

Abstract
The 90-kDa heat shock protein (HSP90) is a molecular chaperone that maintains the proper folding of its client proteins including protein kinases and steroid hormone receptors. Helicases are a group of nucleic acid-binding ATPases that can unwind DNA and/or RNA and function in almost every aspect of nucleic acid metabolism. Not much, however, is known about the interactions between HSP90 and helicase proteins. Herein, we developed a parallel-reaction monitoring (PRM)-based targeted proteomic method that allows for quantifying >80% of the human helicase proteome. By employing this method, we demonstrated that a large number of helicase proteins exhibited diminished expression in cultured human cells upon treatment with two small-molecule inhibitors of HSP90. We further introduced a tandem affinity tag to the C-terminus of endogenous HSP90β protein by using the CRISPR-Cas9 genome editing method. Affinity purification followed by LC-PRM analysis revealed an enrichment of 40 out of the 66 quantified helicases from the lysate of cells expressing tagged HSP90β. Together, we developed a high-throughput targeted proteomic method for assessing quantitatively the human helicase proteome, and our results support that helicases may constitute an important group of client proteins for HSP90.
Graphical Abstract
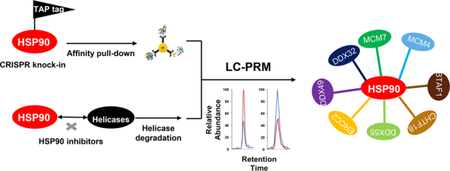
Nucleic acid unwinding is an important biological process that regulates nearly all aspects of nucleic acid metabolism, including DNA replication, transcription, and repair, as well as mRNA translation.1,2 Helicases are nucleic acid-dependent ATPases that are capable of unwinding duplex and more complex structures of nucleic acids.2 Owing to these unique roles of helicases, their aberrant functions are frequently associated with genomic instability, cancer development, and premature aging. For instance, FANCJ, BLM, and WRN, which are DNA helicases that can help maintain genomic integrity by resolving guanine-quadruplex (G4) DNA structures, are implicated in genetic disorders of Fanconi anemia, Bloom’s syndrome, and Werner’s syndrome, respectively.3,4
The 90-kDa heat shock protein (HSP90) is a molecular chaperone that ensures the proper folding of proteins (clients), thereby maintaining the homeostasis of the proteome.5 HSP90 has a large group of client proteins, including protein kinases6 and steroid hormone receptors.7 In addition, several helicases, including yeast SSL28 as well as human MCM4 and MCM7,9 were shown by in vitro protein binding assay to be client proteins of HSP90. However, there has been no systematic investigation about the interactions between HSP90 and the helicase proteome.
Targeted proteomic methods, involving LC-MS/MS analyses in the parallel-reaction monitoring (PRM) or multiplereaction monitoring (MRM) mode, exhibit much higher sensitivity and reproducibility in peptide detection than proteomic analysis in the data-dependent acquisition (DDA) mode.10,11 Due to the high mass-resolving power and mass accuracy afforded by a time-of-flight or Orbitrap mass analyzer, the PRM method offers better accuracy and specificity than MRM in quantifying analytes present in complex sample matrices;10,11 hence, it has become increasingly used in bioanalysis.12–14
In this study, we developed, for the first time, an LC-PRM-based targeted quantitative proteomic method for high-throughput analysis of the human helicase proteome. With this method, we assessed the differential expression of helicase proteins in cultured human cells upon treatment with two HSP90 inhibitors. We also introduced a tandem affinity tag to the C-terminus of endogenous HSP90β protein using CRISPR-Cas915,16 and examined the interaction between HSP90β and helicases by employing affinity pull-down in conjunction with LC-PRM analysis.
We first constructed, on the basis of data acquired from shotgun proteomic analyses of the tryptic digestion mixtures of protein lysates of 10 cell lines derived from different human tissue origins,14 a Skyline17 PRM library for helicase proteins. The PRM library contains the retention time, MS, and MS/MS of tryptic peptides of helicases. In this respect, we inspected all the identified helicase peptides and incorporated, into the library, only those peptides that could be uniquely assigned to specific helicases. By incorporating 3–6 such signature peptides with the strongest signal intensity for each helicase, our helicase PRM library encompasses 411 unique peptides, which represent 121 distinct human helicases (Figure 1a, Table S1). Among them, 20, 60, and 41 were DNA helicases, RNA helicases, and other helicases, respectively (Figure 1b). Hence, the library covers approximately 84% of the 95 known DNA and RNA helicases.18 The failure in detecting a small number of RNA or DNA helicases from the above-mentioned 10 cell lines might be attributed to the relatively low levels of expression of these helicase proteins in these cells.
Figure 1.
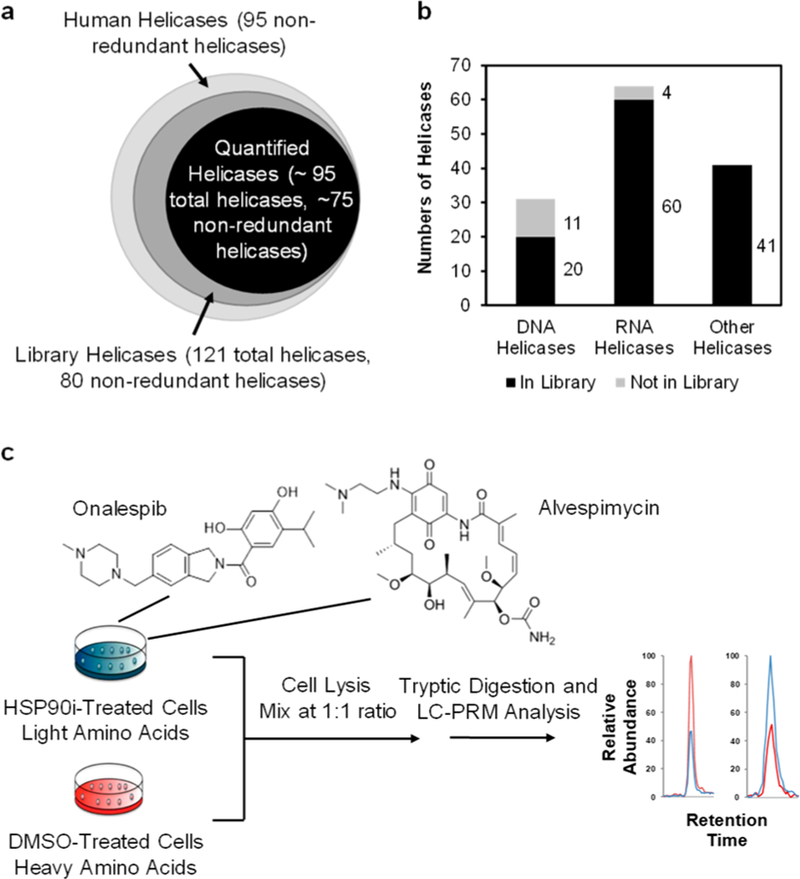
A PRM-based targeted proteomic approach for interrogating the human helicase proteome. (a) A Venn diagram depicting the numbers of helicases included in the PRM library and those that could be quantified by the PRM method. (b) A bar graph showing the coverages of different groups of helicases in the PRM library. (c) The experimental strategy, involving the use of forward SILAC labeling together with the PRM-based targeted proteomic analysis, for assessing the alterations in expression of helicase proteins in human cells upon treatment with the two HSP90 inhibitors.
By adopting scheduled PRM analysis, where we set up the mass spectrometer to collect the MS/MS for the precursor ions from a limited number of peptides in each 10 min retention time (RT) window, we were able to achieve high-throughput detection of peptides derived from helicase proteins. In this vein, we calculated the normalized RT (iRT) value for each peptide on our target list according to previously published methods.19,20 In particular, we employed 10 tryptic peptides of bovine serum albumin (BSA) as standards for establishing the iRT scale and for calibrating the retention time. We subsequently converted the experimentally determined retention times for all the 411 helicase peptides into normalized iRT scores, where the linear relationship of RT vs iRT was redetermined in every 5–7 LC-PRM runs through the analysis of the tryptic digestion mixture of BSA. On the grounds that iRT constitutes an inherent property of a peptide, a substantial difference between the measured RT of a tryptic peptide and that predicted from the linear plot of RT vs iRT is considered false-positive, which is employed together with characteristic fragment ions observed for the peptide as parameters to validate the results obtained from the PRM assay.
Some HSP90 inhibitors can disrupt the binding of HSP90 with ATP and compromise HSP90’s capability of maintaining the proper folding of client proteins, thereby resulting in their degradation via the ubiquitin-dependent proteasomal pathway.21,22 Hence, we next applied the established LC-PRM method, together with stable isotope-labeling by amino acids in cell culture (SILAC),23 to identify helicases as putative client proteins of HSP90. In particular, we assessed the reprogramming of the human helicase proteome in M14 cells upon treatment with two HSP90 inhibitors, i.e., onalespib and alvespimycin (Figure 1c), which are resorcyclic dihydroxybenzamide and bacteria-derived benzoquinone ansamycin derivatives, respectively.22 These two inhibitors occupy the ATP-binding pocket of the N-terminal domain of HSP90 protein and inhibit its chaperone activity.22 Our LC-PRM analysis results showed that 49 out of the 95 quantified helicases were down-regulated by at least 1.5-fold in M14 cells after a 24 h treatment with 100 nM onalespib. A similar treatment with alvespimycin led to decreased expression (by at least 1.5-fold) of 28 out of the 96 quantified helicases (Figure 2 and Table S2). In this context, it is of note that we excluded post-translationally modified peptides in the PRM helicase library; therefore, the failure in detecting some helicases in the library could be attributed to their low levels of expression in M14 cells and/or the lack of generation of the target tryptic peptides arising from post-translational modification(s).
Figure 2.

Alterations in expression levels of helicase proteins in M14 cells after treatment with two small-molecule HSP90 inhibitors, onalespib (a) and alvespimycin (b). The cells were treated with 100 nM inhibitor for 24 h. Displayed are the ratios of expression of helicase proteins in HSP90 inhibitor-treated over mock-treated M14 cells, where the Y-axis was plotted in log10 scale. The data represent the average ratios obtained from two biological replicates (i.e., one forward and one reverse SILAC labeling experiments). The red and blue bars designate those helicases that were up- and down-regulated, respectively, by at least 1.5-fold upon the inhibitor treatment.
We also confirmed the diminished expression of four helicases (i.e., DDX11, DDX34, MCM4, and MCM7) by Western blot analysis (Figure 3c–f), where ARAF kinase, a known client protein of HSP90, was employed as a positive control (Figures 3c and S1).6
Figure 3.
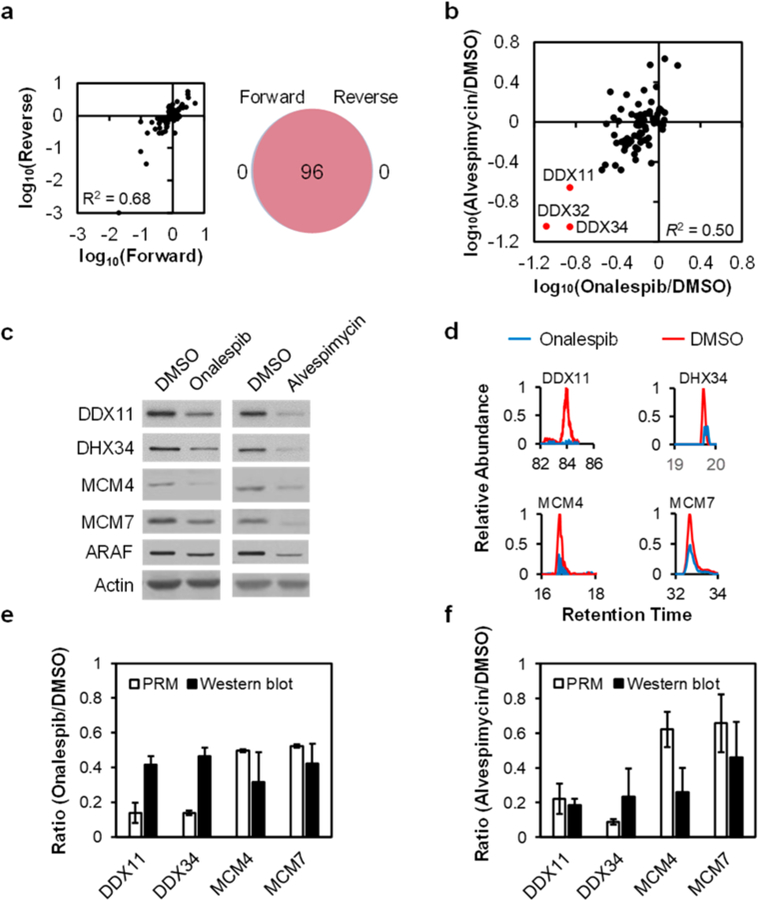
Analytical performance of the PRM method. (a) A scatter plot displaying the correlation between the ratios obtained from forward and reverse SILAC labeling experiments (left), and a Venn diagram showing the overlap between quantified helicases from the forward and reverse SILAC labeling experiments in M14 cells with or without alvespimycin treatment (right). (b) A scatter plot showing the correlation between the alterations in expression levels of helicase proteins in M14 cells treated with onalespib or alvespimycin. (c) Western blot for the validation of the expression levels of representative helicases in M14 cells with vs without HSP90 inhibitor treatment, where ARAF was used as a positive control. (d) PRM traces for the relative quantifications of representative helicases. (e, f) Quantitative comparisons of the ratios obtained from PRM (n = 2, one forward and one reverse SILAC labelings) and Western blot analyses (n = 3) for representative helicases in M14 cells with vs without a 24 h treatment with 100 nM of onalespib (e) or alvespimycin (f).
All the quantified peptides for helicases exhibit a reasonably good linear fit between the observed retention time and iRT in the library (Figure S2), lending support for the robust detection of helicases. In addition, all 4–6 transitions used for the relative quantification of each peptide from helicases exhibited the same retention time with a dot product (dotp)24 of >0.7 when compared to the same fragment ions found in the MS/MS acquired from shotgun proteomic analysis (representative results are shown in Figure S3), further confirming the reliability of the method in peptide identification.
Our results showed that all the quantified helicases could be detected in both forward and reverse SILAC labeling experiments (Figure 3a). In addition, the ratios for the quantified peptides obtained from forward and reverse SILAC labeling experiments are consistent (Figures 3a, S4, and S5). Furthermore, the average relative standard deviation (RSD) for the tryptic peptides quantified for the same helicases was 13.1% (Table S2). These results underscored the reasonably good reproducibility of the analytical method (Table S2).
We also analyzed the same samples by using LC-MS/MS in the DDA mode, where the peptide mixture was first separated into 20 fractions using a strong cation-exchange (SCX) column, and the resultant fractions were individually subjected to LC-MS/MS analysis in the DDA mode. The resulting LC-MS/MS data only led to the relative quantification of 60 helicases. In contrast, our scheduled PRM method enabled the relative quantification of ∼95 helicases in only 4–5 LC-PRM runs without any prefractionation, revealing the superior sensitivity and throughput of the PRM method (Figure S4b).
Linear regression analysis of the ratios for the commonly quantified helicases in cells treated with the two HSP90 inhibitors yielded an R2 value of 0.50 (Figures 3b and S4d), suggesting that the two inhibitors led to similar reprogramming of a large number of helicases. Nevertheless, notable differences were observed for the two HSP90 inhibitors, particularly for those helicases shown in the top-left quadrant of Figure 3b. The latter finding is in accordance with the fact that the two inhibitors possess different chemical scaffolds (Figure 1c) and the previous notion that HSP90 inhibitors, albeit targeting the same ATP-binding pocket of HSP90, may modulate differently the interaction of HSP90 with its client proteins.22 Considering that HSP90 inhibition can lead to improper folding and proteasomal degradation of its client proteins,21 those helicases that were down-regulated upon the inhibitor treatment are considered candidate client proteins for HSP90.
We reason that those helicases that are client proteins of HSP90 should also interact with HSP90. Hence, we embarked on an interactome study by assessing, at the proteome-wide scale, the interactions between helicases and HSP90. To this end, we employed the CRISPR-Cas9 genome-editing method to introduce a tandem affinity tag (3 × Flag, 2 × Strept) to the C-terminus of endogenous HSP90β6 protein in HEK293T cells.16 Screening using Western blot with anti-Flag antibody led to the identification of two positive clones (Figure 4a, clone 8 and clone 15). The different intensities for the anti-Flag band for the two clones could be attributed to heterozygous (for clone 8) and homozygous (for clone 15) integration of the tandem affinity tag to the C-terminus of HSP90β, and we employed Clone 15 for all the subsequent experiments (referred to as the Flag-HSP90β cells). The successful incorporation of the tandem affinity tag was also validated by Western blot analysis with the use of an anti-HSP90 antibody (Figures 4a and S6a). The HSP90 antibody can recognize all the HSP90 isoforms, and the higher molecular-weight band corresponds to HSP90β incorporated with the tandem affinity tag. Affinity purification of HSP90β using anti-Flag M2 beads from the lysate of CRISPR-engineered cells, followed by tryptic digestion and LC-MS/MS analysis in the DDA mode, gave rise to the identification of HSP90β with a 65% sequence coverage (Figure S6b).
Figure 4.
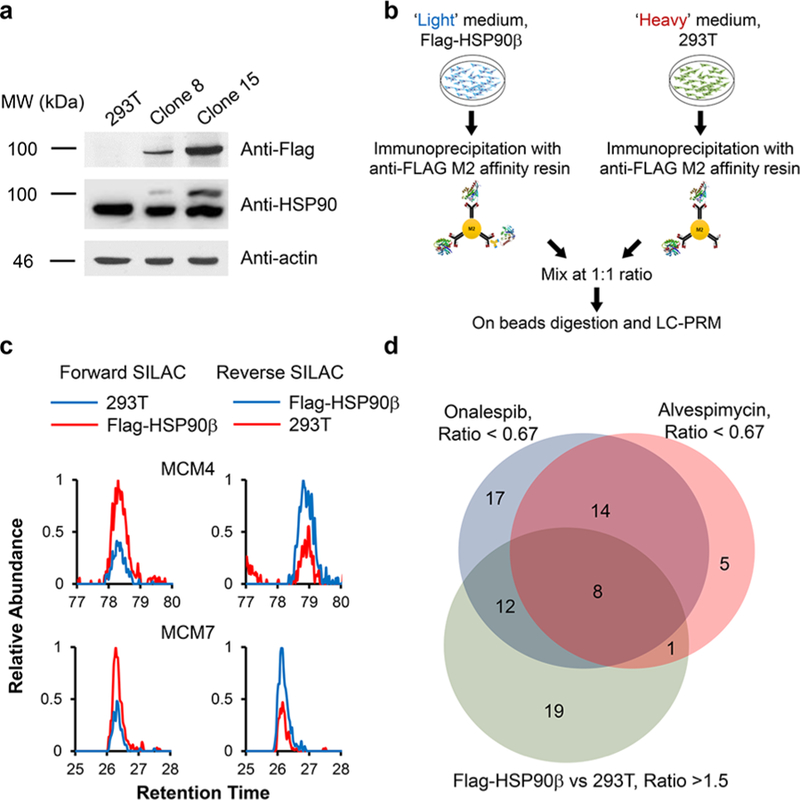
Targeted integration of a tandem affinity tag to the C-terminus of HSP90β and affinity pull-down in conjunction with LC-PRM analysis for assessing the interaction between HSP90β and helicase proteins. (a) Western blot confirmed the targeted integration of the tandem affinity tag to endogenous HSP90β protein with the CRISPR-Cas9 method. (b) Experimental strategy for combining forward SILAC labeling with the LC-PRM-based targeted proteomic approach for the identification of cellular proteins that can interact with HSP90β. (c) Representative PRM traces showing the relative quantification results of MCM4 and MCM7 from the anti-Flag pull-down mixture in HEK293T cells with or without the integration of tandem affinity tag to the C-terminus of HSP90β protein from both forward and reverse SILAC labeling experiments. (d) A Venn diagram depicting the number of helicases that could bind with HSP90β, i.e., those helicases that could be enriched from affinity pull-down from Flag-HSP90β cells and that could be down-regulated upon HSP90 inhibitor treatment.
Relative quantification using SILAC labeling, affinity pull-down with anti-Flag beads, and LC-PRM analysis revealed the interactions between HSP90β and a number of helicases (Figure 4b–d). In particular, we were able to quantify 66 helicases, among which 40 were enriched by at least 1.5-fold with the lysate of the Flag-HSP90β cells relative to the use of the lysate from parental HEK293T cells (Figure S7, Table S2). The ratios obtained from forward and reverse SILAC experiments are again consistent (Figure 4c, Table S2). Together with the data from the above-mentioned inhibitor experiment, 21 helicases were both enriched from Flag-HSP90β cells over the parental HEK293T cells and downregulated upon treatment with at least one of the two HSP90 inhibitors (Figures 4d and S8). These 21 helicases are considered strong candidates for client proteins of HSP90β. Among them, eight (BTAF1, CHTF18, DDX32, DDX49, DDX55, ERCC2, MCM4, and MCM7) were down-regulated upon treatment with both HSP90 inhibitors (Figures 4d, 5a, and S8). In this vein, MCM4 and MCM7 were previously shown to interact with HSP90 through its cochaperone, FKBP51.9 Moreover, we validated the interaction between HSP90 and MCM4/MCM7 by immunoprecipitation followed by Western blot analysis (Figure 5b). In this regard, similar affinity pull-down followed by Western blot analysis confirmed the interaction between HSP90β and ARAF kinase, a previously validated client protein of HSP906 (Figure S6a). On the other hand, we failed to detect EGFR, which is not a client protein of HSP90β,6 in the affinity pull-down mixture. It is worth noting that, among the helicases that are putative clients of HSP90β, FANCJ, ERCC2 (a.k.a. XPD), and DDX11 (a.k.a. CHIR1) are iron–sulfur cluster proteins that are known to be important in DNA replication and repair.25
Figure 5.
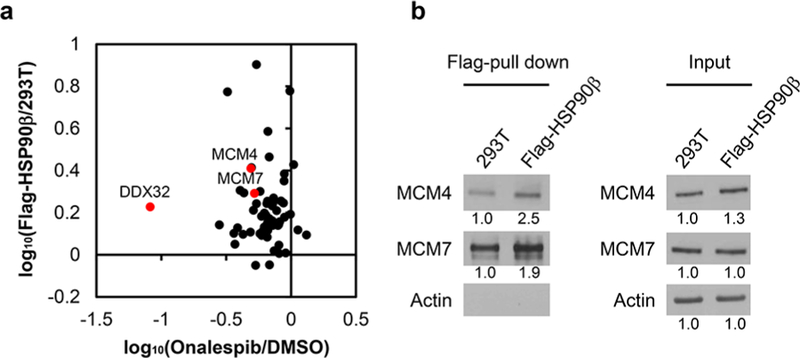
Interactions between HSP90 and helicases. (a) Correlation between the ratios obtained from affinity pull-down and onalespib treatment. (b) Immunoprecipitation followed by Western blot analysis for validating the interactions between HSP90β and MCM4/MCM7.
In summary, we developed a PRM-based targeted proteomic approach to fulfill large-scale relative quantification of helicase proteins. We applied this method to interrogate quantitatively the interaction between HSP90 and helicases. We found that the expression levels for a large number of helicases were diminished in human cells upon treatment with two small-molecule inhibitors of HSP90. By incorporating a tandem affinity tag to endogenous HSP90β using CRISPR-Cas9, we found that 40 out of the 66 quantified helicases could be enriched by affinity pull-down of HSP90β from the engineered cells, supporting the interactions between helicases and HSP90. Hence, the results from these studies support that helicases are a group of client proteins for HSP90.
Supplementary Material
Table S1. Proteins and peptides included in the Skyline human helicase library (XLSX)
Table S2. Relative expression levels of helicase proteins in M14 cells with or without treatment with HSP90 inhibitors (XLSX)
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b03142.
Detailed experimental procedures; quantitative result of the ratios of Western blot analyses; scatter plots showing the correlations between iRT in the library and the measured RTs from LC-PRM analyses of tryptic digestion mixtures of protein lysates; extracted-ion chromatograms for monitoring representative helicases; analytical performance of the PRM method; heatmap showing the differences in expression levels of helicases in cells with or without alvespimycin treatment; validation for the incorporation of Flag-tag in endogenous HSP90β protein; bar graph depicting the ratios of helicase proteins; helicases as putative client proteins of HSP90 (PDF)
Notes
The authors declare no competing financial interest.
The Skyline PRM library (including the transition list for quantification and the iRT file) for helicases and the raw files for LC-PRM analyses of helicases were deposited into PeptideAtlas with the identifier number of PASS01191 (http://www.peptideatlas.org/PASS/PASS01191).
REFERENCES
- (1).Brosh RM Nat. Rev. Cancer 2013, 13, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Singleton MR; Dillingham MS; Wigley DB Annu. Rev. Biochem 2007, 76, 23–50. [DOI] [PubMed] [Google Scholar]
- (3).Wu Y; Shin-ya K; Brosh RM Mol. Cell. Biol 2008, 28, 4116–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mohaghegh P; Karow JK; Brosh RM Jr.; Bohr VA; Hickson ID Nucleic Acids Res 2001, 29, 2843–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Taipale M; Jarosz DF; Lindquist S Nat. Rev. Mol. Cell Biol 2010, 11, 515–528. [DOI] [PubMed] [Google Scholar]
- (6).Taipale M; Krykbaeva I; Koeva M; Kayatekin C; Westover KD; Karras GI; Lindquist S Cell 2012, 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Manninen T; Purmonen S; Ylikomi TJ Steroid Biochem. Mol. Biol 2005, 96, 13–18. [DOI] [PubMed] [Google Scholar]
- (8).Flom G; Weekes J; Johnson JL Curr. Genet 2005, 47, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Taipale M; Tucker G; Peng J; Krykbaeva I; Lin ZY; Larsen B; Choi H; Berger B; Gingras AC; Lindquist S Cell 2014, 158, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shi T; Song E; Nie S; Rodland KD; Liu T; Qian WJ; Smith RD Proteomics 2016, 16, 2160–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon J Mol. Cell. Proteomics 2012, 11, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou J; Liu H; Liu Y; Liu J; Zhao X; Yin Y Anal. Chem 2016, 88, 4478–4486. [DOI] [PubMed] [Google Scholar]
- (13).Guo L; Wang Q; Weng L; Hauser LA; Strawser CJ; Rocha AG; Dancis A; Mesaros C; Lynch DR; Blair IA Anal. Chem 2018, 90, 2216–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Miao W; Li L; Wang Y Anal. Chem 2018, 90, 6835–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cong L; Ran FA; Cox D; Lin S; Barretto R; Habib N; Hsu PD; Wu X; Jiang W; Marraffini L; Zhang F Science 2013, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dalvai M; Loehr J; Jacquet K; Huard CC; Roques C; Herst P; Cote J; Doyon Y Cell Rep 2015, 13, 621–633. [DOI] [PubMed] [Google Scholar]
- (17).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Umate P; Tuteja N; Tuteja R Commun. Integr. Biol 2011, 4, 118–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Miao W; Xiao Y; Guo L; Jiang X; Huang M; Wang Y Anal. Chem 2016, 88, 9773–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Butler LM; Ferraldeschi R; Armstrong HK; Centenera MM; Workman P Mol. Cancer Res 2015, 13, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Taldone T; Ochiana SO; Patel PD; Chiosis G Trends Pharmacol. Sci 2014, 35, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (24).de Graaf EL; Altelaar AF; van Breukelen B; Mohammed S; Heck AJ J. Proteome Res 2011, 10, 4334–4341. [DOI] [PubMed] [Google Scholar]
- (25).Wu Y; Brosh RM Nucleic Acids Res 2012, 40, 4247–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteins and peptides included in the Skyline human helicase library (XLSX)
Table S2. Relative expression levels of helicase proteins in M14 cells with or without treatment with HSP90 inhibitors (XLSX)