239).
RESULTS AND DISCUSSION
Design of the GTP Probes.
Similar to ATP-binding proteins, most GTP-binding proteins carry a consensus phosphate-binding loop (P-loop), which is responsible for their nucleotide binding and GTPase activities.25 In many GTPases, a highly conserved Walker A sequence motif of GxxxxGKT/S is often found in the P-loop region of nucleotide-binding proteins (“x” denotes any amino acid residue). Based on the nucleophilic properties of lysine, nucleotide acyl phosphate probes that target the conserved lysine residue(s) located at or near the nucleotide binding site have been developed for the affinity-based profiling of ATP-and GTP-binding proteins at the proteome-wide scale.15 In general, the designed probe can be recognized by targeted proteins, and the lysine residue(s) at or near the binding sites is then covalently modified by reacting with the acyl phosphate moiety (Figure 1A).
Figure 1.
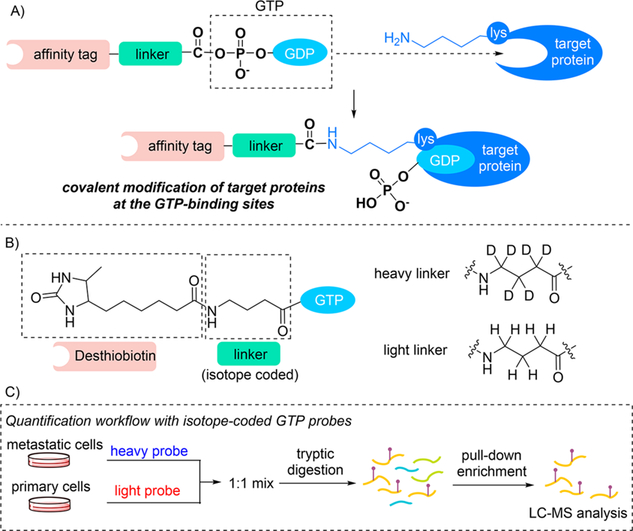
(A) GTP-binding protein affinity probe design; (B) Chemical structures of the isotope-coded GTP probes; (C) General workflow for quantitative analysis using isotope-coded GTP probes.
Our goal is to improve the coverage of GTP-binding proteins with GTP probe enrichment and apply this method for quantitative proteomic analysis. To this end, we prepared a desthiobiotin-GTP probe, with desthiobiotin being the affinity tag.16 Using this probe, about 60 GTP-binding proteins were labeled in HEK293T or HeLa cell lysates and identified with 1D LC-MS/MS analysis on an LTQ Orbitrap Velos mass spectrometer. Because the affinity tag may potentially influence the binding between the probe and target proteins, we introduced γ-aminobutyric acid (GABA) as a linker to increase the distance between GTP and the affinity tag (Figure 1B). With the incorporation of the linker, we were able to identify approximately 80 GTP-binding proteins from each of the aforementioned cell lines. In addition, deuterium-labeled linker can be employed to enable quantitative analysis.26 Unlike SILAC-based protein quantification, which requires labeling of cells with isotope-coded amino acids for a few weeks, the isotope-coded GTP probes facilitate the quantification of GTP-binding proteins without metabolic labeling. Hence, our approach not only simplified sample preparation (Figure 1C), it is also potentially useful for samples that are not readily amenable to SILAC labeling (e.g., tissue samples). In addition, the identification of desthiobiotin-modified peptides also provides information about the lysine residue(s) that are at or near the GTP binding sites of these proteins. In this context, it is of note that enrichment can also be carried out at the protein level prior to tryptic digestion, which may improve the sequence coverage and reveal more unique peptides for some targeted GTP-binding proteins. However, an additional step of metabolic or chemical labeling would be necessary for robust protein quantification.
Shotgun Proteomics for Profiling GTP-Binding Proteins.
In large-scale shotgun proteomic studies, due to the complex composition of peptide samples, peptides of low abundance might be overridden by highly abundant peptides, which reduced their possibility to be detected in the DDA mode. By employing online 2D-LC-MS/MS analysis with strong-cation exchange separation followed by separation using a reverse-phase C18 column, we were able to identify up to 167 GTP-binding proteins from the lysate of a single human cell line. To further improve the coverage, we consolidated the data obtained from 96 LC-MS/MS runs, which were acquired from shotgun proteomic analyses of 12 different cell lines derived from different human tissue origins.
By performing gene ontology (GO) analysis of the protein list, we found that several GO terms were highly enriched, including nucleotide-binding, ATP-binding, GTP-binding, DNA-binding, and RNA-binding proteins (Table S1). Given the structural similarity of these nucleotides, we could infer that some proteins might harbor relatively flexible nucleotide-binding pockets. Among the 6984 identified proteins, 233 were categorized as GTP-binding proteins, which represent 64% of the 365 annotated GTP-binding proteins in the human proteome. Since GTP-binding proteins are often present in GTP- or GDP-bound states, the affinity probe labeling efficiency is largely affected by the nucleotide exchange rates. Similar to previous reports,15 we employed EDTA to accelerate the dissociation of bound nucleotides and added Mg2+ to promote the binding of the GTP probes during the labeling step. In this context, it is of note that the probe-labeling efficiency of some GTP-binding proteins might be impaired owing to their strong GTP-binding affinity and, as a result, the poor removal of the bound endogenous GTP.
Different types of GTP-binding proteins are effectively labeled and identified with our shotgun proteomic approach, as depicted in Figure 2. A major group of the identified proteins is the Ras superfamily of small GTPases, which constitutes a wide and diverse class of intracellular signaling proteins that are highly conserved throughout evolution.27 Other families of GTP-binding proteins are also detected, including the α-subunit of large G proteins,28 GBP family,29 IMAP family,30 tubulins,31 dynamins,32 septins,33 and initiation and elongation factors.34
Figure 2.
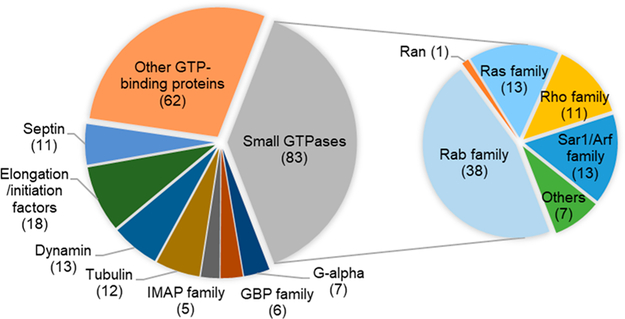
Summary of GTP-binding proteins identified with the ABPP probe.
Development of Skyline Spectral Library and MRM Assay.
With the data from shotgun proteomics, we constructed a Skyline peptide library that encompasses 217 GTP-binding proteins. After selecting up to six top-ranking peptides for each protein and checking manually the uniqueness of each peptide (i.e., those peptides that can be attributed to a specific GTP-binding protein), we included 605 peptides in the library, among which 85 are derived from Walker A motif. A small number of peptides from the NKXD motif, which is conserved and crucial for guanosine recognition, are also included.
One limitation in applying the ABPP-based approach in profiling GTP-binding proteins resides in the high sequence conservation of the P-loop, which hampers the selection of peptides that can be uniquely attributed to a specific GTP-binding protein. For instance, one probe-labeled peptide, LLLLGAGESGK*STIVK, could be assigned to 10 different GTP-binding proteins (mostly G-α subunits) in the human proteome. This is also a common scenario for several small GTPases and tubulins. Peptide derived from the Walker A motif are among the most enriched peptides identified from our probe labeling and enrichment experiments.
The lack of sequence uniqueness hindered us from evaluating the expression of those GTP-binding proteins with only nonunique peptides. In this vein, 22 peptides in our library are not unique (most of them are derived from the P-loop) and 17 proteins only contain nonunique peptides. These peptides are highlighted in Table S2 and are excluded from our quantification results. Additional efforts are needed for quantifying these proteins by combining with other techniques.
For each peptide in the library, we selected at least three top-ranking y-ions for the quantification. Combining peptides that are labeled with light and heavy GTP probes, over 4000 transitions are included. To achieve efficient quantification, scheduled MRM, where the mass spectrometer is programmed to detect targeted peptides in predefined retention time windows, is highly desirable. To enable the accurate prediction of retention time, we utilized iRT calibration for our MRM assay by using 10 selected tryptic peptides from bovine serum albumin (BSA) as external standards. By employing the calculated iRT values of peptides derived from LC-MS/MS data acquired in the DDA mode, we were able to predict the corresponding retention time window for MRM detection.
Scheduled MRM Analysis of Differential Expression of GTP-Binding Proteins in Matched Metastatic and Primary Colon Cancer Cells.
Metastasis is the major cause of cancer mortality.35 Understanding the regulatory mechanisms underlying metastasis is very important to control cancer progression.36 To evaluate the potential functions of GTP-binding proteins in colon cancer metastasis, we applied our method for the quantitative analysis of these proteins in a pair of primary (SW480) and metastatic (SW620) colon cancer cells derived from the same patient. In the forward labeling experiment, the lysates of SW480 and SW620 cells were labeled with light and heavy GTP probes, respectively, and the reverse labeling experiment was conducted in the opposite way. After importing all data files to the Skyline library, we manually checked all the quantified peptides to ensure that the distributions of selected transitions match with those in the spectral library (Figure 3A). The correlation between detected retention time and calculated iRT was found to be excellent for the 189 quantified peptides (Figure 3B). For most quantified peptides, the peptide intensity ratios in SW620 over SW480 cells obtained from the forward experiment is consistent with the ratios obtained from the reverse labeling experiment (Figure 3C).
Figure 3.
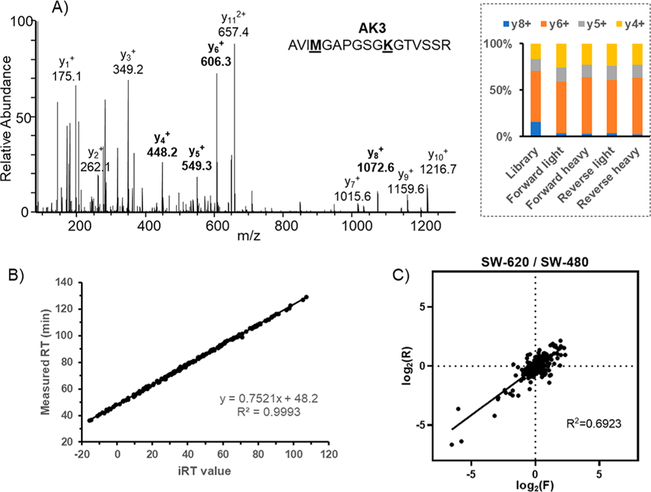
Performance of the LC-MRM-based method for the quantitative profiling of GTP-binding proteins. (A) Representative MS/MS of a probe-labeled peptide from AK3, where the distributions of relative abundances of four y ions (y4–y8) found from DDA and MRM analyses are shown in the inset; (B) Scatter plot depicting the correlation of measured retention times with calculated iRT values for all the quantified peptides; (C) Comparison of quantification results obtained from forward and reverse labeling experiments.
A total of 97 GTP-binding proteins were quantified with our MRM method (Figure 4, Table S2). Among them, 14 and 7 proteins are up- and down-regulated, respectively, by at least 1.75-fold in metastatic over primary colon cancer cells. We also validated the expression levels of several differentially expressed proteins (AK3, AK4, AGAP3, GLUD1, RHOG, SEPT10, and TGM2) with Western blot analyses, which showed the same trends as our proteomics data. The MS/MS obtained from forward and reverse labelings, the MRM traces and the quantification data from Western and proteomic analyses for these proteins are depicted in Figures 5 and S3–S9.
Figure 4.
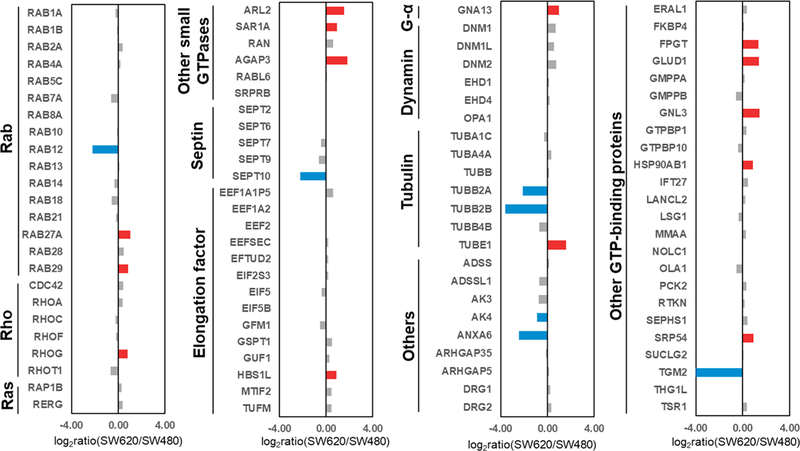
Summary of quantification results of GTP-binding proteins in SW480 and SW620 cells. Shown are the mean of results obtained from one forward and one reverse labeling experiments. Red and blue bars designate those GTP-binding proteins that were up- and down-regulated, respectively, by at least 1.75-fold in metastatic (SW620) over primary (SW480) colon cancer cells.
Figure 5.
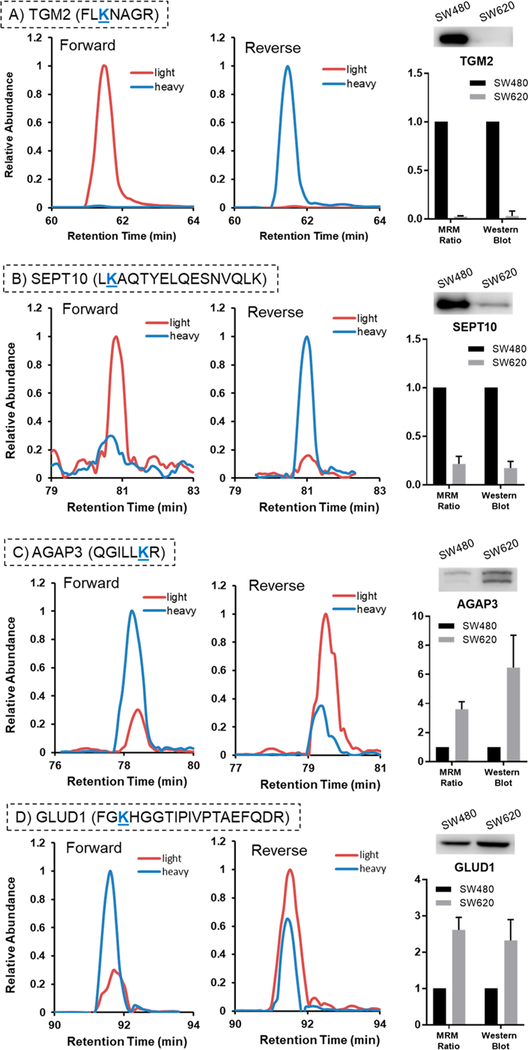
Validation of expression levels of selected GTP-binding proteins in SW480 and SW620 cells by Western blot analysis. Displayed are the selected-ion chromatograms for the light and heavy forms of the probe-labeled peptides obtained from forward and reverse isotope-labeling experiments, and the quantification results obtained from LC-MRM and Western blot analyses. The data represent the mean and standard deviation of results from LC-MRM analyses of one forward and one reverse labeling experiment and three biological replicates of Western blot experiments.
Among the differentially expressed proteins, several were previously found to modulate cancer progression. For instance, the GTP-binding protein with the most pronounced decrease in expression in the metastatic line, transglutaminase 2 (TGM2), was found to be important in drug resistance and metastasis of breast cancer,37 pancreatic cancer,38 ovarian cancer,39 and colon cancer.40 Cellura et al.41 reported that miR-19-mediated inhibition of TGM2 could enhance invasion and metastasis in colorectal cancer. Consistent with our quantification results, the expression of TGM2 was previously shown to be lower in SW620 (metastatic) cells than in SW480 (primary) cells.41 In addition, TGM2 expression is inversely correlated with invasiveness of colon cancer cells;41 upon knockdown of TGM2, the invasion of SW480 cells is enhanced, whereas its overexpression diminishes the invasiveness of SW620 cells.41
Several GTP-binding proteins that are up-regulated in metastatic relative to primary colon cancer cells were previously assessed for their functions in colon cancer metastasis. For instance, upregulation of GNL3 was found to promote colon cancer cell proliferation, migration, invasion and epithelial-mesenchymal transition via the Wnt/β-catenin signaling pathway.42 In addition, overexpression of GNA13 could promote the migration and invasion of colon cancer cells in vitro through the β-catenin pathway.43 High GLUD1 expression was also found to be associated with tumor aggressiveness, including depth of tumor invasion, lymph node and distant metastasis of colon cancer.44 Thus, our quantitative proteomic data, along with these previous studies, underscored the power of our MRM-based method for the high-throughput identification of GTP-binding proteins as novel biomarkers for prognosis and potential therapeutic targets in cancer or other diseases.
CONCLUSIONS
In this study, we developed a high-throughput targeted quantitative proteomic method for profiling GTP-binding proteins using isotope-coded GTP probes. With the desthiobiotin-GTP acyl phosphate probes, we were able to label the lysine residues in the nucleotide-binding sites of GTP-binding proteins and enrich the desthiobiotin-labeled tryptic peptides. To improve the coverage of labeled GTP-binding proteins, we constructed a Skyline library based on proteomic data acquired from 2D LC-MS/MS of desthiobiotin-labeled peptides derived from the lysates of 12 different cell lines. The library encompasses desthiobiotin-conjugated peptides that are representative of 217 GTP-binding proteins. We then introduced an isotope-tagged GABA linker to the GTP probe to facilitate quantifications of the desthiobiotin-labeled peptides derived from GTP-binding proteins. Using this MRM-based targeted proteomic method, we successfully quantified 97 GTP-binding proteins in a pair of primary/ metastatic colon cancer cell lines (SW480/SW620). More than 20 quantified GTP-binding proteins exhibited substantially different (by at least 1.5-fold) expression levels in metastatic and primary cancer cells. These proteins could be potential drivers or suppressors for colon cancer metastasis.
Compared with DDA methods, the MRM-based targeted proteomic approach offers much better sensitivity and reproducibility, rendering it a powerful approach for quantitative analysis of GTP-binding proteins. It is worth noting that this targeted proteomic method is also applicable for the quantification of GTP-binding proteins in clinical samples (e.g., biological fluids and tissues), which are more difficult to achieve with metabolic labeling. Thus, it can be envisaged that the analytical method developed here can be generally employed for assessing the alterations of GTPase signaling in cells and tissues triggered by intracellular cues and extracellular stimuli.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072), and M.H. was supported in part by an NRSA T32 Institutional Training Grant (T32 ES018827).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b03727.
Detailed experimental conditions for probe synthesis and purification (PDF).
Table S1: Shotgun proteomics analysis of GTP-binding proteins (XLSX).
Table S2: Quantitative profiling of GTP-binding proteins in SW480 and SW620 cells (XLSX).
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Lemmon MA; Freed DM; Schlessinger J; Kiyatkin A Cell 2016, 164, 1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhuin T; Roy JK Exp. Cell Res 2014, 328, 1–19. [DOI] [PubMed] [Google Scholar]
- (3).Shimobayashi M; Hall MN Nat. Rev. Mol. Cell Biol 2014, 15, 155–162. [DOI] [PubMed] [Google Scholar]
- (4).Kasahara A; Scorrano L Trends Cell Biol 2014, 24, 761–770. [DOI] [PubMed] [Google Scholar]
- (5).Filomeni G; De Zio D; Cecconi F Cell Death Differ 2015, 22, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vigil D; Cherfils J; Rossman KL; Der CJ Nat. Rev. Cancer 2010, 10, 842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Samatar AA; Poulikakos PI Nat. Rev. Drug Discovery 2014, 13, 928–942. [DOI] [PubMed] [Google Scholar]
- (8).Wittinghofer A; Vetter IR; Kornberg RD; Raetz CRH; Rothman JE; Thorner JW Annu. Rev. Biochem 2011, 80, 943–971. [DOI] [PubMed] [Google Scholar]
- (9).Lim SM; Westover KD; Ficarro SB; Harrison RA; Choi HG; Pacold ME; Carrasco M; Hunter J; Kim ND; Xie T; Sim T; Jänne PA; Meyerson M; Marto JA; Engen JR; Gray NS Angew. Chem., Int. Ed 2014, 53, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cravatt BF; Wright AT; Kozarich JW Annu. Rev. Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- (11).Korlach J; Baird DW; Heikal AA; Gee KR; Hoffman GR; Webb WW Proc. Natl. Acad. Sci. U. S. A 2004, 101, 2800–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kaneda M; Masuda S; Tomohiro T; Hatanaka Y ChemBioChem 2007, 8, 595–598. [DOI] [PubMed] [Google Scholar]
- (13).George Cisar EA; Nguyen N; Rosen HJ Am. Chem. Soc 2013, 135, 4676–4679. [DOI] [PubMed] [Google Scholar]
- (14).Patricelli MP; Szardenings AK; Liyanage M; Nomanbhoy TK; Wu M; Weissig H; Aban A; Chun D; Tanner S; Kozarich JW Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- (15).Qiu H; Wang Y Anal. Chem 2007, 79, 5547–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xiao Y; Guo L; Jiang X; Wang Y Anal. Chem 2013, 85, 3198–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Xiao Y; Ji D; Guo L; Wang Y Anal. Chem 2014, 86, 4550–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hunter JC; Gurbani D; Ficarro SB; Carrasco MA; Lim SM; Choi HG; Xie T; Marto JA; Chen Z; Gray NS; Westover KD Proc. Natl. Acad. Sci. U. S. A 2014, 111, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang M; Qi TF; Li L; Zhang G; Wang Y Cancer Res 2018, 78, 5431–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Nat. Methods 2009, 6, 359–962. [DOI] [PubMed] [Google Scholar]
- (21).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sherwood CA; Eastham A; Lee LW; Risler J; Vitek O; Martin DB J. Proteome Res 2009, 8, 4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Leipe DD; Wolf YI; Koonin EV; Aravind LJ Mol. Biol 2002, 317, 41–72. [DOI] [PubMed] [Google Scholar]
- (26).Xiao Y; Guo L; Wang Y Anal. Chem 2013, 85, 7478–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wennerberg K; Rossman KL; Der CJ J. Cell Sci 2005, 118, 843–846. [DOI] [PubMed] [Google Scholar]
- (28).Downes GB; Gautam N Genomics 1999, 62, 544–552. [DOI] [PubMed] [Google Scholar]
- (29).Vestal DJ; Jeyaratnam JA J. Interferon Cytokine Res 2011, 31, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nitta T; Takahama Y Trends Immunol 2007, 28, 58–65. [DOI] [PubMed] [Google Scholar]
- (31).Schappi JM; Krbanjevic A; Rasenick MM Biochim. Biophys. Acta, Biomembr 2014, 1838, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Praefcke GJK; McMahon HT Nat. Rev. Mol. Cell Biol 2004, 5, 133–147. [DOI] [PubMed] [Google Scholar]
- (33).Akhmetova KA; Chesnokov IN; Fedorova SA Mol. Biol 2018, 52, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maracci C; Rodnina MV Biopolymers 2016, 105, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brabletz T; Kalluri R; Nieto MA; Weinberg RA Nat. Rev. Cancer 2018, 18, 128–134. [DOI] [PubMed] [Google Scholar]
- (36).Punt CJA; Koopman M; Vermeulen L Nat. Rev. Clin. Oncol 2017, 14, 235–246. [DOI] [PubMed] [Google Scholar]
- (37).Mehta K; Fok J; Miller FR; Koul D; Sahin AA Clin. Cancer Res 2004, 10, 8068–8076. [DOI] [PubMed] [Google Scholar]
- (38).Verma A; Guha S; Diagaradjane P; Kunnumakkara AB; Sanguino AM; Lopez-Berestein G; Sood AK; Aggarwal BB; Krishnan S; Gelovani JG; Mehta K Clin. Cancer Res 2008, 14, 2476–2483. [DOI] [PubMed] [Google Scholar]
- (39).Shao MH; Cao LY; Shen CY; Satpathy M; Chelladurai B; Bigsby RM; Nakshatri H; Matei D Cancer Res 2009, 69, 9192–9201. [DOI] [PubMed] [Google Scholar]
- (40).Lentini A; Abbruzzese A; Provenzano B; Tabolacci C; Beninati S Amino Acids 2013, 44, 25–32. [DOI] [PubMed] [Google Scholar]
- (41).Cellura D; Pickard K; Quaratino S; Parker H; Strefford JC; Thomas GJ; Mitter R; Mirnezami AH; Peake NJ Mol. Cancer Res 2015, 13, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tang X; Zha L; Li H; Liao G; Huang Z; Peng X; Wang Z Oncol. Rep 2017, 38, 2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang JX; Mai SJ; Huang XX; Wang FW; Liao YJ; Lin MC; Kung HF; Zeng YX; Xie D Ann. Oncol 2014, 25, 2196–2204. [DOI] [PubMed] [Google Scholar]
- (44).Miyo M; Konno M; Nishida N; Sueda T; Noguchi K; Matsui H; Colvin H; Kawamoto K; Koseki J; Haraguchi N; Nishimura J; Hata T; Gotoh N; Matsuda F; Satoh T; Mizushima T; Shimizu H; Doki Y; Mori M; Ishii H Sci. Rep 2016, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.