

“To call in the statistician after the experiment is done may be no more than asking him to perform a post-mortem examination - he may be able to say what the experiment dies of.”
R.A. Fisher
The delivery of an intervention whether drug, a dietary change, a lifestyle change, or a psychological therapy session counts as an intervention and hence must be dealt as a clinical trial [Figure 1]. Clinical trial design is an important aspect of interventional trials that serves to optimize, ergonomise and economize the clinical trial conduct. The purpose of the clinical trial is assessment of efficacy, safety, or risk benefit ratio. Goal may be superiority, non-inferiority, or equivalence. A well-conducted study with a good design based on a robust hypothesis evolved from clinical practice goes a long way in facilitating the implementation of the best tenets of evidence-based practice. A robust well-powered trial adds to the meta-analyzable evidence base and contributes huge quanta to our knowledge of dermatological practice. This article sets out to describe the various trial designs and modifications and attempts to delineate the pros and cons of each design and attempts to provide illustrative samples for the same where possible.
Figure 1.
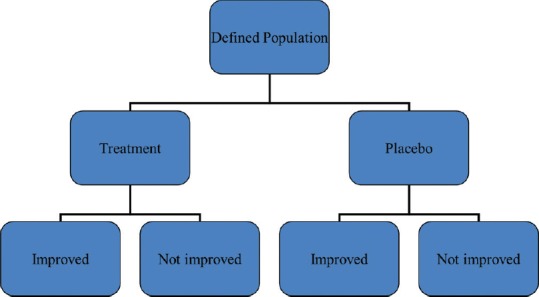
Basic framework of clinical trials
Uncontrolled Trials
This design incorporates no control arm. This design is usually utilized to determine pharmacokinetic properties of a new drug (Phase 1 trials). Uncontrolled trials are known to produce greater mean effect estimates than a controlled trial, thereby inflating the expectations from the intervention. There is a threat of inherent bias and results are considered less valid than RCT. Another issue is use of this design in spontaneously resolving maladies that might again overstate the effect [Figure 2].
Figure 2.
Single arm trial schematic
Illustrative example
In immunotherapy in warts, it is imperative to avoid an uncontrolled study. Warts can be self-resolving and hence the efficacy of immunotherapy as opposed to the self-resolution compromises the validity of the results.
Control Arm Options in Controlled Trials
Controlled trials allow discrimination of the patient outcome from an outcome caused by other factors (such as natural history or observer or patient expectation). Choosing a right control at the right dose and right frequency is pivotal to trial success. The controls which can be used are:
Placebo concurrent control – Placebo is a form of inert substance, or an intervention designed to simulate medical therapy, without specificity for the condition being treated. The placebo must share the same appearance, frequency, and formulation as the active drug. Placebo control helps to discriminate outcomes due to intervention (new product) from outcomes due to other factors. This design is used to demonstrate superiority or equivalence. This design must be adopted only when no effective treatment exits, and it will be deemed unethical to use a placebo control if an effective standard of care exits. Placebo must only be used if no permanent harm (death or irreversible morbidity) accrues by delaying available active treatment for the duration of the trial and is preferable for a minimal risk, short-term study
“No treatment” concurrent control – No intervention will be administered in control arm in this design. Study end points must be objective in this design. The downsides are potential for observer bias and difficulty in blinding in this design
Active treatment concurrent control – This design involves comparison of a new drug to a standard drug or compare combination of new and standard therapies vis a vis standard therapy alone. A therapeutic modality that should preferably be the current standard of care against which the active drug to be studied is compared with. This design can be used to demonstrate equivalence, non-inferiority, and superiority. This design is most ethical whenever approved drugs are available for the disease under study. The Declaration of Helsinki mandates the use of standard treatment as controls
Dose-comparison concurrent control – Different doses or regimens of same treatment are used as active arm and control arm in this design. The purpose is to establish a relationship between dose and efficacy/safety of the intervention. This design may include active and placebo groups also in addition to the different dose groups. This design may be inefficient if the therapeutic range of the drug is not known
Historical control (external and non-concurrent) – Source of controls are external to the present study and were treated at an earlier time (earlier therapeutic gold standard) or in a different setting. The advantage of historical controls is in studying rare conditions where sample size generation is difficult. The downside is that no randomization or blinding is possible in this design. A disadvantage is that the co-interventions evolve in due course of time thereby reducing the comparability of the present intervention versus historic control. Another deficiency of this design is the difference between baseline characteristics of subjects in trial arm versus historical arm.
For example, toxic epidermal necrolysis, where clinical outcomes in cyclosporine treated patients can be compared with historical controls treated in the same center with IVIg in the past.
Variants of Placebo Controlled Trial Designs
Add-on design – This design denotes a placebo-controlled comparison on top of a standard treatment given to all patients. If the improvement that is achievable in addition to that obtained from the standard treatment is small, the size of such trial may need to be very large
Early escape design – The early escape design using a placebo control allows a patient to be withdrawn from the study as soon as a predefined negative efficacy criterion has been attained. This reduces the time on placebo or in treatment failure. This design analyses failure rate, so minimizes exposure to ineffective treatment. The time for withdrawal is then used as the primary outcome variable. The patient could then be switched over to another therapy, including the test treatment if appropriate. The attendant limitations are sacrifice of study power with increased “escape” cases and evaluation of only short-term efficacy. If the drug has a slow and deliberate effect on long-term use then that might be missed in this design
Unbalanced assignment of patients to placebo and test treatment. By this design it is implied that a smaller number of patients could be assigned to the placebo group compared to the test treatment group (e.g., 2/3 case arm, 1/3 placebo arm)
-
Double-dummy design [Figure 3] – This design is of great utility if the comparator interventions are of different nature
Illustrative example – Comparison of oral acitretin versus injection purified protein derivative (PPD) in extensive verruca vulgaris. So, blinding of patients is not feasible in this scenario. But this issue can be circumvented by administering acitretin orally with a dummy injection like normal saline to one study group and injection PPD along with placebo capsule identical in size and appearance to the acitretin capsule to the comparator arm
Placebo run-in design – Placebo run-in period is a period before a clinical trial is commenced, when placebo is administered for all study subjects. The clinical data from this stage of a trial are only occasionally of value but can serve a valuable role in screening out ineligible or non-compliant participants, in ensuring that participants are in a stable condition, and also helps in providing baseline observations. After the run-in phase, randomisation is done, patients are randomized into study arms where different active interventions are added to the placebo in each study arm [Figure 4].
Figure 3.
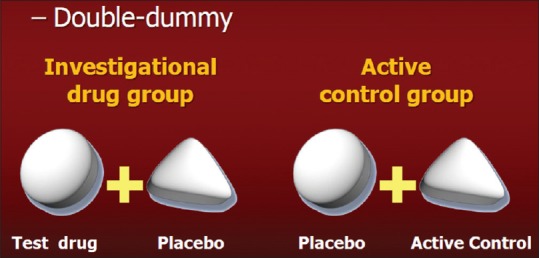
Double dummy trial design
Figure 4.
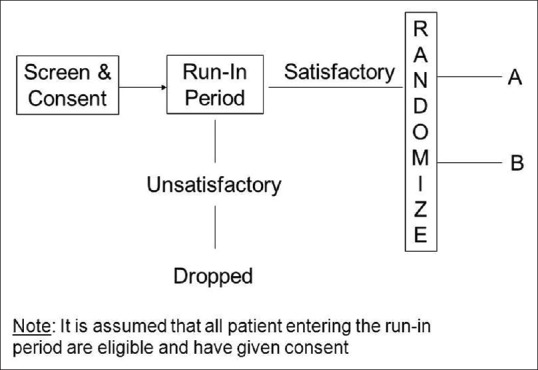
Run in design
Randomized Clinical Trials (RCT)
In randomized controlled trials, trial participants are randomly assigned to either treatment or control arms. The process of randomly assigning a trial participant to treatment or control arms is called “randomization”. Different tools can be used to randomize (closed envelopes, computer generated sequences, random numbers). There are two components to randomization: the generation of a random sequence and the implementation of that random sequence, ideally in a way that keeps participants unaware of the sequence (allocation concealment). Randomization removes potential for systematic error or bias. The biggest upside of an RCT is the balancing of both the known and unknown confounding factors which leads to wrong conclusions.
Randomization Schemes in Randomized Controlled Trials to Eliminate Confounding Factors
Stratified randomization – This refers to the situation in which strata are constructed based on values of prognostic variables and a randomization scheme is implemented separately within each stratum. The objective of stratified randomization is to ensure balance of the treatment groups with respect to the various combinations of the prognostic variables. This method can be used to achieve balance among groups in terms of subjects' baseline characteristics (covariates). Specific covariates must be identified by the researcher who understands the potential influence each covariate has on the dependent variable. To avoid strata with very less patients, the number of strata should be kept minimum. After all the subjects have been identified and assigned into strata, simple randomization is performed within each stratum to assign subjects to either case or control groups
Block randomization – Blocking is the arranging of experimental units in groups (blocks) that are similar to one another. Typically, a blocking factor is a source of variability that is not of primary interest to the experimenter. An example of a blocking factor might be the sex of a patient; by blocking on sex, this source of variability is controlled for, thus leading to greater accuracy. The block randomization method is designed to randomize subjects into groups that result in equal sample sizes. This method is used to ensure a balance in sample size across groups over time. Blocks are small and balanced with predetermined group assignments, which keeps the numbers of subjects in each group similar at all times
Randomization by body halves or paired organs (Split Body trials) – This is a scenario most often used in dermatology and ophthalmic practice where one intervention is administered to one half of the body and the comparator intervention is assigned to other half of the body. This can be implemented only if experimental treatment acts locally. Randomization is used to select which side of the body receives which drug. The upside is the elimination of confounding factors between trial arms, as the baseline characteristics of both arms are the same. The downside is difficulty in blinding the investigator, statistical analysis, and influence of therapy administered in one half of the body influencing disease on the other side as the halves of the human body is a continuum and not entirely independent entities (carryover of the experimental treatment to control half). Allocation between paired organs/split skin can obscure systemic adverse events. Paired data statistical analytic tests need to be done in this scenario
Cluster randomization – Study patients and treating interventionists do not exist in isolation. Sometimes interventions need to be applied at ward level, village level, hospital level, or group practice level. Hence intervention is administered to clusters by randomization to prevent contamination. Each cluster forms a unit of the trial and either active or comparator intervention is administered for each cluster
Allocation by randomized consent (Zelen trials) – Eligible patients are allocated to one of the two trial arms prior to informed consent. This is utilized when informed consent process acts as an impediment to study subject accrual. However, this design raises serious ethical uncertainties and must only be used in severely flagging trials in terms of insufficient sample size of great public health importance and is not recommended in routine clinical trial design
Minimization – Stratification based on multiple co-variates (age, sex, gender, baseline severity of disease, personal habits, co-morbidities, treatment naivety, etc.) leads to excessive number of strata with smaller number of patients at times in each strata. Hence, an alternate strategy to control for prognostic variables to avoid such small strata is minimization. After identification of these variables, they are dichotomized at some break point in case of continuous variables or based on presence or absence of a categorical variable. Then each dichotomized half is given a value of 0 or 1 (e.g., male = 0, female = 1; age <50 years = 0, age >50 years = 1). Thus, in a female of age 55, the total will be 1 + 1 = 2. A male of age 65 will be allocated 0 + 1 = 1 point, a female of age 45 will have score of 1 + 0 = 1 point, etc. For example, patient number 1 with score 2 is randomized to control arm. Patient no. 2 has 1 point and to minimize the difference in total scores between the study arms, he is allocated to case arm. So now the control arm total score is 2 and case arm score is 1. Patient 3 is a female with score 1 and will be allocated to case arm and thus the cumulative score in both groups will be balanced at 2 points. Once the running scores in both arms are tied, the next recruited subject is again randomly allocated and the whole cycle repeats. Thus, minimization is a viable alternative to randomization for known prognostic factors, but does not factor in the unknown prognostic confounding variables. Hence, it can be considered a platinum standard to the gold standard of random allocation.
RCT Designs
a. Parallel group trial design
Parallel arm design is the most commonly used study design. In this design, subjects are randomized to one or more study arms and each study arm will be allocated a different intervention. After randomization each participant will stay in their assigned treatment arm for the duration of the study [Figure 5]. Parallel group design can be applied to many diseases and allows running experiments simultaneously in a number of groups, and groups can be in separate locations. The randomized patients in parallel groups should not inadvertently contaminate the other group by unplanned co-interventions or cross-overs.
Figure 5.

Parallel arm design
Steps involved in a parallel arm trial design
Eligibility of study subject assessed → Recruitment into study after consent → Randomization → Allocation to either test drug arm or control arm
Illustrative example – A comparative trial of Acitretin and Apremilast in palmoplantar psoriasis, where there exits clinical equipoise as regarding efficacy can be conducted as a randomized controlled (acitretin as active control) parallel arm trial design.
b. Cross over design
Another advantage is requirement of a smaller sample size [Figure 6]. The ethical limitations of a placebo control are partially overcome by a cross over design in which each patient receives both interventions but in a different order. The order in which patient receives interventions is randomized. Each person serves as his/her own control results in balancing the covariates in treatment and control arms. Another advantage is requirement of a smaller sample size. In this design, some participants start with drug A and then switch to drug B (AB sequence) in one trial arm, while subjects in other trial arm start with drug B and then switch to drug A (BA sequence). Adequate washout period must be given before crossover to eliminate the effects of the initially administered intervention. Outcomes are then compared within the same subject (effect of A vs. effect of B). The requirements are two fold. (a) The disease must be chronic, stable, and incurable and characteristics must not vary for the duration of the two study periods and the interim wash out period and (b) the effect of each drug must not be irreversible. Bioequivalence and biosimilar equivalence studies usually utilize a cross over design. The duration of follow-up for the patient is longer than for a parallel design, and there is a risk that a significant number of patients do not complete the study and drop out leading to compromised study power. Salient points regarding cross over trials are summarized in Table 1.
Figure 6.

Cross over trial design
Table 1.
Points to be factored in during cross over design
| Effects of intervention during first period should not carry over into second period. In case of suspected carry over effects more complex sequences are needed which increase study duration and thus the chance of drop outs |
| The treatment effect should be relatively rapid in onset with rapid reversibility of effect |
| The disease has to be chronic, stable, and non-self-resolving. This design is usually avoided in vaccine trials because immune system is permanently affected. Patient’s health status must be identical at the beginning of each treatment period |
| Period effect - the changes in patient characteristics due to the effect of the first drug or extraneous factors to which the patients are exposed to over time leads to what is called the ‘period effect’.Internal and external trial environment must remain constant over time. This reduces ‘period effect’ |
| Before the cross over is implemented, a drug free wash out period for complete reversibility of drug effect administered in the first period in a trial arm needs to be ensured so as to avoid a cumulative or substractive effect which piggy backs on to the drug administered in the second period. An accepted convention for the wash out period is five half lives of the drug involved |
| Each treatment period must give adequate time for the intervention to act meaningfully |
| Sensitivity of trial power to drop outs due to longer anticipated duration of trial |
| Identification of culprit drug for delayed adverse events during later period of the study becomes difficult |
Variations of crossover designs
(i) Switch back design (ABA vs BAB arms) – Drug A → Drug B → Drug A in one arm versus Drug B → Drug A → Drug B in other arm. The switch back and multiple switchback designs are of emerging relevance with the advent of biosimilars where switchability and interchangeability of a biosimilar to a bio-originator molecule can only be confirmed by such trial designs.
(ii) N of 1 design – N of 1 trials or “single-subject” or “structured within-patient randomized controlled multi-crossover trial design” are used to evaluate all interventions in a single patient. A typical single patient trial consists of experimental/control treatment periods repeated a number of times. The order of treatment is randomly assigned within each treatment period pair. Usually, the primary objective of such a trial is to determine the treatment preference for the individual patient and this design is gaining popularity in recent times. The advantage of this design is its flexibility such that it can be continued until a definitive conclusion can be reached for the particular subject being studied. The utility also rests in analyzing treatments that elicit heterogenous responses in different subjects. Data from many N-of 1 subjects can be even combined to derive population effect sizes by meta-analysis or Bayesian methods.
c. Factorial Design (2 × 2 design)
This is a design suited for the study of two or more interventions in various combinations in one study setting and helps in the study of interactive effects resulting from combination of interventions. Both use versus non-use of agent and different dose intensities of one agent can be studied as illustrated in Figure 7. This design can answer two or more research questions with one trial and delivers more “bang for the buck” with limited sample sizes. In a 2 × 2 factorial design with placebo, patients are randomized into four groups: (i) to treatment A plus placebo; (ii) treatment B plus placebo; (iii) both treatments A and B; or (iv) neither of them, placebo only. Outcomes are analyzed using two-way analysis of variance (ANOVA) comparing all patients who receive treatment A (groups 1 and 3) with those not treated with A (groups 2 and 4), and all patients who receive treatment B (groups 2 and 3) with those not treated with B (groups 1 and 4). The sample size requirement reduces by almost 50% as compared to carrying out drug A and drug B comparison with placebo in 2 different trials. However, a prerequisite requirement is that there is no interaction between treatments A and B. If interaction exits, then loss of power is possible in case of separate analyses of the four different combinations. If an interaction is anticipated, then that has to be factored into the sample size in addition to estimated sample size. Hence, it is not suited for rare diseases where interaction between A and B are likely. The limitations of this trial design are complexity of trial, difficulty in meeting inclusion criteria of both drugs during study subject recruitment, inability to combine two incompatible interventions, complex protocols, and statistical analytical complexities. Incomplete factorial designs are used when it is deemed unethical to exercise a non-intervention option and here the placebo only arm is eliminated.
Figure 7.

Factorial design
d. Randomized withdrawal design [Enrichment enrolment randomized withdrawal (EERW)]
In this design, after an initial open label period (enrichment period) during which all subjects are assigned to receive intervention, the non-responders are dropped from the trial and the responders (the enriched population) are randomized to receive intervention or placebo in the second phase of the trial. Thereby only responders are carried forward and randomized. Study analysis is conducted using only data from the withdrawal phase and outcome is usually relapse of symptoms. The enrichment phase increases statistical power for a given sample size. The randomized withdrawal design aims to evaluate the optimal duration of a treatment in patients who respond to the treatment. The advantage is reduction in the time on placebo since only responders are randomized to placebo thereby giving an ethical advantage. This ensures acceptability to trial subjects and hence facilitates recruitment. This design can assess if treatment needs to be continued or can be stopped. This design allows subjects who have therapy withdrawn (control arm subjects) to re-start effective therapy after they have reached the study end-point (e.g., relapse of disease), thereby addressing both ethical concerns and patient preferences about placebo assignment by shortening the time that patients are on placebo [Figure 8].
Figure 8.
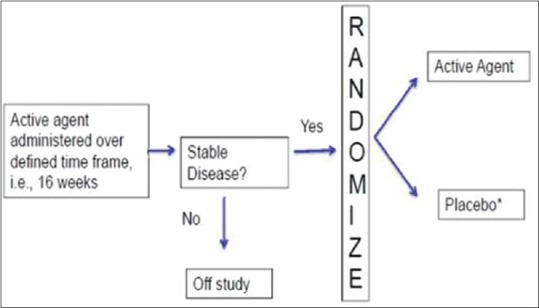
Enrichment Enrolment Randomised withdrawal design
Illustrative example. Subjects of psoriasis vulgaris are initiated on a biological and a group of patients attain PASI 75 response at 16 weeks. Subsequently the non-PASI 75 achievers are dropped from the trial due to lack of efficacy. PASI 75 responders are continued on the drug or are assigned to placebo and retention of PASI 75 response at 1 year after randomization can be compared between two arms. If there is no statistically significant difference in outcomes between the arms, an expensive biological can be administered till PASI 75 is achieved and then omitted, thereby reducing cost of therapy.
The disadvantages of this design are (1) Missing data due to withdrawals that can be countered by imputation or using time to event analysis. (2) Carry over effects from enrichment phase. (3) Restriction to responders can affect external validity of results. (4) The treatment effect is overestimated since only responders are included. (5) Not suitable for unpredictable diseases (e.g., spontaneous remission) or those with slow evolution; hence, can be used only for chronic stable diseases. (6) Correct duration of enrichment phase is important.
Newer Study Designs
a. Adaptive randomization methods (play the winner, drop the loser designs)
This paradigm is useful only for studies with binary outcomes and are most useful when the anticipated effect size being evaluated is large. The play-the-winner and the drop-the-loser designs aim to favor the group with the best chance of success by increasing the probability of patients being randomized to that group. The probability of being randomized to one group or another is modified according to the results obtained with previous patients. The response of each patient after treatment plays an essential role in the determination of subsequent compositions of the study population. In “play the winner” design, more study subjects are randomized to the effective intervention. In “drop the loser” strategy, study subjects are removed from the ineffective intervention arm. The advantage is enhanced exposure of subjects to an effective intervention and increased chances of recruitment and this can also result in unequal group sizes, which can adversely affect statistical power. The adaptation can be based on the following methods, which can be combined in select trial situations [Table 3].
Table 3.
Rarer designs used in clinical trials
| Trial design | Description |
|---|---|
| Matched pairs design | Patients with similar characteristics who are expected to respond similarly are grouped into matched pairs and then the members of each pair are randomized to receive either drug or control. The confounding factors are eliminated and intra-group variability is reduced |
| Delayed start design (DS) | This design has initial placebo-controlled phase (patients randomized to treatment or placebo) followed by active control phase (all patients receive treatment). Those in the initial placebo group have a delayed start on active drug. Disease progression as well as disease relapses can be studied and design evaluates the effect of the treatment on the symptoms and the evolution of the disease. There must be a sufficient number of follow-up visits to measure the treatment effect. Upside of design is that every subject is exposed to active therapy. Downsides are evaluation bias and carry over effect. |
| Randomized placebo phase design (RPPD) | Subjects are first randomly allocated to either an experimental or a control group. However, after a short, fixed time period (called the placebo-phase), all patients in the control group are switched to the experimental treatment. All patients receive the tested treatment in the end - but have varying lengths of time on placebo. This design assumes that if treatment is effective, then those administered drug earlier will respond sooner. At the end of the trial, average time-to-response is compared between the two groups, most often using survival analysis methods. Upside is that all patients receive active drug by the end of trial. There is a need to establish an effective placebo phase duration (i.e., short enough to ensure no change in condition over time and long enough to ensure valid outcome measurement). A longer placebo-phase duration will decrease the required sample size but increase the chance of dropouts. |
| Stepped wedge design (SWS) | Intervention allocated sequentially to participants either as individuals or clusters of individuals is called stepped wedge design. In the first step all patients are initiated on control interventions and subsequently over 4 time periods, individual or clusters of individuals are randomized to treatment arm with all patients receiving treatment by the end of the study period. The utility rests in testing medicines that are anticipated to cause harm. It can be used when therapy cannot be initiated concurrently to all subjects simultaneously |
| Three stage design (3S) | Constitutes initial randomized placebo-control phase, a randomized withdrawal stage for responders from Stage 1, and a third phase when all placebo non-responders (from Stage 1) are first prescribed active treatment and then the responders (from Stage 3) are randomly assigned to placebo or treatment. It is an extension of randomized withdrawal design. This suits only to study chronic conditions where both response to therapy and withdrawal of therapy can be assessed. The withdrawal phase has to be sufficiently long so that the drug can be completely washed out and the clinical effects of therapy reversed. Requires less sample size than parallel arm design. Helps to evaluate the efficacy of a therapeutic agent in a particular patient subpopulation when efficacy in the general patient population has already been established |
-
(i)
Response adaptive – This design reduces patient recruitment to ineffective intervention arms. It requires rapidly available measurable responses. It is infeasible in diseases and therapies with a prolonged time to outcome. This design can compromise allocation concealment and result in selection bias as trial progresses. It can also be affected by changes in patient or treatment characteristics over time that are associated with the treatment received resulting from inherent chances of prolonged recruitment schedule (temporal drift)
-
(b)
Ranking and selection –First phase of this adaptive design has subjects randomized to many interventions and placebo. The best therapy from Phase 1 is compared with placebo in a randomized parallel or adaptive design in Phase II. The final comparison is between all subjects receiving the selected intervention versus all the subjects receiving placebo in both phases combined. It is best suited for multiple intervention comparison in low sample size scenarios. However, there is a chance that wrong selection of the most efficacious therapy in phase I will vitiate the trial results
-
(c)
Sequential adaptive design – This design allows repeated interim analysis and stoppage once end point of efficacy, safety or futility is achieved. In contrast to traditional trials, the final number of participants needed for a sequential trial is unknown at initiation. The trial ends at the first interim analysis which meets pre-set stopping criteria thereby potentially limiting the number of subjects exposed to an inferior, unsafe, or futile treatment or one that is already proven efficacious. Analysis can be performed after each patient (continuous sequential) or after a fixed or variable number of patients (group sequential). This design is only effective when study enrolment is expected to be prolonged and treatment outcomes occur relatively soon after recruitment, so that outcomes can be measured before the next patient or group of patients is likely to be recruited. Challenges include the complexity of analyzing multiple treatments, Power calculation complexities, and appropriate selection of timing and number of interim analyses.
b. Seamless design
There is also growing interest in developing a type of continuous trial process by “connecting” different trial phases, especially from Phase II to Phase III, called seamless design. Scientists combine the learning stage of Phase II trials and confirmatory stage of Phase III trials. In the beginning of Phase II, subjects are randomized into the treatment arms of A, B, combined therapy of A and B, or control. An interim analysis is then performed to determine which active arm should be dropped. In the confirmatory stage of Phase III study, the treatment groups with the residual effective active arm and control arm will be investigated. This design has two variants: “inference seamless” and “operational seamless”. In the inference seamless approach, the subjects will carry their treatment arm from learning phase to confirmatory phase, and the data in both phases will be analyzed together. For the operational seamless variant, the data in two phases are analyzed separately.
c. Internal pilot design
In a conventional pilot study, participants are often ineligible for analysis along with cases in future definitive studies due to concerns about selection bias, carry-over, and training effects. Where patients are few in number as in case of rare diseases, allocating them to a pilot study rather than the definitive study could be seen as a wasteful approach. In an internal pilot study, the first phase of the study is designated a “pilot phase,” and the study is continued till this sample size is achieved (definitive phase) and analysis incorporates the pilot subjects also. In contrast to external pilots, internal pilots can be large, as they do not “use up” eligible patients and do not require additional time or funds.
Selection of Trial Designs
Although there is no perfect all-encompassing study design for each trial situation, an overarching algorithm is depicted in Tables 2 and 4.
Table 2.
Factors to consider in design selection
| Number and characteristics of treatments to be compared |
| Characteristics of the disease under study |
| Study objectives |
| Timeframe |
| Treatment course and duration |
| Carry over effects |
| Duration of the study which is linked with drop-out rates |
| Cost and logistics |
| Patient convenience |
| Ethical considerations |
| Statistical considerations |
| Study subject availability (disease rarity) |
| Inter and intra subject variability |
Table 4.
Algorithm for choice of study design
| Trial situation | Appropriate suggested design |
|---|---|
| To demonstrate superiority, equivalence, and non-inferiority | Randomized parallel arm design |
| If the intervention has a predictable and rapidly reversible effect | EERW (if efficacy is expected only in a subset), crossover (if enough sample size and stability of disease for 2 study periods), or n-of-1 design (if sufficient sample not available for crossover) |
| If the time between study inclusion and outcome assessment is short compared with recruitment time of study subjects | Adaptive design If preliminary data favors one drug - response adaptive randomizationIf multiple options exis -> Ranking and selection |
| Where the therapy is likely to provide a lasting response and the possibility of randomization to placebo is major impediment to recruitment | Randomized placebo phase design (RPPD) |
| If there are two or more therapiesof interest that can be given concurrently or in cases of rare disease | Factorial design, parallel arm |
| If the outcomes of intervention are irreversible | Parallel group, factorial, randomized placebo phase (RPPD), Stepped Wedge (SWS), adaptive design (not for drugs with slow response on disease) |
| (a) Minimizes exposure to placebo (b) No minimization of exposure time to placebo |
RPPD, SW, Adaptive design Parallel arm, Factorial |
| If the outcomes of intervention are reversible | Parallel group, Crossover, Factorial, N of 1, RPPD, Stepped Wedge (SWS), Enrichment Enrolment Randomised Withdrawal (EERW), Early Escape (EE), Delayed start (DS), Three Stage (3S), Adaptive |
| (a) Rapid response of disease to intervention - Minimizes exposure to placebo - No minimization of exposure time to placebo - Intra-patient comparison - Inter-patient comparison (b) Slow response of disease to intervention - Minimize time on placebo - No minimization of time on placebo |
Any design above can be used DS, RPPD, SWS, EERW, EE, 3S, Adaptive design Parallel arm, Crossover, Factorial, Latin square, N of 1 Cross over, Latin Square, N of 1 Parallel, Factorial Parallel arm, Factorial, DS, RPPD, SWS, EERW DS, RPPD, SWS, RW Parallel arm, factorial |
Conclusion
Advances in statistical software and computing power continue to allow for increasingly more complex study designs and analytical techniques, and researchers should take best advantage of these advances. There is a concern regarding acceptability of evidence generated by alternative trial designs by regulatory authorities and peer reviewers. It is imperative to understand that same research question may be tackled through alternative designs and that there is no definitive trial design for every research question. The time frame, logistics involved, and availability of study subjects are key to selection. Factors, such as objective of the trial, number of patients needed, length of trial, and how the variability is handled, could be important in the choice of the most suitable trial design. The readers are reminded of the fact that no trial design is perfect, and no design provides optimum answer to all research questions. In this imperfect milieu, all the above-mentioned contingencies must guide the researchers to study the most optimum design among a clutch of options and they must incorporate biostatistician in initial trial design and post-trial analysis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Suggested Reading
- 1.Spilker B. Guide to Clinical Trials. New York: Raven; 1991. [Google Scholar]
- 2.Kianifard F, Islam MZ. A guide to the design and analysis of small clinical studies. Pharm Stat. 2010;10:363–8. doi: 10.1002/pst.477. [DOI] [PubMed] [Google Scholar]
- 3.Feldman B, Wang E, Willanc A, Szalai JP. The randomized placebo-phase design for clinical trials. J Clin Epidemiol. 2001;54:550–7. doi: 10.1016/s0895-4356(00)00357-7. [DOI] [PubMed] [Google Scholar]
- 4.Jones B, Kenward MG. Design and Analysis of Cross-Over Trials. London: Chapman and Hall; 2003. [Google Scholar]
- 5.Senn S. Cross-over trials in statistics in medicine: The first '25' years. Stat Med. 2006;25:3430–42. doi: 10.1002/sim.2706. [DOI] [PubMed] [Google Scholar]
- 6.Guyatt GH, Heyting A, Jaeschke R, Keller J, Adachi JD, Roberts RS. N of 1 randomised trials for investigating new drugs. Control Clin Trials. 1990;11:88–100. doi: 10.1016/0197-2456(90)90003-k. [DOI] [PubMed] [Google Scholar]
- 7.Scuffham PA, Nikles J, Mitchell GK, Yelland MJ, Vine N, Poulos CJ, et al. Using N-of-1 trials to improve patient management and save costs. J Gen Intern Med. 2010;25:906–13. doi: 10.1007/s11606-010-1352-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couper DJ, Hosking JD, Cisler RA, Gastfriend DR, Kivlahan DR. 2005. Factorial designs in clinical trials: Options for combination treatment studies. J Stud Alcohol Suppl. 2005:24–32. doi: 10.15288/jsas.2005.s15.24. [DOI] [PubMed] [Google Scholar]
- 9.Brown CH, Ten Have TR, Jo B, Dagne G, Wyman PA, Muth_en B, et al. Adaptive designs for randomized trials in public health. Annu Rev Public Health. 2009;30:1–25. doi: 10.1146/annurev.publhealth.031308.100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berry DA. Adaptive clinical trials in oncology. Nat Rev Clin Oncol. 2011;9:199–207. doi: 10.1038/nrclinonc.2011.165. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Rosenberger WF. Response adaptive randomization for clinical trials with continuous outcomes. Biometrics. 2006;62:562–9. doi: 10.1111/j.1541-0420.2005.00496.x. [DOI] [PubMed] [Google Scholar]
- 12.Karrison TG, Huo D, Chappell R. A group sequential, response-adaptive design for randomized clinical trials. Control Clin Trials. 2003;24:506–22. doi: 10.1016/s0197-2456(03)00092-8. [DOI] [PubMed] [Google Scholar]
- 13.Stallard N, Rosenberger WF. Exact group-sequential designs for clinical trials with randomized play-the-winner allocation. Stat Med. 2002;21:467–80. doi: 10.1002/sim.998. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberger W. Randomized play-the-winner clinical trials: Review and recommendations. Control Clin Trials. 1999;20:328–42. doi: 10.1016/s0197-2456(99)00013-6. [DOI] [PubMed] [Google Scholar]
- 15.Jennison C, Turnbull BW. Adaptive seamless designs: Selection and prospective testing of hypotheses. J Biopharm Stat. 2007;17:1135–61. doi: 10.1080/10543400701645215. [DOI] [PubMed] [Google Scholar]
- 16.Katz N. Enriched enrollment randomized withdrawal trial designs of analgesics: Focus on methodology. Clin J Pain. 2009;25:797–807. doi: 10.1097/AJP.0b013e3181b12dec. [DOI] [PubMed] [Google Scholar]
- 17.Gupta S, Faughnan ME, Tomlinson GA, Bayoumi AM. A framework for applying unfamiliar trial designs in studies of rare diseases. J Clin Epidemiol. 2011;64:1085–94. doi: 10.1016/j.jclinepi.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 18.Brown CA, Lilford RJ. The stepped wedge trial design: A systematic review. BMC Med Res Method. 2006;6:54. doi: 10.1186/1471-2288-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honkanen VE, Siegel AF, Szalai JP, Berger V, Feldman BM, Siegel JN. A three-stage clinical trial design for rare disorders. Stat Med. 2001;20:3009–21. doi: 10.1002/sim.980. [DOI] [PubMed] [Google Scholar]
- 20.Wittes J, Brittain E. The role of internal pilot studies in increasing the efficiency of clinical trials. Stat Med. 1990;9:65–71. doi: 10.1002/sim.4780090113. [DOI] [PubMed] [Google Scholar]
- 21.Cornu C, Kassai B, Fisch R, Chiron C, Alberti C, Guerrini R, et al. Experimental designs for small randomised clinical trials: An algorithm for choice. Orphanet J Rare Dis. 2013;8:48. doi: 10.1186/1750-1172-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abrahamyan L, Feldman BM, Tomlinson G, Faughnan ME, Johnson SR, Diamond IR, et al. Alternative designs for clinical trials in rare diseases. Am J Med Genet C Semin Med Genet. 2016;172:313–31. doi: 10.1002/ajmg.c.31533. [DOI] [PubMed] [Google Scholar]