

Abstract
The lymphatic system is essential for the maintenance of tissue fluid homeostasis, gastrointestinal lipid absorption, and immune trafficking. Whereas lymphatic regeneration occurs physiologically in wound healing and tissue repair, pathological lymphangiogenesis has been implicated in a number of chronic diseases such as lymphedema, atherosclerosis, and cancer. Insight into the regulatory mechanisms of lymphangiogenesis and the manner in which uncontrolled inflammation promotes lymphatic dysfunction is urgently needed to guide the development of novel therapeutics: These would be designed to reverse lymphatic dysfunction, either primary or acquired. Recent investigation has demonstrated the mechanistic role of leukotriene B4 (LTB4) in the molecular pathogenesis of lymphedema. LTB4, a product of the innate immune response, is a constituent of the eicosanoid inflammatory mediator family of molecules that promote both physiological and pathological inflammation. Here we provide an overview of lymphatic development, the pathophysiology of lymphedema, and the role of leukotrienes in lymphedema pathogenesis.
Keywords: leukotriene B4, 5-lipoxygenase, lymphedema
INTRODUCTION
The lymphatic system is characterized as a unidirectional circulatory network that complements the blood vascular circulation; it provides a conduit for reabsorption of the interstitial fluid that escapes from the arteriovenous circulation. Additional vital lymphatic functions include gastrointestinal lipid absorption and the trafficking of immune cells. In the face of heritable defects or acquired lymphatic vascular insults, the resultant lymphatic dysfunction can produce metabolic derangements, loss of normal immune responses, or the development of lymphatic vascular insufficiency, known as lymphedema. Lymphedema is a common disease state that is estimated to afflict more than 120 million individuals globally; this disease burden has enormous functional and economic implications. At present, there are few, if any, therapeutic options to minimize the impact of this pathology. Molecular insight into the pathogenesis of lymphedema is necessary to facilitate the identification of novel therapeutic targets for this disease that currently lacks a meaningful pharmacology.
Recent investigation of a murine model of acquired lymphedema has disclosed the central role of leukotriene B4 (LTB4), an inflammatory lipid mediator, in the pathogenesis and maintenance of the disease, thereby implicating tissue inflammation as the mechanistic platform for the development of acquired lymphedema. This review, accordingly, provides an overview of leukotriene biology, lymphatic vascular development, lymphatic function, and lymphedema pathophysiology. This facilitates an exploration of the role of LTB4 in experimental murine lymphedema and permits a discussion of emerging novel therapeutic targets for human lymphedema.
LEUKOTRIENE BIOLOGY
Leukotrienes are a collection of short-lived lipid mediators produced primarily by proinflam-matory immune cells, such as macrophages, neutrophils, eosinophils, mast cells, and dendritic cells. In response to diverse immune and inflammatory stimuli, these lipid mediators elicit potent inflammatory responses through binding to, and activation of, their cognate G protein-coupled receptors.
Synthesis of Leukotrienes
Biosynthesis of leukotrienes is initiated by cytosolic phospholipase A2 (cPLA2)-mediated membrane phospholipid catalysis into arachidonic acid (AA), which is then guided to the enzyme 5-lipoxygenase (5-LO) by 5-LO-activating protein (FLAP) and subsequently converted to leukotriene A4 (LTA4). LTA4 can be further metabolized to form LTB4 in the nucleus or cytosol by LTA4 hydrolase (LTA4H) or LTC4 in the cytosol by LTC4 synthase. LTB4 and LTC4 are then transported across the cell membrane by multidrug resistance-associated protein 4 (MRP4) or 1 (MRP1) transporter proteins, respectively. LTC4 can also be further hydrolyzed to LTD4 and LTE4 in a sequential fashion (1). Whereas LTB4 elicits its biological effects through its receptors, BLT1 and BLT2, LTC4, LTD4, and LTE4 act on CysLT1 and CysLT2 (Figure 1). Nonleukocytes generally do not contain the complete enzymatic machinery required for leukotriene production. However, those cells, including endothelial cells, are able to absorb LTA4 produced by neighboring immune cells and convert this molecule to LTB4; this mechanism is called transcellular biosynthesis.
Figure 1.
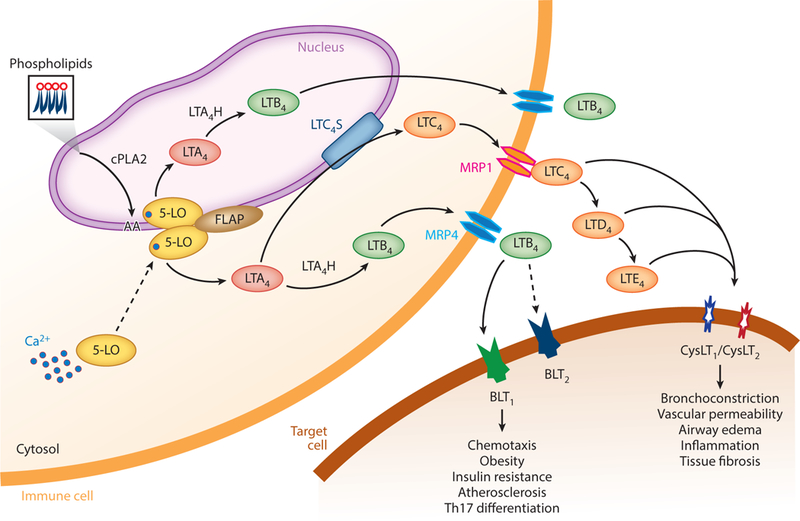
Overview of the synthesis and actions of leukotrienes. Ca2+-activated 5-LO translocates to either the inner or outer nuclear membrane and converts arachidonic acid to LTA4 under the influence of FLAP. LTA4 is further metabolized to LTB4 or LTC4. These are transported out of the cell by MRP4 or MRP1. LTC4 can be sequentially metabolized into LTD4 and LTE4. The molecules bind to BLT or CysLT receptors and elicit biological or pathological functions. Abbreviations: 5-LO, 5-lipoxygenase; BLT, LTB4 receptor; cPLA2, cytosolic phospholipase A2; CysLT, cysteinyl LT receptor; FLAP, 5-LO-activating protein; LT, leukotriene; LTA4H, LTA4 hydrolase; LTB4, leukotriene B4; LTC4S, LTC4 synthase; MRP, multidrug resistance-associated protein; Th17, T helper 17.
Leukotriene biosynthesis can be regulated at multiple steps, including cPLA2α-mediated AA liberation, 5-LO/FLAP-mediated LTA4 production, LTA4 hydrolase-mediated LTB4 production, and LTC4 synthase-mediated LTC4 synthesis.
5-LO and FLAP.
Human 5-LO is a large gene (71.9 kb) located on chromosome 10. The gene contains 14 exons, with expression restricted chiefly to leukocytes. The mammalian 5-LO gene, including that in the rat, mouse, and hamster, shares more than 90% identity with its human counterpart (2). Cellular 5-LO activity is determined by its localization. In resting cells, 5-LO is located in either the cytoplasm or nucleoplasm. Depending on the cell type, it shuttles between these sites by regulated nuclear import or export processes (1). Upon stimulation, it relocates to the outer or inner membrane of the nucleus (3). This process is thought to be regulated mainly by levels of intracellular calcium as well as the phosphorylation status of the protein. Translocation of 5-LO from nucleoplasm to the inner nuclear membrane appears to maximize the production of LTB4 (4). In the absence of calcium, 5-LO has minimal catalytic activity (5). Calcium is able to increase 5-LO activity through stimulating oxygenation and dehydration reactions (6). The main phosphorylation sites of 5-LO include Ser-271, Ser-523, and Ser-663. Ser-271 appears to be the most important phosphorylation site; located within the nuclear export sequence, this presumably facilitates 5-LO translocation from nucleoplasm to the inner nuclear membrane (4, 7, 8). By contrast, phosphorylation of Ser-523 by protein kinase A appears to suppress 5-LO activity by directing its cytoplasmic localization (9). Researchers demonstrated that FLAP was required for ionophore-induced leukotriene synthesis through its effect as an AA transfer protein (10, 11). FLAP expression is primarily regulated at the transcriptional level (12). Despite the substantial difference between the promoter regions for 5-LO and FLAP, gene expression regulation appears to be parallel (12), which presents a potential mechanism for facilitating leukotriene synthesis throughout the pathway.
LTA4 hydrolase and LTC4 synthase.
LTA4H catalyzes the final step in the LTB4 biosynthesis pathway, utilizing its epoxide hydrolase activity (13). An in vitro assay system has shown that LTA4H is the rate-limiting enzyme for LTB4 formation (14). Therefore, targeting this enzyme might efficiently block LTB4 synthesis. LTA4H is present in various cell types that lack 5-LO activity, such as endothelial cells and erythrocytes. This uncoupled expression of LTA4H and 5-LO provides the mechanistic basis for transcellular biosynthesis of LTB4 (13, 15). Additionally, LTA4H has been identified as a member of the family of zinc metalloproteases, which possess peptide-cleaving activity (16).
LTC4 synthase is the key enzyme in the cysteinyl leukotriene biosynthesis pathway, where it catalyzes the conjugation of LTA4 with glutathione to form LTC4. LTC4 synthase is an integral molecule that is present in both the endoplasmic reticular membrane and the outer nuclear membrane (17). Expression of LTC4 synthase is mainly seen in cells of myeloid origin, such as macrophages, basophils, eosinophils, and mast cells (18). Resembling LTA4H, LTC4 synthase is also expressed in platelets where it can catalyze LTC4 formation by the transcellular mechanism (19).
Leukotrienes in Physiology and Diseases
Leukotrienes elicit physiological and pathophysiological roles through binding to their cognate G protein-coupled seven transmembrane domain receptors, BLT1 or BLT2 for LTB4, and to cys-teinyl leukotriene receptors, CysLT1 and CysLT2 for LTC4, LTD4, and LTE4. CysLT1 is chiefly expressed in the lung smooth muscle cells, lung interstitial macrophages, and the spleen (20); it is commonly recognized to induce bronchoconstriction, mucus production, and airway edema. CysLT2 is also highly expressed in the spleen and peripheral blood leukocytes. It is uniquely expressed in the heart, adrenal gland, and brain and executes important roles in promoting inflammation, vascular permeability, and tissue fibrosis (21–23).
The biological function of LTB4 is mediated primarily by the BLT1 receptor, which has an affinity that is approximately 20-fold higher than that of BLT2 (1, 24). LTB4 is a strong chemoat-tractant and activator of leukocytes and is one of the most potent lipid chemotactic factors for neutrophils. Neutrophils can respond to the bacterial-produced peptide, N-formylmethionine-leucyl-phenylalanine, producing high levels of LTB4 to recruit more neutrophils to the inflammatory site. This maintains the active inflammatory status until the infection is cleared (25). LTB4 also mediates neutrophil swarming once these cells have migrated out of the blood vessels (26). LTB4-mediated monocyte/macrophage recruitment has been linked to a number of chronic diseases, such as obesity, insulin resistance and type 2 diabetes, and atherosclerosis (27–29). The LTB4/BLT1 axis has also been shown to mediate CD8+ cytotoxic T lymphocyte recruitment into inflamed tissue (30). Additionally, LTB4 appears to promote Th17 cell differentiation as well as its migration (31, 32). Therefore, LTB4, acting through BLT1 receptor, regulates migration and activity of cells of both innate and adaptive immunity and plays essential roles not only in physiological defense against infection but also in pathogenesis of a number of chronic diseases.
BIOLOGY OF THE LYMPHATIC VASCULAR SYSTEM
Structure of the Lymphatic Vascular System
The lymphatic vascular system is essential for tissue immune function, tissue fluid homeostasis, and dietary fat absorption. It is a unidirectional circulatory system that begins as blind-ended lymphatic capillaries comprising a single layer of lymphatic endothelial cells (LECs) that are interconnected by discontinuous junctional structures known as buttons. The buttons express abundant levels of the adherens junction protein, VE-cadherin, and tight or gap junction proteins, including claudin, ZO-1, endothelial cell selective adhesion molecule, connexin, and occludin (33). The initial lymphatic LECs are not ensheathed by pericytes or smooth muscle cells and have minimal or no basement membrane coverage. They are connected to the surrounding elastic fibers of the extracellular matrix (ECM) through the anchoring filaments that control interstitial fluid and cell uptake into the initial lymphatics (33, 34). The flaps between the buttons are overlapping and are thought to be able to open and close in response to high interstitial fluid pressure and to facilitate fluid reabsorption (35). Fluid and cells can also enter the lymphatic vessels through a transcellular route (36). Lymphatic capillaries converge into larger collecting vessels. LECs of the collecting vessels connect to one another with continuous junctional structures known as zippers (33). The collecting vessels are invested with smooth muscle cells that provide the pumping force for fluid movement, and they also have intraluminal valves to allow unidirectional flow of lymph (37). Transitional lymphatic vessels between the capillary and collecting vessels are referred to as precollectors, which are characterized by partial smooth muscle cell coverage (38). The collecting lymphatic vessels provide a conduit for lymph through chains of lymph nodes before converging into the thoracic duct(s), through which the lymph is transported into the subclavian vein of the blood circulatory system (Figure 2).
Figure 2.
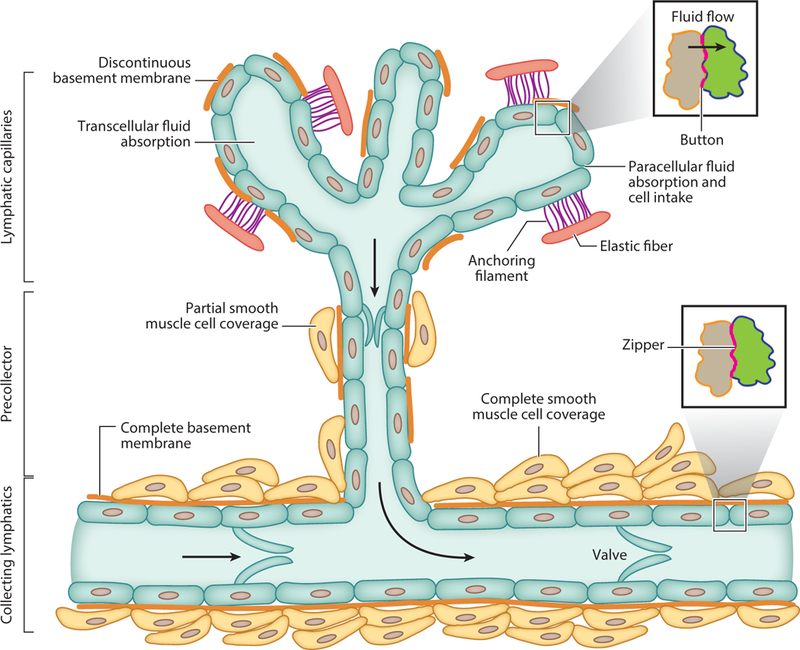
Schematic diagram of the lymphatic vascular tree. Lymphatic capillaries are blind ended. Capillary lymphatic endothelial cells (LECs) are only partially covered by basement membrane (BM). Button structures locate at the capillary LECs, and LECs anchor to the elastic components of the extracellular matrix by anchoring filaments. This facilitates interstitial fluid and cellular entry into the lymphatic capillaries. Interstitial fluid and cells can enter the lymphatic capillary through both paracellular and transcellular routes. Lymphatic capillaries converge into precollectors, which also have incomplete BM and partial smooth muscle cell (SMC) coverage. Precollectors further converge into collecting lymphatics, which have complete BM and SMC layers. Lymphatic valves allow only unidirectional lymph flow. Zippers locate among the collector LECs.
Development of the Lymphatic Vascular System
The last decade has witnessed tremendous progress in deciphering developmental programs of the lymphatic vascular system. Signaling pathways orchestrating tissue or organ development often reactivate to promote the restoration of tissue hemostasis following injury. Furthermore, developmental pathways may remain active in tissue maintenance in adults, and lymphatic dysfunction may also arise from developmental insufficiency. In this section, we summarize key molecular pathways involved in developmental processes of the lymphatic vascular system.
Lymphatic endothelial cell specification.
Despite recognition of the lymphatic vascular system as a circulatory system hundreds of years ago, in-depth study of the lymphatic vasculature has been hampered by the lack of molecular markers that can be used to discern LECs from the blood endothelial cells (39). It was more than a century ago when the lymphatic vasculature was demonstrated to be derived from the embryonic venous anlage (40). This hypothesis was further confirmed nearly 100 years later with the identification and characterization of the role of lymphatic-specific markers, such as vascular endothelial growth factor 3 [VEGFR3 (FLT4)] and prospero homeobox 1 (Proxl) in the formation of the lymphatic vascular system during specified stages of embryonic development (41, 42). Because VEGFR3 knockout also leads to defective blood vascular development, its specific roles in early lymphatic vascular development were not readily identified (43) until it was demonstrated that VEGF-C, a ligand of VEGFR3, is essential for initial lymphatic development (44). Although not required for the initial LEC lineage commitment from blood vascular endothelial cells, VEGF-C is required for LEC sprouting. VEGF-C haploinsufficiency leads to cutaneous lymphatic hypoplasia and lymphedema. Similarly, VEGF-C overexpression leads to lymphatic, but not blood vascular, hyperplasia and enlargement (45). In summary, these observations underscore the essential role of the VEGF-C/VEGFR3 axis in early lymphatic vascular development.
Prox1 is designated as a master regulator of lymphatic vessel development (39). Prox1 is expressed in a subset of cardinal vein endothelial cells. Those Prox1+ cells bud off to form rudimentary lymphatic vessels, known as jugular lymph sacs (42). No LEC budding can be observed during the very early stages of development in Prox1-null mice. In the putative budding region of the cardinal vein, there is no identifiable expression of other lymphatic markers, such as VEGFR3 or LYVE-1 (46), suggesting that Prox1 is a key factor regulating LEC specification and differentiation. Moreover, it has been shown that Proxl has the capacity to induce lymphatic reprogramming from differentiated blood endothelial cells, and Proxl expression is required to maintain the LEC phenotype (47, 48), highlighting the role of Proxl in the maintenance of the postnatal lymphatic vasculature. Notch signaling suppresses early LEC specification by suppressing Proxl expression, presumably through inhibiting chicken ovalbumin upstream promoter-transcription factor 2 expression (49). The fact that loss of Notch activity leads to excessive Prox1+ LEC differentiation from cardinal vein and lymphatic overgrowth suggests that tightly regulated Notch signaling is required for normal lymphatic vessel patterning during early stages of development (49).
Lymphatic tree expansion.
Following specification, mature LECs form lymph sacs and a lymphatic plexus. They subsequently give rise to the mature lymphatic structures, including the collecting vessels and capillaries. This hierarchical lymphatic tree develops mainly through lymphan- giogenesis, a means of sprouting growth from the preexisting structures (40). VEGF-C/VEGFR3 signaling is absolutely required for lymphatic sprouting from the lymph sac (44, 50). Collagen- and calcium-binding epidermal growth factor (EGF) domain 1 (CCBE1) protein is essential for lymphatic tree expansion: Promotion of VEGF-C/VEGFR3 signaling occurs through enhanced VEGF-C cleavage into its active form by the ADAM metallopeptidase with thrombospondin type 1 motif 3 metalloprotease (51–53). The axon guidance receptor, neuropilin 2 (NRP2), is also involved in lymphatic development. NRP2 mutant mice display lymphatic capillary hypoplasia with normal blood vasculature (54). Mechanistically, NRP2 acts as a coreceptor for VEGFR3 and may coordinate an enhanced affinity of VEGF-C for its receptor to provide maximal sensing of ligand gradients. As in blood vascular endothelial cells, Notch pathway activation in the LEC also determines tip-stalk cell specification; LECs with increased Notch activity adopt the stalk cell phenotype and presumably contribute to lymphatic vessel stabilization. Consistent with in vitro data, blockade of Notch signaling promotes lymphangiogenesis in vivo, but whether these excessively sprouted lymphatic vessels are functional is not known (55).
Lymphatic maturation.
Following the establishment of the initial lymphatic tree, the vessels undergo maturation to form the hierarchical lymphatic vascular system, including the capillaries, precollectors, and collecting vessels. One change that characterizes lymphatic capillary maturation appears to be the transition of the LEC junction from zippers to buttons (56). The maturation of collecting lymphatics is characterized by valve formation, mural cell recruitment, and basement membrane deposition (40). Formation of the lymphatic valves is a crucial step for lymphatic vascular maturation. Forkhead box protein C2 (FOXC2) has been identified as one of the major molecular mediators of this process (57). FOXC2 is not required for initial LEC specification, but expression of this gene is absolutely essential for patterning of the lymphatics during later developmental stages (58). FOXC2 deficiency leads to agenesis of the lymphatic valves; it also leads to abnormal mural cell and basement coverage of the lymphatic capillaries through upregulation of platelet-derived growth factor subunit B expression. It appears that VEGFR3 and FOXC2 coordinate this patterning process, with FOXC2 acting downstream to VEGFR3 signaling (58). It was later shown that FOXC2 interacts with the nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) and that this coordination is not only required for lymphatic valve formation during development (59) but is also important for its maintenance in adulthood (60). Further mechanistic analysis suggested that FOXC2 maintains lymphatic integrity, especially around the valve area, through coordination ofcell-cell junction maturation and shear stress responses. This presumably occurs through cross talk between FOXC2 and a Hippo pathway transcriptional coactivator, a transcriptional coactivator with PDZ-binding motif (61). The gap junction protein alpha-4 (Cx37) is also essential to valve formation, and its expression is regulated by oscillatory fluid shear stress in a Proxl- and FOXC2-dependent manner (60). Lastly, lymph flow also regulates collecting vessel maturation through a mechanism that involves induction of maturation-related genes such as GATA2 and FOXC2 (62).
Functions of the Lymphatic Vascular System
The lymphatic vascular system is patterned to function as a unidirectional circulatory network that facilitates tissue fluid reabsorption and transportation. There is also evidence that lymphatic vessels actively participate in dietary lipid absorption. Furthermore, the lymph fluid passes through chains of drainage lymph nodes before it is transported back to the systemic circulation, providing an important anatomic basis for the immune regulatory functions of the lymphatic system.
Tissue fluid balance and dietary lipid absorption.
Although it is traditionally thought that approximately 90% of the capillary ultrafiltrate and associated plasma proteins is reabsorbed at the venous end of the capillary, more recent evidence, to the contrary, supports the view that the bulk of the tissue fluid is taken up by the lymphatic vasculature. There is minimal reabsorption into the venules (63), thus underscoring the importance of the lymphatic vasculature in the maintenance of tissue fluid homeostasis (64). Whereas the unique LEC button-like junction structure, with its tethering to ECM through anchoring filaments, facilitates passive paracellular absorption of the interstitial fluid, macromolecules, and immune cells, LECs also have the capacity to actively transport cholesterol from the interstitium, a process that involves the scavenger receptor class B member 1 (65).
Lymphatic capillaries, located at the center of each of the intestinal villi, known as lacteals, serve as an essential conduit for the drainage of absorbed dietary lipids and fat-soluble vitamins. The mesenteric lymphatic tree is the main conduit that transports those molecules into the systemic circulation (66). Recent intravital imaging studies have demonstrated that the lacteal is not simply acting as a passive conduit; instead, it is able to respond to autonomic nerve stimulation to the surrounding smooth muscle cells and actively transport the absorbed lipid (67). It was also recently revealed that the lacteal undergoes constant remodeling and regeneration: Both VEGF-C and Delta-like protein 4 appear to be essential to the maintenance of its structural and functional integrity in adult mice (68, 69).
Immune regulatory function.
The lymphatic vessels are critical conduits for the trafficking of leukocytes and soluble antigens from the peripheral tissue to the draining lymph nodes, where either immune priming or tolerance can take place, depending on the type of transported antigens and the immune cells (70). Dendritic cells (DCs) are the most well-studied cell type in the context of entry into the lymphatic capillary. Under basal conditions, the DC does not need to undergo adhesion to the ECM component integrin to allow its movement through the matrix before its entry into the lymphatic vessels (71). However, during inflammation, with a loosened collagen network (72), DC migration requires adhesion molecules such as ICAM-1 and VCAM-1 (73, 74), and T cells require αv integrin for movement through the structurally altered ECM network (72). Interestingly, the point of entry for immune cells is often located in areas of the lymph capillary with sparse basement membrane, known as portals (75). Following the migration through the ECM, entry of the interstitial immune cell into the lymphatic capillary occurs, guided by the LEC expression of chemokines (70). In leukocyte homing, the best-studied chemokines are chemokine ligands 21 and 19 (CCL21 and CCL19), which guide and recruit immune cells that express the cognate receptor C-C chemokine receptor type 7 (CCR7). Mice lacking CCR7 cannot mount an adaptive immune response due to impaired DC and T cell homing to lymph nodes (76). LEC-produced semaphorin-3A engages DC-expressed plexin-Al, which promotes the contraction of actomyosin expressed at the trailing edge of the migrating DC and facilitates its entrance into the lymph capillary (77). LYVE-1 is a known hyaluronan receptor, but it does not bind tissue high molecular weight hyaluronic acid (HA) (78). However, it does bind to the cell wall component HA of group A streptococci (79). Therefore, LYVE-1 may serve as a microbial recognition receptor rather than a receptor for degraded tissue HA, as initially thought (70).
An interesting point is that chemotactic guidance gradients, as established by chemokines, are likely able to overcome the challenge of lymph flow reduction; therefore, cellular transport is minimally affected by interstitial flow variation. By contrast, absorption of molecules such as antigens, peptides, and cytokines is sensitive to the changing velocity of lymph flow (70), suggesting that lymph stasis, or slower lymph flow, would have a greater impact on molecular than cellular transport. Beyond clearing inflammatory mediators through their conduit function, the lymphatic vessels also participate in inflammation resolution by a direct LEC-mediated mechanism, namely, the expression by capillary LECs of high levels of nonsignaling G protein-coupled receptor D6, which scavenges a number of proinflammatory cytokines, including CCL2, CCL3, CCL4, and CCL5 (80).
A well-accepted mechanism that induces peripheral tolerance is through cross-presentation of tissue-derived proteins by tissue-resident DCs to self-reactive T cells that have escaped central tolerance, leading to anergy or deletion (81). Recent advances in the field have shown that LECs, as well as the stromal cells of lymph nodes, also contribute to peripheral tolerance (70, 82). Lymph node LECs can express self-antigens and drive the deletion of self-reactive major histocompatibility complex-I (MHC-I)-restricted CD8+ T cells (82). Endocytosed soluble antigen can also be presented by programmed death ligand 1 (PDLl)-expressing LECs to CD8+ T cells and induce deletional tolerance (83). LECs also express MHC-II, but because they do not express human leukocyte antigen DM for peptide exchange onto MHC-II, they are not able to present antigen to CD4+ T cells directly. However, CD4+ T cell tolerance to LEC-expressed antigen still occurs through a process that involves DC binding of antigen from the lymphatics (84). Another study suggested that LECs can serve as antigen-presenting cells by acquiring the MHC-II- peptide complex from DCs (85). These studies suggest that LECs are able to induce tolerance of CD4+ T cells, but this requires the help of DCs.
LYMPHEDEMA
The lymphatic vascular system is increasingly recognized to play an important role in a number of pathological conditions, such as tumor metastasis (86), chronic inflammation (87), metabolic diseases (69, 88), and cardiovascular diseases, such as atherosclerosis and myocardial infarction (40). Lymphedema, characterized by excessive accumulation of interstitial tissue fluid as a result of impaired fluid transport through this vasculature, is the major form of lymphatic dysfunction. Based on etiology, lymphedema can be classified as primary or secondary lymphedema. Secondary lymphedema is the most common form of lymphedema, afflicting more than 120 million patients worldwide. We focus on this disease, with an emphasis on the current understanding of its pathogenesis and on emerging novel therapeutic targets for the disease, including the newly identified role of the 5-LO/LTB4 pathway in its pathogenesis.
Primary Lymphedema
Primary lymphedema results from heritable defects in lymphatic vascular development or function. The majority of these cases still lack an identified mechanism. The first mutation to be identified to cause inherited lymphedema involves the flt4 gene that encodes VEGFR3. Heterozygous mutation of flt4 within the tyrosine kinase domain leads to receptor dysfunction and causes congenital hypoplastic lymphedema, known as Nonne-Milroy lymphedema (89). Mutation of the VEGF-C gene also causes a primary Milroy-like lymphedema that essentially has the same clinical manifestations as those of Nonne-Milroy lymphedema (90). Mutations of a number of other genes that are involved in the VEGF-C/VEGFR3 signaling pathway, such as CCBE1, FOXC2, and PTPN14, have also been shown to cause primary lymphedema (40), highlighting again that the VEGF-C/VEGFR3 axis is essential to lymphatic vessel development. Mutations of upstream transcription factors involved in LEC specification, such as Sox-18 and GATA2, have also been shown to cause primary lymphedema (91, 92). Other genes that have been linked to primary lymphedema include GJC2, GJA1, PIEZO1, HGF, KIF11, PTPN11, KRAS, SOS1, RAF1, IKBKG, RASA1, and HRAS (40).
Secondary Lymphedema
Secondary lymphedema is a result of obstruction or disruption of the lymphatic vascular system, which occurs as a consequence of infection, malignancies, or trauma. The resulting lymphatic insufficiency leads to interstitial fluid accumulation distal to the disrupted lymphatic structure.
Causes.
The most common form of secondary lymphedema worldwide is lymphatic filariasis, a condition caused by lymphatic vessel infiltration and obstruction by the nematode parasite, predominantly Wuchereria bancrofti. The estimated incidence of filariasis ranges between 140 and 200 million people, with those afflicted individuals residing primarily in third world countries (93).
In the United States, secondary lymphedema results mainly from surgical and radiation therapies, either combined or individually administered for malignant conditions. These include not only breast cancer, but also other cancers, such as prostate, testicular, uterine, and ovarian as well as lymphoma, melanoma, and various head and neck tumors (94). Radiation appears to promote surgery-induced lymphedema through promoting tissue fibrosis (95). In fact, extensive injury to the superficial (i.e., dermal) lymphatic system is sufficient to cause lymphedema. Infection, such as cellulitis, has also been shown to be a risk factor for lymphedema development following surgery of certain tumors (96, 97). One of the identified systemic risk factors for lymphedema development in at-risk patient cohorts is obesity (98). Although the mechanism of this obesity risk is not well understood, it is recognized that obesity alone can produce impaired interstitial fluid transport, decreased immune cell migration, decreased pumping ability of the collecting lymphatic vessels, and abnormal lymph node structure (99, 100). These factors predispose obese individuals to lymphedema (101), suggesting that obesity may act as another “hit” to promote lymphedema development in cancer survivors. In parallel to illustrating the role of genetic mutations in the etiology of primary lymphedema, some studies have shown that genetic mutation may also predispose cancer patients to develop lymphedema following surgical and/or radiation treatment. For example, mutations in the hepatocyte growth factor (HGF)/tyrosine-protein kinase Met (MET) pathway and single nucleotide polymorphisms within VEGFR2, VEGFR3, and retinoic acid receptor-related orphan receptor C are associated with an increased likelihood of developing lymphedema (102, 103). Taken together, these observations suggest that multiple factors, both genetic and nongenetic, are involved in promoting the development of secondary lymphedema in those at risk for the development of the disease.
Pathophysiology.
Interstitial fluid accumulation often occurs early during lymphedema development. Accumulated fluid significantly impacts cellular behavior within the affected region and induces subsequent pathological changes, such as immune cell infiltration and activation of the inflammatory cascade, adipose accumulation, and tissue fibrosis. It is hypothesized that early intervention to control tissue fluid accumulation will efficaciously prevent the development of uncontrolled chronic inflammation and lymphedema progression.
Inflammation in filarial lymphedema.
Both innate and adaptive immunity appear to be involved in the development of chronic inflammation following initial fluid accumulation. In human filar-iasis, the pathogenic worms and their products have direct effects on both LECs and cells of the immune system. Activated immune responses elicit the production of proinflammatory molecules, which also impact LECs. These factors act together to promote the development of lymphedema (104). Filarial antigens are able to promote LEC proliferation and lymphangiogenesis. However, they also induce expression of tight junction proteins and likely restrict absorptive capability of the lymphatics through diminishing LEC permeability (105). Infection of athymic mice with Brugia malayi induces lymphedema and increases lymph fluid levels of proinflammatory molecules, such as interleukin (IL)-1, IL-6, tumor necrosis factor-alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (106), suggesting that the cells of the innate immune system, such as macrophages, are likely essential to initiate the development of lymphedema.
The importance of proinflammatory cytokines in promoting human filariatic lymphedema can be further inferred from a series of studies that demonstrate increased circulating levels of tumor TNF-α, IL-6, endothelin-1, IL-2, IL-8, macrophage inflammatory protein (MIP)-1α, MIP-1β, monocyte chemoattractant protein 1, thymus- and activation-regulated chemokine, interferon gamma-induced protein (IP)-10, and soluble TNF receptor (107). Reconstitution with spleen cells in B. malayi-infected severe combined immunodeficiency mice that have already developed lymphangiectasia promotes disease progression (108), suggesting that adaptive immunity is required for lymphedema progression. Several subsets of T cells have been implicated in filarial pathology. CD8+ T cells were found increased in both the peripheral blood (109) and tissue (110). Examination of the T cell receptor Vβ repertoire consistently suggests the existence of distinct and limited T cell populations in the affected tissues (111). A more recent study found augmented Th1 andTh17 activation, accompanied by decreased regulatoryT cell differentiation in peripheral blood mononuclear cells (PBMCs) isolated from patients with filarial lymphedema, but not those from asymptomatic infected individuals (112), which strongly implies that uncontrolled Th1 and Th17 immune responses may act as a driving force for the overt development of lymphedema following filarial infection. However, another study showed that neutralization of regulatory T (Treg) cells promotes Th17 activity, diminishes M2 macrophages, and reduces parasite burden (113), suggesting that Th17 cells may also elicit active immune response against filarial worms. The Th2 subtype, on the other hand, appears to be more frequent in PBMCs isolated from asymptomatic infected individuals than in those from filarial lymphedema (114). Lastly, antigen-specific IL-9 and IL-10-coexpressing Th9 cells have also been implicated in chronic filarial lymphedema (115). In summary, development of filarial lymphedema requires cells of innate immunity such as the macrophage and cells of adaptive immunity, such as CD4+ and CD8+ T cells.
Inflammation in postsurgical lymphedema.
Pathological manifestations in nonfilarial acquired lymphedema also include tissue fluid accumulation, inflammatory changes, fibrosis of the lymphatic vessels and surrounding tissues, and adipose tissue deposition. However, immune responses in filarial and postsurgical lymphedema appear to be different. A comparison of skin tissue from filarial and postsurgical lymphedema discloses much less infiltration of inflammatory cells such as the CD68+ macrophage, CD4+, and CD8+ T cells in the latter (116), suggesting a much heightened inflammatory response in the filarial lymphedema lesion. In mouse tail and axillary lymphedema models, affected skin tissues do display increased CD4+ T cells, macrophages, neutrophils, and DC infiltration. Depletion of CD4+ T cells, but not CD25+ Treg cells, was able to significantly decrease lymphedema, inflammation, fibrosis, and adipose tissue deposition (117), suggesting that the non-Treg population of CD4+ T cells is essential for promoting lymphedema pathology. In concert with this finding, the Th2 response has been shown to be involved in the development of lymph stasis-induced fibrosis through promotion of transforming growth factor (TGF)-β signaling (118). Blocking Th2 differentiation by IL-4 or IL-13 antibody could prevent as well as reverse lymphatic fibrosis and improve lymphatic function (119). Th2 cytokines IL-4 and IL-13 were further shown to suppress LEC LYVE-1 and Prox1 expression as well as to diminish their survival, proliferation, migration, and tube formation, thereby inhibiting lymphangiogenesis (120, 121). Additionally, adoptive transfer of Treg cells was able to improve the major histologic hallmarks of lymphedema pathology, including edema, inflammation, and fibrosis (122). In an axillary lymph node dissection model of lymphedema, both Th1 and Th17 cells were shown to be associated with edema development and tissue fibrosis (123). Taken together, Th1, Th2, and Th17 cells all appear to play influential roles in promoting postsurgical lymphedema (Figure 3).
Figure 3.
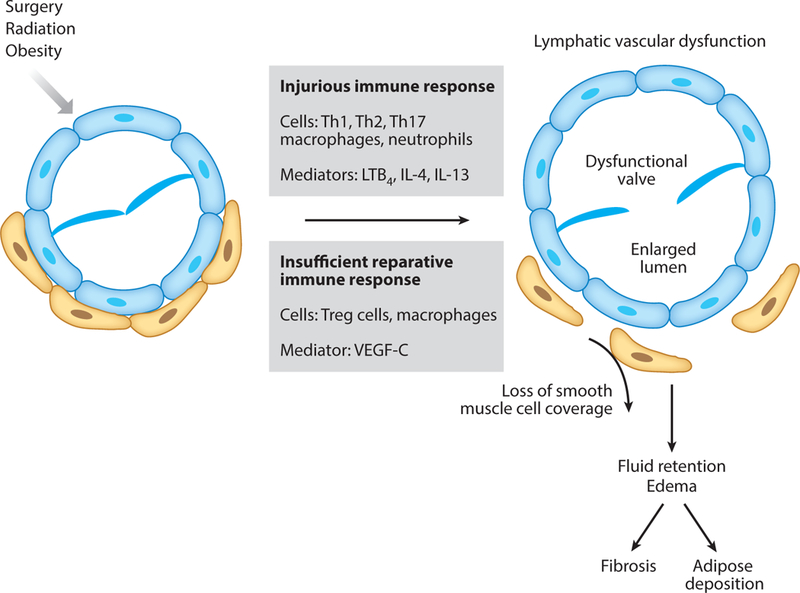
Pathophysiology of acquired lymphedema. Lymphatic injury, caused by surgery, radiation, or obesity, induces an immune response that includes Th1-, Th2-, and Th17-mediated T cell immunity, along with macrophage- and neutrophil-mediated innate immunity. Insufficient regulatory T cell (Treg) activity and insufficient production of lymphatic reparative factors, such as VEGF-C, lead to lymphatic vascular dysfunction, which includes lymphatic vessel lumen enlargement, valve dysfunction, and loss of smooth muscle cell coverage. Prolonged lymphatic vascular dysfunction induces interstitial fluid retention and tissue edema, followed by tissue fibrosis and adipose deposition. Abbreviations: IL, interleukin; LTB4, leukotriene B4;Th1/2/17,T helper 1/2/17; VEGF-C, vascular endothelial growth factor-C.
Macrophages are known to be associated with a number of pathologies, including tissue repair, chronic inflammation, cancer, and infection (124, 125). Macrophages were found to be present in human skin lesions of postsurgical lymphedema (126). In mouse models of lymphedema, macrophages were also shown to be present during disease development (127) and to preferentially differentiate into M2-type macrophages (126). Although macrophages promote lymphedema development during acute phases (123) of edema, depletion of macrophages in chronic lymphedema was not able to diminish swelling, adipose tissue deposition, and overall inflammation, but they actually promoted tissue fibrosis. The enhanced tissue fibrosis response following macrophage depletion may result from increased CD4+ cell accumulation and skewed Th2 differentiation (126). Another study showed that Toll-like receptor deficiency hindered lymphangiogenesis and lymphatic vascular repair in a mouse tail lymphedema model, which is likely a result of decreased macrophage infiltration as well as the diminished capacity of those cells to produce VEGF-C (128). Taken together, these studies suggest that macrophages might play pleiotropic roles during various stages of lymphedema pathogenesis (Figure 3).
LTB4 in postsurgical lymphedema.
In line with the concept that inflammation is a principal factor that promotes lymphedema progression (129), we have previously demonstrated that ketoprofen, a nonsteroidal anti-inflammatory drug (NSAID), reversed edema and the associated alterations in cutaneous histology (130). On the other hand, TNF-α inhibition exacerbated tissue edema (130), implying that beneficial inflammatory responses also exist during lymphedema disease evolution. TNF-α may indirectly promote lymphangiogenesis because LECs do not express TNF-R1 (131). These observations suggest that identification of the deleterious and beneficial inflammatory molecular pathways will be essential to a better understanding of lymphedema pathophysiology and to the identification of novel therapeutic targets. Ketoprofen is known to have inhibitory effects on two enzymes involved in AA metabolism: cyclooxygenase (COX) and 5-LO. Dual pathway inhibition by ketoprofen is a pharmacological feature unique within NSAIDs; this attribute prompted us to examine the roles of prostaglandins and leukotrienes in postsurgical lymphedema (133). We found that the COX inhibitor, ibuprofen, was not only incapable of reversing lymphedema, but it actually exacerbated the edema, suggesting a potential protective role of COX-generated prostaglandins in lymphedema pathogenesis. Decreased circulating levels of prostaglandin E2 (PGE2) were consistently found in both human and mouse lymphedema, suggesting that PGE2 may have the capacity to contribute to lymphedema resolution. Subsequent series of investigations with pharmacological inhibitors and genetic manipulation, all targeting the 5-LO pathway, led to the conclusion that blockade of LTB4, but not the cysteinyl leukotriene signaling pathway, is effective in reversing edema and improving lymphatic vascular function. Although the macrophage is a cell type that produces large amounts of LTB4 (1), macrophage depletion was not able to halt disease progression, albeit with partial beneficial effects during the early phases of edema progression. There are various potential explanations for this observation: LTB4 produced by other immune cells, such as neutrophils, may overwhelm this effect and continue to promote lymphedema progression, or macrophages may produce other beneficial factors that contribute to lymphedema resolution. Nevertheless, we consistently observed that LTB4 promotes lymphedema development (Figure 3).
We further explored the mechanisms that explain the impact of LTB4 in acquired lymphatic vascular insufficiency. Lower concentrations of LTB4 (10 nM) were surprisingly demonstrated to have a prolymphangiogenic effect, suggesting a possible role for this eicosanoid in lymphatic repair at more physiological levels (133). Higher concentrations of LTB4 (400 nM) inhibit both VEGFR3 mRNA expression and VEGFR3 protein phosphorylation. Additionally, higher LTB4 concentrations inhibit Notch signaling, a pathway known to be important for both lymphatic development and maintenance (134). Furthermore, we used an LEC-specific Notch1 conditional knockout model to show that LEC Notch signaling deficiency renders the lymphedema unable to be rescued by LTB4 signaling antagonism, which highlights an essential role for LEC Notch signaling in the reversal of postsurgical lymphedema. This result also suggests that LTB4 suppression of LEC Notch signaling in the untreated disease contributes to lymphedema progression (133).
We also showed that blockade of LTB4 signaling diminishes macrophage, neutrophil, and CD4+ T cell infiltration into the lymphedematic tissue (133). These results indicate that LTB4 influences not only the innate immunity but also T cell pathophysiology during lymphedema development. These findings are consistent with prior studies showing that LTB4 mediates effector T cells’ (including both CD4+ and CD8+ T cells’) recruitment to inflammatory tissues (30, 135, 136). Given that LTB4 promotes Th17 cell differentiation and migration (31, 32), it is possible that LTB4 also stimulates Th17 differentiation and activation during lymphedema progression. LTB4 therefore may act as a molecular link between the innate and adaptive immunity in postsurgical lymphedema.
In summary, our study further corroborated the notion that lymph stasis-induced inflammation is the principal factor that promotes postsurgical lymphedema progression. Our data also demonstrated pleiotropic roles for LTB4 during the evolution ofpostsurgical lymphedema. LTB4 acts as an important prowound healing molecule during the initial stages of postsurgical tissue repair, yet when the concentration rises into an elevated pathological range, LTB4 has the capacity to diminish LEC function, exacerbate lymphatic vascular dysfunction, and promote disease progression. LTB4 also appears to regulate both innate and adaptive immunity in lymphedema. Lastly, serum LTB4 levels are significantly elevated in human lymphedema patients, indicating that LTB4 is clinically relevant in postsurgical lymphedema pathophysiology (133).
Fibrotic remodeling in lymphedema.
Fibroblast activation and enhanced production of collagen are major steps leading to excessive ECM accumulation and tissue fibrotic remodeling. A Th1/Th2 paradigm in tissue fibrosis has been established (137). It is widely accepted that Th2 cells are involved in wound healing, but they also simultaneously promote tissue fibrotic remodeling and therefore display both beneficial and potentially deleterious effects. They can promote fibrosis by producing profibrotic cytokines such as IL-13 or by indirectly promoting monocyte differentiation toward alternatively activated macrophages (137). Mechanisms associated with tissue fibrotic remodeling in postsurgical lymphedema are coming under scrutiny. Blockade of Th2 cell differentiation by neutralizing IL-4 or IL-13 was able to decrease edematous skin fibrosis (119). Th2 cells have been shown to promote tissue fibrosis in lymphedema through production of TGF-β (118). A recent study showed that hyaluronidase was able to reverse tissue fibrosis in a hind limb postsurgical lymphedema model through a mechanism that promotes Th1 but diminishes Th2 cell activity (138). Together, these studies suggest that Th2 cells are essential in promoting tissue fibrosis in postsurgical lymphedema.
Although we have not yet determined the role of LTB4 in tissue fibrosis in the postsurgical lymphedema model, the LTB4/BLT1 axis has been shown to mediate bleomycin-induced lung fibrosis through a mechanism in which LTB4 induces TGF-p expression in macrophage or epithelial cells, and TGF-β further activates fibroblasts to produce excessive collagen (139). We have also previously demonstrated that LTB4 activates the lung fibroblast (140). It is therefore possible that LTB4 may also play a similar role in promoting tissue fibrotic remodeling in lymphedema.
Although it is commonly accepted that lymph stasis and chronic inflammation eventually cause tissue fibrotic remodeling, there is evidence that fibrosis can also, conversely, impact edema and inflammation. In one study, administration of bleomycin around the postsurgical wound site significantly exacerbates tissue swelling, lymphatic vessel regeneration, and lymph drainage (141). Thus, edema-induced chronic inflammation and fibrosis may act bidirectionally to promote lymphedema progression. Simultaneously targeting both inflammation and fibrosis might therefore be the most efficacious therapeutic strategy.
Adipose tissue deposition in lymphedema.
Although obesity has been identified as a risk factor for postsurgical lymphedema (98), progressive lymphedema also causes abnormal adipose tissue deposition, a pathology that might be considered as regional obesity. The mechanisms through which adipose tissue deposition occurs in lymphedema are poorly understood. It has been shown that lymphatic injury and lymph stasis rapidly and significantly activate adipocytes and upreg-ulate fat differentiation genes, including CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ in both mouse tail and axillary lymphadenectomy models (142, 143). The degree of adipose deposition is associated with the severity of lymphatic dysfunction and inflammation, as depletion of CD4+ T cells or inhibition of Th2 differentiation has a decremental effect (119). These studies suggest that edema and inflammation are the likely initiating factors for excessive adipose tissue deposition. Intriguingly, whereas the proinflammatory cytokine IL- 6 is elevated in mouse tail lymphedema and involved in tissue inflammation, inhibition of this cytokine increases adipose deposition, revealing a unique role of IL-6 in limiting adipose tissue deposition during lymphedema progression (144). Whether and the extent to which LTB4 might affect adipose deposition are not known; however, given recent studies showing the role of LTB4 in promoting insulin resistance in obese mice (28), it is highly likely that LTB4 may also play roles in adipose deposition in postsurgical lymphedema.
CONCLUSIONS
Recent research advances into the lymphatic vascular system have progressively implicated pathological lymphangiogenesis within a variety of states, such as cancer, cardiovascular diseases, and the response to traumatic injury. Our current understanding of pathological lymphangiogenesis as well as the constituent molecular processes is incomplete. In this review, we have highlighted some recent intriguing observations regarding the role of leukotrienes in the initiation and progression of tissue pathology in lymphedema. Continued exploration of the role of these and other mediators of the uncontrolled inflammation in lymph stasis and of the relationship of these mechanisms to the pathogenesis of tissue fibrosis and adipose hypertrophy holds great promise. Enhanced insights into the interplay between inflammation and pathological tissue remodeling will be required to pave new therapeutic avenues in lymphedema and other forms of lymphatic pathology.
ACKNOWLEDGMENTS
Grant support for the authors includes Stanford Startup Funds and Endowed Chair funds (to M.R.N. and S.G.R.).
Footnotes
DISCLOSURE STATEMENT
X.J., M.R.N., W.T., and S.G.R. are inventors of a patent (pending; PCT/US2016/022132) licensed by Eiger BioPharmaceuticals from Stanford University/VA Palo Alto Health Care System that covers LTB4 inhibition to prevent and treat human lymphedema. W.T. and M.R.N. have equity interest in Eiger BioPharmaceuticals, which develops drugs targeting LTB4 for the treatment of secondary lymphedema.
LITERATURE CITED
- 1.Peters-Golden M, Henderson WR Jr. 2007. Leukotrienes. N. Engl.J. Med. 357:1841–54 [DOI] [PubMed] [Google Scholar]
- 2.Radmark OP. 2000. The molecular biology and regulation of 5-lipoxygenase. Am. J. Respir. Crit. Care Med. 161:S11–15 [DOI] [PubMed] [Google Scholar]
- 3.Peters-Golden M, Brock TG. 2001. Intracellular compartmentalization of leukotriene synthesis: unexpected nuclear secrets. FEBS Lett. 487:323–26 [DOI] [PubMed] [Google Scholar]
- 4.Luo M, Jones SM, Peters-Golden M, Brock TG. 2003. Nuclear localization of 5-lipoxygenase as a determinant ofleukotriene B4 synthetic capacity. PNAS 100:12165–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rouzer CA, Samuelsson B. 1985. On the nature of the 5-lipoxygenase reaction in human leukocytes: enzyme purification and requirement for multiple stimulatory factors. PNAS 82:6040–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Percival MD, Denis D, Riendeau D, Gresser MJ. 1992. Investigation of the mechanism ofnon-turnover-dependent inactivation of purified human 5-lipoxygenase. Inactivation by H2O2 and inhibition by metal ions. Eur. J. Biochem. 210:109–17 [DOI] [PubMed] [Google Scholar]
- 7.Hanaka H, Shimizu T, Izumi T. 2005. Stress-induced nuclear export of 5-lipoxygenase. Biochem. Biophys. Res. Commun. 338:111–16 [DOI] [PubMed] [Google Scholar]
- 8.Flamand N,Luo M,Peters-Golden M, Brock TG. 2009. Phosphorylation of serine 271 on 5-lipoxygenase and its role in nuclear export. J. Biol. Chem. 284:306–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo M, Jones SM, Flamand N, Aronoff DM, Peters-Golden M, Brock TG. 2005. Phosphorylation by protein kinase A inhibits nuclear import of 5-lipoxygenase. J. Biol. Chem. 280:40609–16 [DOI] [PubMed] [Google Scholar]
- 10.Dixon RA, Diehl RE, Opas E, Rands E, Vickers PJ, et al. 1990. Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis. Nature 343:282–84 [DOI] [PubMed] [Google Scholar]
- 11.Abramovitz M, Wong E, Cox ME, Richardson CD, Li C, Vickers PJ. 1993. 5-lipoxygenase-activating protein stimulates the utilization of arachidonic acid by 5-lipoxygenase. Eur. J. Biochem. 215:105–11 [DOI] [PubMed] [Google Scholar]
- 12.Peters-Golden M, Brock TG. 2003. 5-lipoxygenase and FLAP. Prostaglandins Leukot. Essent. Fatty Acids 69:99–109 [DOI] [PubMed] [Google Scholar]
- 13.Haeggstrom JZ. 2000. Structure, function, and regulation of leukotriene A4 hydrolase. Am. J. Respir. Crit. Care Med. 161:S25–31 [DOI] [PubMed] [Google Scholar]
- 14.Jakschik BA, Kuo CG. 1983. Characterization of leukotriene A4 and B4 biosynthesis. Prostaglandins 25:767–82 [DOI] [PubMed] [Google Scholar]
- 15.Zhou S, Stark JM, Leikauf GD. 1995. Leukotriene B4 formation: human neutrophil-airway epithelial cell interactions. J. Appl. Physiol. 78:1396–403 [DOI] [PubMed] [Google Scholar]
- 16.Haeggstrom JZ, Wetterholm A, Vallee BL, Samuelsson B. 1990. Leukotriene A4 hydrolase: an epoxide hydrolase with peptidase activity. Biochem. Biophys. Res. Commun. 173:431–37 [DOI] [PubMed] [Google Scholar]
- 17.Murphy RC, Gijon MA. 2007. Biosynthesis and metabolism of leukotrienes. Biochem. J. 405:379–95 [DOI] [PubMed] [Google Scholar]
- 18.Lam BK. 2003. Leukotriene C4 synthase. Prostaglandins Leukot. Essent. Fatty Acids 69:111–16 [DOI] [PubMed] [Google Scholar]
- 19.MacloufJA Murphy RC. 1988. Transcellular metabolism ofneutrophil-derived leukotriene A4 by human platelets. A potential cellular source of leukotriene C4. J. Biol. Chem. 263:174–81 [PubMed] [Google Scholar]
- 20.Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, et al. 1999. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399:789–93 [DOI] [PubMed] [Google Scholar]
- 21.Hui Y, Cheng Y, Smalera I, Jian W, Goldhahn L, et al. 2004. Directed vascular expression of human cys-teinyl leukotriene 2 receptor modulates endothelial permeability and systemic blood pressure. Circulation 110:3360–66 [DOI] [PubMed] [Google Scholar]
- 22.Beller TC, Maekawa A, Friend DS, Austen KF, Kanaoka Y. 2004. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J. Biol. Chem. 279:46129–34 [DOI] [PubMed] [Google Scholar]
- 23.Kanaoka Y, Boyce JA. 2004. Cysteinyl leukotrienes and their receptors: cellular distribution and function in immune and inflammatory responses. J. Immunol. 173:1503–10 [DOI] [PubMed] [Google Scholar]
- 24.Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. 2000. A second leukotriene B4 receptor, BLT2: a new therapeutic target in inflammation and immunological disorders. J. Exp. Med. 192:421–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, et al. 2012. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell 22:1079–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, et al. 2013. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498:371–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNelis JC, Olefsky JM. 2014. Macrophages, immunity, and metabolic disease. Immunity 41:36–48 [DOI] [PubMed] [Google Scholar]
- 28.Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S,et al. 2015. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 21:239–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subbarao K, Jala VR, Mathis S, Suttles J, Zacharias W, et al. 2004. Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler. Thromb. Vasc. Biol. 24:369–75 [DOI] [PubMed] [Google Scholar]
- 30.Goodarzi K, Goodarzi M, Tager AM, Luster AD, von Andrian UH. 2003. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 4:965–73 [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Qin J, Wei P, Zhang J, Li Q, et al. 2009. Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+ T regulatory cells and Th17 cells. Prostaglandins Leukot. Essent. Fatty Acids 80:195–200 [DOI] [PubMed] [Google Scholar]
- 32.Lee W, Su Kim H, Lee GR. 2015. Leukotrienes induce the migration of Th17 cells. Immunol. Cell Biol. 93:472–29 [DOI] [PubMed] [Google Scholar]
- 33.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, et al. 2007. Functionally specialized junctions between endothelial cells oflymphatic vessels. J. Exp. Med. 204:2349–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. 2014. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 14:159–72 [DOI] [PubMed] [Google Scholar]
- 35.Yao LC, McDonald DM. 2014. Plasticity of airway lymphatics in development and disease. Adv. Anat. Embryol. Cell Biol. 214:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA. 2010. Transmural flow modulates cell and fluid transport functions oflymphatic endothelium. Circ. Res. 106:920–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makinen T, Norrmen C, Petrova TV. 2007. Molecular mechanisms of lymphatic vascular development. Cell. Mol. Life Sci. 64:1915–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerjaschki D 2014. The lymphatic vasculature revisited. J. Clin. Investig. 124:874–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi I, Lee S, Hong YK. 2012. The new era of the lymphatic system: no longer secondary to the blood vascular system. Cold Spring Harb. Perspect. Med. 2:a006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. 2016. Lymphatic system in cardiovascular medicine. Circ. Res. 118:515–30 [DOI] [PubMed] [Google Scholar]
- 41.Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, et al. 1995. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. PNAS 92:3566–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wigle JT, Oliver G. 1999. Prox1 function is required for the development of the murine lymphatic system. Cell 98:769–78 [DOI] [PubMed] [Google Scholar]
- 43.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, et al. 1998. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282:946–49 [DOI] [PubMed] [Google Scholar]
- 44.Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, et al. 2004. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5:74–80 [DOI] [PubMed] [Google Scholar]
- 45.Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, et al. 1997. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 276:1423–25 [DOI] [PubMed] [Google Scholar]
- 46.Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, et al. 2002. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBOJ. 21:1505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, et al. 2004. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36:683–85 [DOI] [PubMed] [Google Scholar]
- 48.Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, et al. 2008. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 22:3282–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murtomaki A, UhM K, Choi YK, Kitajewski C, Borisenko V, et al. 2013. Notch1 functions as a negative regulator of lymphatic endothelial cell differentiation in the venous endothelium. Development 140:2365–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hägerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, et al. 2013. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 32:629–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.BosFL CauntM, Peterson-MaduroJ Planas-PazL, Kowalski J, et al. 2011. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ. Res. 109:486–91 [DOI] [PubMed] [Google Scholar]
- 52.Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppanen VM, et al. 2014. CCBE1 enhances lymphan- giogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation 129:1962–71 [DOI] [PubMed] [Google Scholar]
- 53.Le Guen L, Karpanen T, Schulte D, Harris NC, Koltowska K, et al. 2014. Ccbe1 regulates Vegfc-mediated induction ofVegfr3 signaling during embryonic lymphangiogenesis. Development 141:1239–49 [DOI] [PubMed] [Google Scholar]
- 54.Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, et al. 2002. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 129:4797–806 [DOI] [PubMed] [Google Scholar]
- 55.Zheng W, Tammela T, Yamamoto M, Anisimov A, Holopainen T, et al. 2011. Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood 118:1154–62 [DOI] [PubMed] [Google Scholar]
- 56.Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. 2012. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 180:2561–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y, Oliver G. 2014. Transcriptional control of lymphatic endothelial cell type specification. Adv. Anat. Embryol. CellBiol. 214:5–22 [DOI] [PubMed] [Google Scholar]
- 58.Petrova TV, Karpanen T, Norrmen C, Mellor R, Tamakoshi T, et al. 2004. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10:974–81 [DOI] [PubMed] [Google Scholar]
- 59.Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, et al. 2009. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 185:439–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hägerling R, et al. 2012. Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell 22:430–45 [DOI] [PubMed] [Google Scholar]
- 61.Sabine A, Bovay E, Demir CS, Kimura W, Jaquet M, et al. 2015. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 125:3861–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sweet DT, Jiménez JM, Chang J, Hess PR, Mericko-Ishizuka P, et al. 2015. Lymph flow regulates collecting lymphatic vessel maturation in vivo. J. Clin. Investig. 125:2995–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levick JR, Michel CC. 2010. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 87:198–210 [DOI] [PubMed] [Google Scholar]
- 64.Mortimer PS, Rockson SG. 2014. New developments in clinical aspects of lymphatic disease. J. Clin. Investig. 124:915–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, et al. 2013. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 17:671–84 [DOI] [PubMed] [Google Scholar]
- 66.Tammela T, Alitalo K. 2010. Lymphangiogenesis: molecular mechanisms and future promise. Cell 140:460–76 [DOI] [PubMed] [Google Scholar]
- 67.Choe K, Jang JY, Park I, Kim Y, Ahn S, et al. 2015. Intravital imaging of intestinal lacteals unveils lipid drainage through contractility. J. Clin. Investig. 125:4042–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernier-Latmani J, Cisarovsky C, Demir CS, Bruand M, Jaquet M, et al. 2015. DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. J. Clin. Investig. 125:4572–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nurmi H, Saharinen P, Zarkada G, Zheng W, Robciuc MR, Alitalo K. 2015. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol. Med. 7:1418–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP. 2016. The lymphatic system: integral roles in immunity. Annu. Rev. Immunol. 35:31–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, et al. 2008. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453:51–55 [DOI] [PubMed] [Google Scholar]
- 72.Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, et al. 2013. Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin αv. Nat. Immunol. 14:949–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma J, Wang JH, Guo YJ, Sy MS, Bigby M. 1994. In vivo treatment with anti-ICAM-1 and anti-LFA-1 antibodies inhibits contact sensitization-induced migration of epidermal Langerhans cells to regional lymph nodes. Cell Immunol. 158:389–99 [DOI] [PubMed] [Google Scholar]
- 74.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. 2006. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 203:2763–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pflicke H, Sixt M. 2009. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 206:2925–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Förster R, Davalos-Misslitz AC, Rot A. 2008. CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8:362–71 [DOI] [PubMed] [Google Scholar]
- 77.Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, et al. 2010. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat. Immunol. 11:594–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gale NW, Prevo R, Espinosa J, Ferguson DJ, Dominguez MG, et al. 2007. Normal lymphatic development and function in mice deficient for the lymphatic hyaluronan receptor LYVE-1. Mol. Cell. Biol. 27:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lynskey NN, Banerji S, Johnson LA, Holder KA, Reglinski M,et al. 2015. Rapid lymphatic dissemination of encapsulated group A streptococci via lymphatic vessel endothelial receptor-1 interaction. PLOS Pathog. 11:e1005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weber M, Blair E, Simpson CV, O’Hara M, Blackburn PE, et al. 2004. The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol. Biol. Cell 15:2492–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hernandez J, Aung S, Redmond WL, Sherman LA. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, et al. 2010. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 207:681–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirosue S, Vokali E, Raghavan VR, Rincon-Restrepo M, Lund AW, et al. 2014. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: a new role for lymphatic endothelial cells. J. Immunol. 192:5002–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rouhani SJ, Eccles JD, Tewalt EF, Engelhard VH. 2014. Regulation of T-cell tolerance by lymphatic endothelial cells. J. Clin. Cell. Immunol. 5:1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, et al. 2014. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4+ T cell tolerance. J. Exp. Med. 211:1153–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swartz MA, Lund AW. 2012. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat. Rev. Cancer 12:210–19 [DOI] [PubMed] [Google Scholar]
- 87.Kim H, Kataru RP, Koh GY. 2014. Inflammation-associated lymphangiogenesis: a double-edged sword? J. Clin. Investig. 124:936–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karaman S, Hollmén M, Robciuc MR, Alitalo A, Nurmi H, et al. 2015. Blockade of VEGF-C and VEGF-D modulates adipose tissue inflammation and improves metabolic parameters under high-fat diet. Mol. Metab. 4:93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, et al. 2000. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 25:153–59 [DOI] [PubMed] [Google Scholar]
- 90.Gordon K, Schulte D, Brice G, Simpson MA, Roukens MG, et al. 2013. Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant Milroy-like primary lymphedema. Circ. Res. 112:956–60 [DOI] [PubMed] [Google Scholar]
- 91.Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, et al. 2003. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms ofhypotrichosis-lymphedema-telangiectasia. Am. J. Hum. Genet. 72:1470–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, et al. 2011. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 43:929–31 [DOI] [PubMed] [Google Scholar]
- 93.Babu S, Nutman TB. 2012. Immunopathogenesis of lymphatic filarial disease. Semin. Immunopathol. 34:847–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cormier JN, Askew RL, Mungovan KS, Xing Y, Ross MI, Armer JM. 2010. Lymphedema beyond breast cancer: a systematic review and meta-analysis of cancer-related secondary lymphedema. Cancer 116:5138–49 [DOI] [PubMed] [Google Scholar]
- 95.Avraham T, Yan A, Zampell JC, Daluvoy SV, Haimovitz-Friedman A, et al. 2010. Radiation therapy causes loss of dermal lymphatic vessels and interferes with lymphatic function by TGF-p1-mediated tissue fibrosis. Am.J. Physiol. Cell Physiol. 299:C589–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gould N, Kamelle S, Tillmanns T, Scribner D, Gold M, et al. 2001. Predictors of complications after inguinal lymphadenectomy. Gynecol. Oncol. 82:329–32 [DOI] [PubMed] [Google Scholar]
- 97.Vignes S, Arrault M, Dupuy A. 2007. Factors associated with increased breast cancer-related lymphedema volume. Acta Oncol. 46:1138–42 [DOI] [PubMed] [Google Scholar]
- 98.Treves N 1957. An evaluation of the etiological factors of lymphedema following radical mastectomy. An analysis of1,007 cases. Cancer 10:444–59 [DOI] [PubMed] [Google Scholar]
- 99.Weitman ES, Aschen SZ, Farias-Eisner G, Albano N, Cuzzone DA, et al. 2013. Obesity impairs lymphatic fluid transport and dendritic cell migration to lymph nodes. PLOS ONE 8:e70703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blum KS, Karaman S, Proulx ST, Ochsenbein AM, Luciani P, et al. 2014. Chronic high-fat diet impairs collecting lymphatic vessel function in mice. PLOS ONE 9:e94713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Greene AK, Grant FD, Slavin SA. 2012. Lower-extremity lymphedema and elevated body-mass index. N. Engl. J. Med. 366:2136–37 [DOI] [PubMed] [Google Scholar]
- 102.Finegold DN, Schacht V, Kimak MA, Lawrence EC, Foeldi E, et al. 2008. HGF and MET mutations in primary and secondary lymphedema. Lymphat. Res. Biol. 6:65–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Newman B, Lose F, Kedda MA, Francois M, Ferguson K, et al. 2012. Possible genetic predisposition to lymphedema after breast cancer. Lymphat. Res. Biol. 10:2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nutman TB. 2013. Insights into the pathogenesis of disease in human lymphatic filariasis. Lymphat. Res. Biol. 11:144–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bennuru S, Nutman TB. 2009. Lymphangiogenesis and lymphatic remodeling induced by filarial parasites: implications for pathogenesis. PLOS Pathog. 5:e1000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rao UR, Vickery AC, Kwa BH, Nayar JK. 1996. Regulatory cytokines in the lymphatic pathology of athymic mice infected with Brugia malayi. Int. J. Parasitol. 26:561–65 [DOI] [PubMed] [Google Scholar]
- 107.Babu S, Nutman TB. 2014. Immunology of lymphatic filariasis. Parasite Immunol. 36:338–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nelson FK, Greiner DL, Shultz LD, Rajan TV. 1991. The immunodeficient scid mouse as a model for human lymphatic filariasis. J. Exp. Med. 173:659–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lal RB, Kumaraswami V, Krishnan N, Nutman TB, Ottesen EA. 1989. Lymphocyte subpopulations in Bancroftian filariasis: activated (DR+) CD8+ T cells in patients with chronic lymphatic obstruction. Clin. Exp. Immunol. 77:77–82 [PMC free article] [PubMed] [Google Scholar]
- 110.Freedman DO, Horn TD, Maia e Silva CM, Braga C, Maciel A. 1995. Predominant CD8+ infiltrate in limb biopsies ofindividuals with filarial lymphedema and elephantiasis. Am. J. Trop. Med. Hyg. 53:633–38 [PubMed] [Google Scholar]
- 111.Freedman DO, Plier DA, de Almeida A, Miranda J, Braga C, et al. 1999. Biased TCR repertoire in infiltrating lesional T cells in human Bancroftian filariasis. J. Immunol. 162:1756–64 [PubMed] [Google Scholar]
- 112.Babu S, Bhat SQ, Pavan Kumar N, Lipira AB, Kumar S, et al. 2009. Filarial lymphedema is characterized by antigen-specific Th1 and Th17 proinflammatory responses and a lack of regulatory T cells. PLOS Negl. Trop. Dis. 3:e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pathak M, Sharma P, Sharma A, Verma M, Srivastava M,Misra-Bhattacharya S. 2016. Regulatory T-cell neutralization in mice during filariasis helps in parasite clearance by enhancing T helper type 17-mediated pro-inflammatory response. Immunology 147:190–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Anuradha R, George PJ, Hanna LE, Chandrasekaran V, Kumaran PP, et al. 2014. Parasite-antigen driven expansion of IL-5- and IL-5+ Th2 human subpopulations in lymphatic filariasis and their differential dependence on IL-10 and TGFβ. PLOS Negl. Trop. Dis. 8:e2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anuradha R, George PJ, Hanna LE, Chandrasekaran V, Kumaran P, et al. 2013. IL-4-, TGF-β-, and IL-1-dependent expansion of parasite antigen-specific Th9 cells is associated with clinical pathology in human lymphatic filariasis. J. Immunol. 191:2466–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Olszewski WL, Jamal S, Manokaran G, Lukomska B, Kubicka U. 1993. Skin changes in filarial and non-filarial lymphoedema of the lower extremities. Trop. Med. Parasitol. 44:40–44 [PubMed] [Google Scholar]
- 117.Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. 2012. CD4+ cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PLOS ONE 7:e49940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, et al. 2010. Blockade of transforming growth factor-β1 accelerates lymphatic regeneration during wound repair. Am. J. Pathol. 177:3202–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Avraham T, Zampell JC, Yan A, Elhadad S, Weitman ES, et al. 2013. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEBJ. 27:1114–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Savetsky IL, Ghanta S, Gardenier JC, Torrisi JS, Garcia Nores GD, et al. 2015. Th2 cytokines inhibit lymphangiogenesis. PLOS ONE 10:e0126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shin K, Kataru RP, Park HJ, Kwon BI, Kim TW, et al. 2015. TH2 cells and their cytokines regulate formation and function oflymphatic vessels. Nat. Commun. 6:6196. [DOI] [PubMed] [Google Scholar]
- 122.Gousopoulos E, Proulx ST, Bachmann SB, Scholl J, Dionyssiou D, et al. 2016. Regulatory T cell transfer ameliorates lymphedema and promotes lymphatic vessel function. JCI Insight 1:e89081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ogata F, Fujiu K, Matsumoto S, Nakayama Y, Shibata M, et al. 2016. Excess lymphangiogenesis cooperatively induced by macrophages and CD4+ T cells drives the pathogenesis of lymphedema. J. Investig. Dermatol. 136:706–14 [DOI] [PubMed] [Google Scholar]
- 124.Vannella KM, Wynn TA. 2017. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 79:593–617 [DOI] [PubMed] [Google Scholar]
- 125.Sica A, Erreni M, Allavena P, Porta C. 2015. Macrophage polarization in pathology. Cell. Mol. Life Sci. 72:4111–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ghanta S, Cuzzone DA, Torrisi JS, Albano NJ, Joseph WJ, et al. 2015. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am. J. Physiol. Heart Circ. Physiol. 308:H1065–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. 2006. Secondary lymphedema in the mouse tail: lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc. Res. 72:161–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zampell JC, Elhadad S, Avraham T, Weitman E, Aschen S, et al. 2012. Toll-like receptor deficiency worsens inflammation and lymphedema after lymphatic injury. Am. J. Physiol. Cell Physiol. 302:C709–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tabibiazar R, Cheung L, Han J, Swanson J, Beilhack A, et al. 2006. Inflammatory manifestations of experimental lymphatic insufficiency. PLOS Med. 3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nakamura K, Radhakrishnan K, Wong YM, Rockson SG. 2009. Anti-inflammatory pharmacotherapy with ketoprofen ameliorates experimental lymphatic vascular insufficiency in mice. PLOS ONE 4:e8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baluk P, Yao LC, Feng J, Romano T, Jung SS, et al. 2009. TNF-α drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J. Clin. Investig. 119:2954–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ambrus JL Jr., Haneiwich S, Chesky L, McFarland P, Engler RJ. 1991. Improved in vitro antigen-specific antibody synthesis in two patients with common variable immunodeficiency taking an oral cyclooxygen-ase and lipoxygenase inhibitor (ketoprofen). J. Allergy Clin. Immunol. 88:775–83 [DOI] [PubMed] [Google Scholar]
- 133.Tian W, Rockson SG, Jiang X, Kim J, Begaye A, et al. 2017. Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci. Transl. Med. 9:eaal3920. [DOI] [PubMed] [Google Scholar]
- 134.Murtomaki A, Uh MK, Kitajewski C, Zhao J, Nagasaki T, et al. 2014. Notch signaling functions in lymphatic valve formation. Development 141:2446–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, et al. 2003. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat. Immunol. 4:982–90 [DOI] [PubMed] [Google Scholar]
- 136.Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. 2003. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat. Immunol 4:974–81 [DOI] [PubMed] [Google Scholar]
- 137.Wynn TA. 2004. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 4:583–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cho S, Roh K, Park J, Park YS, Lee M, et al. 2017. Hydrolysis of hyaluronic acid in lymphedematous tissue alleviates fibrogenesis via TH1 cell-mediated cytokine expression. Sci. Rep. 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lv J, Xiong Y, Li W, Yang W, Zhao L, He R. 2017. BLT1 mediates bleomycin-induced lung fibrosis independently of neutrophils and CD4+ T cells. J. Immunol. 198:1673–84 [DOI] [PubMed] [Google Scholar]
- 140.Qian J, Tian W, Jiang X, Tamosiuniene R, Sung YK, et al. 2015. Leukotriene B4 activates pulmonary artery adventitial fibroblasts in pulmonary hypertension. Hypertension 66:1227–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lynch LL, Mendez U, Waller AB, Gillette AA, Guillory RJ 2nd, Goldman J. 2015. Fibrosis worsens chronic lymphedema in rodent tissues. Am. J. Physiol. Heart Circ. Physiol. 308:H1229–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Aschen S, Zampell JC, Elhadad S, Weitman E, De Brot M, Mehrara BJ. 2012. Regulation of adipogenesis by lymphatic fluid stasis part II: expression of adipose differentiation genes. Plast. Reconstr. Surg. 129:838–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zampell JC, Aschen S, Weitman ES, Yan A, Elhadad S, et al. 2012. Regulation of adipogenesis by lymphatic fluid stasis part I: adipogenesis, fibrosis, and inflammation. Plast. Reconstr. Surg. 129:825–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cuzzone DA, Weitman ES, Albano NJ, Ghanta S, Savetsky IL, et al. 2014. IL-6 regulates adipose deposition and homeostasis in lymphedema. Am. J. Physiol. Heart Circ. Physiol. 306:H1426–34 [DOI] [PMC free article] [PubMed] [Google Scholar]