

Abstract
Recent advances in chromatography and mass spectrometry (MS) have made rapid and deep proteomic profiling possible. To maximize the performance of the recently produced Orbitrap hybrid mass spectrometer, we have developed a protocol that combines improved sample preparation (including optimized cellular lysis by extensive bead beating) and chromatographic conditions (specifically, 30-cm capillary columns packed with 1.7-μm bridged ethylene hybrid material) and the manufacture of a column heater (to accommodate flow rates of 350–375 nl/min) that increases the number of proteins identified across a single liquid chromatography–tandem MS (LC-MS/MS) separation, thereby reducing the need for extensive sample fractionation. This strategy allowed the identification of up to 4,002 proteins (at a 1% false discovery rate (FDR)) in yeast (Saccharomyces cerevisiae strain BY4741) over 70 min of LC-MS/MS analysis. Quintuplicate analysis of technical replicates reveals 83% overlap at the protein level, thus demonstrating the reproducibility of this procedure. This protocol, which includes cell lysis, overnight tryptic digestion, sample analysis and database searching, takes ~24 h to complete. Aspects of this protocol, including chromatographic separation and instrument parameters, can be adapted for the optimal analysis of other organisms.
INTRODUCTION
Ideally, MS techniques would permit the rapid identification of every protein in the proteome. Historically, deep sampling of proteomes has been laborious and difficult to achieve. A major limitation has been the tandem MS (i.e., MS/MS) sampling speed. MS/MS acquisition rates have ranged from 1 to 10 Hz and often could not keep pace with the number of unique, co-eluting peptide species found in complex mixtures. The resulting undersampling led to stochastic precursor sampling, and it meant that there was a lot of variability in the proteins identified from different HPLC runs of the same sample1–3. Identifying low-abundance proteins in such experiments was difficult because these low-level species were often not sampled when their peaks were close to large, wide peaks associated with more-abundant peptides3. A common solution is fractionation of the sample before LC-MS/MS analysis3. Although effective, this approach is timeconsuming for both the analyst and the instrument, which limits the throughput of proteomics.
Advances in the MS/MS acquisition rate of mass spectrometers have increased the percentage of proteomes identifiable in a single experiment, while decreasing instrument analysis time. A major boost in acquisition speed was realized with the introduction of a novel Orbitrap hybrid mass spectrometer consisting of a mass filter, a collision cell, a high-field Orbitrap and a dual cell linear ion trap analyzer (Q-OT-qlT, Orbitrap Fusion)4,5. Here, precursor ions are selected by the quadrupole and fragmented by higher-energy collisional dissociation (HCD), collisional-activated dissociation (CAD) or electron transfer dissociation, and they are analyzed with respect to m/z either by the ion trap or Orbitrap mass analyzer. Precursor selection using the quadrupole mass filter allows the ion trap and Orbitrap analyzers to operate in parallel, which substantially improves the acquisition speed over previous-generation instruments. When product ions are analyzed in the ion trap, the Fusion enables MS/MS scan rates of up to 20 Hz (ref. 5).
As the first eukaryote with a sequenced genome6, S. cerevisiae is commonly used as a model organism is systems biology. Several proteomics studies have identified the estimated 4,500 proteins7 that are expressed in yeast under standard laboratory conditions8–12. Building on the successes of these earlier studies, we sought to improve both the depth of proteome coverage and the speed at which proteins can be identified. Although the very fast acquisition speed of the Fusion instrument greatly increases the number of peptides selected for MS/MS sequencing, we also sought to increase peptide identifications through improved sample preparation and chromatography, and through the optimization of instrument parameters. Specifically, improvements include lysing yeast cells by extensive bead beating, the use of a 30-cm capillary column packed with 1.7-μm bridged ethylene hybrid material (Waters Corporation) operating at flow rates of 350–375 nl/min, and the addition of DMSO to the mobile phase. Similar to the findings in previously published reports13,14, we find that this additive substantially increases the number of peptides identified in each run.
This optimized sample preparation, coupled with MS analysis performed in the Orbitrap and low-resolution HCD MS/MS performed by ion trap rapid scan, allowed us to identify up to 34,535 yeast peptides over a 70-min LC-MS/MS run. This translates to up to 4,002 proteins, or identification of ~90% of the expressed yeast proteome. Further, this system is capable of identifying 67 proteins per min (ref. 4). This protocol describes cellular lysis, trypsin digestion, column packing, LC gradient setup and Orbitrap Fusion instrument parameters that lead to comprehensive yeast proteome coverage with minimal instrument time. If you are beginning from yeast lysis, this protocol can be completed in 24 h. This protocol is easily adaptable to other organisms; for samples that require less forceful lysing (for example, human cell lines), the bead-beating steps may be omitted.
Experimental design
Although the protocol may be modified on the basis of the sample of interest or available instrumentation, we note that the following details have provided us with the best results:
Yeast lysis.
A vigorous bead beating procedure for yeast cell lysis, in which proteins are lysed via a ball mill, produces the optimal extracted protein mixture for downstream bottom-up processing. Yeast pellets are milled at 30 Hz eight times for 4 min each, as these lysates produced more complete proteome coverage compared with either lysing at 30 Hz for fewer rounds or lysing at a lower frequency. As others have also noted15, we observe a further increase in the number of peptides identified when the cellular debris is not cleared before analysis. Preserving the cellular debris resulted in a 5% increase in protein identifications when compared with an identical analysis in which the precipitated lysate was discarded before digestion. Gene ontology analysis reveals enrichment in membrane proteins when the cellular debris is included during digestion, which results in more complete proteome coverage. After digestion, the samples are centrifuged and only the supernatant is processed.
Chromatography column and column heater.
Ultra-high-performance nano-LC setups using 30–35-cm columns packed with 1.7-μm C18 particles produced optimal chromatography for increased peptide identifications. 1.7-μm particles improve chromatographic resolution peaks, and they increase the peak capacity as compared with packing materials made up of larger particles. This allows for more unique identifications. This setup requires a column heater to operate at suitable pressures, but higher temperatures provide additional chromatographic improvements. We found it economical to manufacture a column heater in-house (Fig. 1; plus computer-aided design (CAD) CAD files included in the Supplementary Data), but there are several excellent models that are commercially available, such as the PRSO-V1 from Sonation or several options from Phoenix S&T, among other options. If a column heater is not available, we recommend using 3-μm C18 particles or packing a shorter column (~15 cm) with1.7-μm particles. This is because both smaller particle size and increased column length can provide more theoretical plates and thus better chromatographic separation.
Figure 1|.
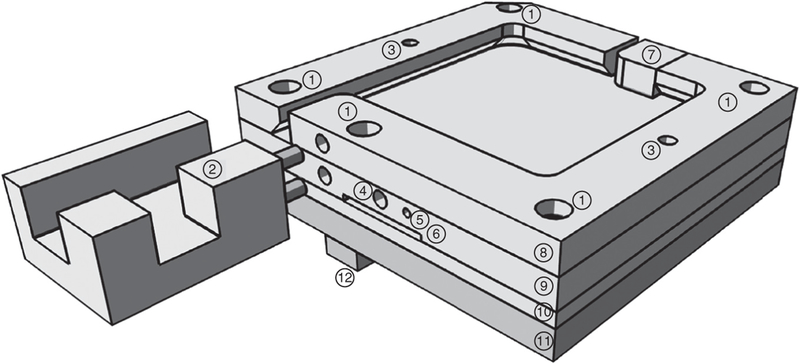
Structure of the in-house-manufactured column heater. Labeled rendering shows the column oven without a lid. (1) Holes for 4–40 ×¾-inch screws to attach the four layers to one another. (2) Polycarbonate micro-tee retainer. (3) Holes to accept pins from lids (not shown). (4) Hole for resistance thermometer. (5) Hole for grounding wire. (6) Slot for foil heater connections. (7) Spring-loaded column tip retainer. (8) Undercut top layer. (9) Layer containing thermometer, heater (against bottom), and grounding wire. (10) Aluminum layer between heater and polycarbonate base plate. (11) Polycarbonate base plate. (12) Polycarbonate attachment to the nanospray ion (NSI) source.
Commercial heaters may be more suitable for some research groups, as their manufacture requires basic familiarity with computer-aided design drawing software, electronics and metal machining. In our laboratory, the grounded column oven was manufactured from aluminum and polycarbonate, and it relies on a resistive heater that is driven by a solid-state relay connected to a proportional-integral derivative (PID) controller receiving feedback from a resistance temperature detector. We set the PID controller to allow temperature control from 25 to 80 °C. Note that the design is intrinsically safe against runaway overheating in the case of a closed relay failure, as the 20-W heating element is too weak to heat the oven to the glass transition temperature of polycarbonate (~145 °C). The oven fits completely within the Flex Ion Source without any changes to the source, and it can be controlled from the instrument computer via a USB interface. Columns 10 cm or longer fit into the 5.1 × 5.1 cm heater cavity whose edges are undercut by 3.5 mm to securely retain columns, and the emitter tip is held in place by a spring-loaded closure. In addition to a list of materials and components needed, complete production drawings and an electrical schematic drawing are available in the Supplementary Data.
Chromatography mobile phase.
As previously16 noted, the addition of 5% (vol/vol) DMSO to mobile phases A and B increases the ionization efficiency of peptides, which results in an increase in the intensity of peptide precursors13,14. To examine this shift toward higher precursor intensities, duplicate injections of 1 μg of tryptic yeast peptides were analyzed with and without the addition of 5% (vol/vol) DMSO in the mobile phases. A total of 24,067 peptides were common across all the four separations; their log2 intensities are plotted in Supplementary Figure 1. The addition of DMSO increased the average precursor signal from ~1.8 × 107 to 3.3 × 107 (arbitrary units). This shift toward higher intensities contributes to an increase in unique peptide identifications, from an average of 32,481 to 34,246 when DMSO is included. Note that this advantage may be different for each instrument type, and it may also depend on other instrumental variables such as duty cycle, column width and/or ionization source. By using the highest-purity DMSO commercially available, we have observed no differences in the cleaning cycles or maintenance of our instruments.
Mass spectrometry.
A survey scan in the Orbitrap, followed by rapid-scan ion trap HCD MS/MS, affords the highest number of unique peptide identifications. The Q-OT-qIT geometry of the Orbitrap Fusion permits ion injection and HCD fragmentation, but not CAD fragmentation, to occur simultaneously with ion trap mass analysis. This translates to ~10% reduction in duty cycle and thus a 6% increase in MS/MS scans for HCD compared with CAD fragmentation. On average, ion trap analyses take 30–35 ms, and setting a low max injection time of 25–35 ms ensures that ion injection and fragmentation are optimally parallelized with the ion trap analysis. When it is optimized, ion trap MS/MS reduces the duty cycle by ~20% compared with Orbitrap MS/MS, while maintaining an identification rate above 50% for all MS/MS scans. In addition, quadrupole isolation of 0.7 Th width around the target precursors works optimally for ion trap MS/MS, presumably by reducing coisolation of off-target precursors, while maintaining adequate flux for MS/MS analysis.
Although the number of MS/MS scans taken at MS resolving power settings of 30,000, 60,000 and 120,000 (at m/z 200) is comparable, the most unique peptide identifications were observed when using the 60,000 or 120,000 MS1 scans. Further, although more MS/MS events and peptide spectral matches (PSMs) are recorded at an MS1 resolving power of 15,000, an increase in the resolving power to 60,000 gives ~20% boost in unique peptide identifications. We reason that with increased resolving power comes a corresponding increase in the number of observable peptidic features, which allows deeper sampling of the proteome. An increase in MS resolving power above 120,000, however, yields no further improvement in identifications, and the 450,000 resolving power slows down the duty cycle enough to reduce the number of identifications.
Setting the MS1 automatic gain control (AGC) to 5 × 105, the highest allowable, provided the highest number of scans and unique peptide identifications.
Optimal values for some of the parameters described above will differ with instrumentation. Specifically, optimal AGC values differ between the Orbitrap Elite and the Orbitrap Fusion, and they should be adjusted accordingly. In addition, these parameters should not be treated as independent variables; changing any of these parameters may affect how well other parameters work. For example, if the MS/MS max inject time was set to 100 ms, then one would probably observe little difference between the Orbitrap and ion trap MS/MS performance.
Limitations
Results may vary greatly depending on the type, the generation and the operational state of the mass spectrometer used, as well as of the nano-LC and chromatography setup. Any added multiplexing labels for isobaric tags, including TMT and iTRAQ, will have a negative effect on the observed proteomic coverage, as this will require Orbitrap analysis.
The results are likely to change substantially depending on the protease used. For example, when using LysC as a protease, we observe ~30% decrease in unique peptide identifications. Similar or greater decreases in identifications are expected when other proteases, including GluC, chymotrypsin and AspN, are used. Proteomic coverage will vary with the amount of peptide sample being analyzed. Typically, injecting much less than 1 μg of sample reduces the sampling depth. Similarly, injecting much more than ~1.5 μg can reduce the column lifetime.
Organisms of higher complexity, containing greater numbers of protein-encoding genes than yeast (for example, mammalian or plant species), will have an increased dynamic range of protein abundance. With such organisms, it may be unrealistic to achieve full proteome coverage in a single run, which necessitates offline fractionation. However, following the protocols outlined in this manuscript will help minimize the number of fractions and instrument time required.
MATERIALS
REAGENTS
Acetonitrile, HPLC grade (ACN; Fisher Scientific, cat. no. A955–4)
Agar (Sigma-Aldrich, cat. no. 9002–18-0)
Ammonium formate (Sigma-Aldrich, cat. no. 516961)
Bicinchoninic acid (BCA) protein assay kit (Pierce, cat. no. 23227)
Calcium chloride (Sigma-Aldrich, cat. no. 223506–500g)
cOmplete protease inhibitor cocktail tablets mini, EDTA-free (Roche, cat. no. 11846 170 001)
Dextrose (Fisher Scientific, cat. no. D16500)
DMSO (≥99.9%; Sigma-Aldrich, cat. no. 472301)
DTT (Sigma-Aldrich, cat. no. 43819)
Formic acid (FA; Thermo Scientific, cat. no. 28905)
Glycerol (Sigma-Aldrich, cat. no. G5516)
Iodoacetamide (IAA; Acros Organic, cat. no. 122270250)
-
Hydrofluoric acid (HF; 48%; Sigma-Aldrich, cat. no. 7664–39-3)
 CAUTION Take appropriate safety precautions when using HF.
CAUTION Take appropriate safety precautions when using HF.Wear proper protective equipment, and perform work in a properly vented fume hood.
Methanol, HPLC grade (Fisher Scientific, cat. no. A454SK-4)
Nicotinamide (Sigma-Aldrich, cat. no. 98–92-0)
Peptone (Fisher Scientific, cat. no. DF0118–17-0)
PhosStop phosphatase inhibitor cocktail tablets (Roche, cat. no. 4906837001)
RP-HPLC solvent A (0.2% (vol/vol) FA, 5% (vol/vol) DMSO in HPLC grade water)
RP-HPLC solvent B (0.2% (vol/vol) FA, 5% (vol/vol) DMSO in ACN)
Sep-Pak C18 cartridge (50 mg sorbent; Waters, cat. no. WAT054955)
Sequencing-grade modified trypsin (Promega, cat. no. V5111 or V5113)
Sodium butyrate (Na butyrate; Sigma-Aldrich, cat. no. 156–54-7)
Sodium chloride (NaCl; Sigma-Aldrich, cat. no. S3014–500g)
Trifluoroacetic acid (TFA; Thermo Scientific, cat. no. 28904)
Tris base (EP154–1, cat. no. EP154–1)
Urea (Sigma-Aldrich, cat. no. U5378–1kg)
Water, HPLC grade (Sigma-Aldrich, cat. no. 270733–4 L)
Water, nanopure
Yeast extract (granulated; Fisher Scientific, cat. no. BP9727)
Yeast protein extract (Promega, cat. no. V7341)
EQUIPMENT
Autoclave
Autodesk Inventor 2014
Beaker, recommended 1 liter
Bunsen burner
Centrifuge, refrigerated, capable of centrifugation at 4,000g (Thermo Scientific, cat. no. 75004380)
Centrifuge buckets, 50 ml conical (Thermo Scientific, cat. no. 75–003-683)
Ceramic scoring wafer (Restek, cat. no. 20116)
Compressed helium tank
Cuvettes
Filter units, 250-ml capacity (Millipore, cat. no. SCGP-U02-RE)
Flask, recommended 4 liter
Glass beads, 425–600 μm (Sigma-Aldrich, cat. no. G8772–500G)
Graduated cylinder, recommended 4 liter
Heatblock (VWR)
High-pressure micro tee union for 360-μm outer diameter (o.d.) tubing (IDEX Health and Science, cat. no. UH-750)
HPLC system capable of nl/min flow rates (nanoAcquity UPLC; Waters)
Incubation shaker
Incubator
Laser-based micropipette puller (Sutter Instrument, cat. no. P-2000/F)
LTQ Orbitrap Fusion mass spectrometer (Thermo Fisher, cat. no. IQLAA EGAAPFADBMBCX)
Magnetic stir plate (IKA, cat. no. 0003907500)
Magnetic stir bar, miniature (VWR, cat. no. 58948–400)
Magnetic stir bar (VWR, cat. no. 58947–114)
Microscope (Zeiss, cat. no. 495005–0004-000)
MM4000 mixer mill (Retsch, cat. no. 20.745.0001)
Petri dishes, sterile, 9-cm diameter (Sigma-Aldrich, cat. no. SIAL506CC0SnV)
pH paper strips, pH range 0–2.5 (EMD Millipore, cat. no. 1.09540.0001)
pH paper strips, pH range 6.5–10 (EMD Millipore, cat. no. 1.09543.0001)
Pressure bomb, connected to a helium tank (Next Advance, cat. no. PC77)
Proteome Discoverer software, version 1.4.0.288 (Thermo Fisher Scientific)
Screw-top grinding jar (Retsch, cat. no. 01.462.0231)
Serological pipettes, 1 ml (Fisher Scientific, cat. no. 13–678-11B)
Serological pipettes, 5 ml (Fisher Scientific, cat. no. 13–678-11D)
Spectrophotometer
SpeedVac (Savant Refrigerated Vapor Trap; Thermo Scientific, cat. no. RVT5105–115)
Test tubes (Sigma-Aldrich, cat. no. CL56982516X)
Test tube rack
Tube rocker (VWR, cat. no. 10159–754)
TX-400 swinging bucket centrifuge rotor (Thermo Scientific, cat. no. 75–003-181)
Vortex (Vortex Genie 2; Scientific Industries, cat. no. SI-0236)
1.5-ml microcentrifuge tubes (Sarstedt, cat. no. 72.692.005)
2-ml microcentrifuge tubes (Sarstedt, cat. no. 72.694.005)
2-ml cryogenic vial (Corning, cat. no. 430662)
50-ml conical centrifuge tube (Thermo Scientific, cat. no. 14–432-22)
75 × 360 μm fused silica (Polymicro Technologies, cat. no. TSP075375)
1.7-μm BEH C18 packing material (Waters cat. no. 186002350)
3.5-μm BEH C18 packing material (Waters, cat. no. 186003034)
Column heater
1/4-inch and 1/8-inch aluminum plate stock
1/4-inch inch Polycarbonate plate stock
1-inch polycarbonate tube stock
1/8-inch stainless steel dowel pins (4)
PID controller (Model 32B, Dwyer Instruments, cat. no. 32B-23)
Adhesive backing polyimide film heater (Omega Engineering, cat. no. KH-202/5-P)
120 V solid-state DC controlled relay (Opto22 P120D2 or equivalent, Newark, cat. no. 18M9301)
DIN 5-pin Deltron plug (Newark, cat. no. 69K6079)
DIN 5-pin Deltron jack (Newark, cat. no. 69K6045)
RS-485 to USB converter (GridConnect, ATC-820, cat. no. GC-ATC-820)
Love Link 3 control software (Dwyer Instruments, cat. no. LOVELINK III)
Line switch
Resistive temperature detector (Omega Engineering, cat. no. PR-10–2-100–1/8–2-E )
22-Ga wire, solder, machine screws (4–40)
Power inlet receptacle, fused (Digikey, cat. no. Q303-ND)
Fuses, 500 mA (2)
Box (Digi-Key, cat. no. HM1123-ND)
REAGENT SETUP
Yeast cells
The procedure starts by extracting a yeast cell pellet. This could be obtained by following standard procedures17,18, by using the protocol described in http://www.nature.com/doifinder/10.1038/protex.2015.030 (ref. 17) or by purchasing a commercially available yeast protein extract (e.g., Promega, cat. no. V7341). Growing yeast cells requires dedicated equipment, including an autoclave, spectrophotometer, incubator and incubator shaker. If these are not available, we recommend using a commercially available yeast protein extract. As the steps described in ref. 17 can yield large quantities of yeast protein, they will not need to be performed each time this protocol is followed. Times and expected outcomes are for S. cerevisiae strain BY4741 grown in yeast extract peptone dextrose agar (YPD) medium.
DTT and IAA solutions
Prepare DTT stock solution by dissolving DTT in water to a final concentration of 0.25 M.  CRITICAL DTT is oxygen sensitive and should be prepared freshly before use. Prepare IAA stock solution by dissolving IAA in water to a final concentration of 0.25 M.
CRITICAL IAA is light sensitive and should be prepared freshly before use.
CRITICAL DTT is oxygen sensitive and should be prepared freshly before use. Prepare IAA stock solution by dissolving IAA in water to a final concentration of 0.25 M.
CRITICAL IAA is light sensitive and should be prepared freshly before use.
Lysis buffer
Lysis buffer is prepared in water, and it includes 8 M urea, 75 mM NaCl, 50 mM Tris (pH 8), 75 mM NaCl, one tablet of protease inhibitor cocktail (cOmplete mini, Roche) per 10 ml of lysis buffer, one tablet of PhosStop phosphatase inhibitor cocktail tablets (Roche) per 10 ml of lysis buffer, 100 mM sodium butyrate and 10 mM nicotinamide.
CRITICAL Fresh lysis buffer should be prepared immediately before use. Concentrated stocks of 1 M Na butyrate and nicotinamide can be prepared in water and stored at 3–80 °C for up to 6 months.
Sep-Pak solvents
Wash buffer is 0.1% (vol/vol) TFA in water. Elution buffer is 50% (vol/vol) ACN and 0.1% (vol/vol) TFA in water, followed by 75% (vol/vol) ACN and 0.1% (vol/vol) TFA in water. Mobile phase can be stored at ambient temperature, and it should be replaced every 2 months.
BSA peptide mixture
A mixture of tryptic BSA peptides can be prepared and used to monitor elution times and column degradation. Peak shape should be monitored for instances of tailing and fronting. An ideal peak shape should be Gaussian, with a retention time of <30 s (for a 60-min LC gradient). Stock solutions of BSA can be digested, divided into aliquots and stored at –80 °C. Prepare a 1 μg/pl solution of BSA in 50 mM Tris and vortex for 1 min. Add 250 mM stock solution of DTT to a final concentration of 5 mM DTT. Incubate the sample for 45 min at 57 °C to reduce the disulfide bonds. Incubate the sample with 15 mM IAA for 30 min in the dark at ambient temperature. Incubate the solution with 5 mM DTT for 15 min at ambient temperature to quench any remaining IAA. Add trypsin at a 1:50 enzyme:protein ratio and incubate the mixture for ~16 h at ambient temperature. To quench the digestion, add a minimum amount of 10% (vol/vol) TFA to reduce the sample to a pH <2. Dry down the tryptic BSA peptides in the SpeedVac. For MS/MS analysis, the tryptic BSA solution is resuspended in 0.2% (vol/vol) FA, with 100–300 fmol injected on the column.
LC-MS/MS solvents
Mobile phase A is 0.2% (vol/vol) FA and 5% DMSO in HPLC-grade water. Mobile phase B is 0.2% FA (vol/vol) and 5% DMSO in ACN. Mobile phase can be stored at ambient temperature, and it should be replaced every 2 months.
EQUIPMENT SETUP
Reversed-phase analytical columns
These columns are prepared in-house using the protocol described in Box 1 (see also Figs. 2 and 3 and Supplementary Table 1). In our setup, a 25-μm inner diameter (i.d.) capillary is plumbed directly to the injection port of a nanoAcquity UPLC (Waters) and to an in-house-prepared reversed-phase column through an ultra-high-pressure stainless steel union formatted for 360-μm o.d. capillaries (IDEX). When you attach an analytical column, set the LC flow rate to 0 μl/min. Care should be taken to tighten the union enough to prevent leaks, but not so tight as to crush the fused silica. When the column is attached, switch the LC flow rate to 100 μl/min at 100% solvent A; at this flow rate, the pressure should be between 4,000 and 6,000 psi. Continue flowing solvent A for 10–15 min. Ramp the flow rate down to 0 μl/min. Remove the analytical column and inspect the end under a light source; it is common for packing material to shift upward at higher pressures. If necessary, trim the end of the column so that the entire length of the column is filled with packing material. Reconnect the column, and equilibrate it by flowing 100% solvent A for 20 min at a flow rate of 100 μl/min. The 25-μm capillary should be kept as short as possible while still allowing the column to reach the MS inlet (<50 cm). Note that the use of an excessively long 25-μm capillary or a wider-i.d. capillary will cause the peptides to come out later because the delivery of the gradient will be delayed. To maintain a suitable back-pressure throughout the analysis (<10,000 psi), the analytical column is heated to 60 °C throughout the run.
Box 1 | Column fabrication  TIMING 2 h
TIMING 2 h
The setup of a pressure bomb, which is attached to a tank of compressed helium, is presented in Figure 2c. A commercially available pressure bomb is recommended in the Equipment section.
Analytical columns with integrated emitter tips are manufactured using 360-μm o.d. × 75 μm i.d. fused silica capillary tubing. To form the emitter tip, use a butane lighter to remove ~2.54 cm of polyimide coating 5 cm from one end of the fused silica.
-
Clean this area with methanol to remove any remaining charred polyimide coating before inserting the silica into the puller (Fig. 2a).
 TROUBLESHOOTING
TROUBLESHOOTING -
Using the manufacturer’s recommended settings for the laser puller, pull a 15-μm tip, by avoiding the polyimide coating. A recommended program for the Sutter Instrument Company P-2000 laser-based micropipette puller is described in Supplementary Table 1. However, as the optimum settings for individual laser-based pullers may differ, this should be considered a guideline.
TROUBLESHOOTING -
Inspect the tip under a microscope; if the tip is closed, etch with HF to create an opening. In the fume hood, transfer 50 μl of HF to a microcentrifuge tube and fill another tube with 0.5 M ammonium formate. Dip the electrospray tip in HF for 1–2 min. Quench any remaining HF present by immersing the tip in ammonium formate for 30 s. Thoroughly rinse the tip with water. Once the HF has been removed from the tip, use a pressure bomb placed on a magnetic stir plate to rinse the analytical column with several column volumes of methanol (Fig. 2c).
CAUTION Take appropriate safety precautions when using HF. Wear proper protective equipment, and perform the work in a properly vented fume hood. Wear safety goggles when you are working with the pressure bombs.
TROUBLESHOOTING Place a 1.5-ml conical tube containing 1 ml of methanol into the pressure bomb.
-
Insert the analytical column, with pulled tip pointing upward, through the high-pressure bomb. Be careful when you are handling the column, as the tip is fragile and easily damaged. Slowly adjust the position of the column until it touches the bottom of the tube, and then raise it so that the bottom of the column is 1–2 mm above the bottom of the tube.
TROUBLESHOOTING -
Set the pressure regulator on the helium tank to 1,000 psi.
TROUBLESHOOTING -
Increase the pressure in the pressure bomb by slowly turning the valve to the open position.
TROUBLESHOOTING -
Rinse the column with several column volumes of methanol (30 s–1 min).
TROUBLESHOOTING Release the pressure by turning the valve to the closed position.
Prepare a slurry of 3.5 μm BEH packing material by transferring the packing material to a glass vial containing a mini magnetic stir bar. Add ACN to the vial. As the 3.5-μm BEH packing material is added only to keep the smaller 1.7-μm material in the column, a very dilute slurry is recommended (0.1 mg, 3.5-μm packing material, 1 ml ACN).
Place the glass vial containing the 3.5-μm BEH slurry into the pressure bomb (Fig. 2d).
Insert the analytical column, with pulled tip pointing upward, through the ferrule and into the high-pressure bomb, so that it is almost touching the bottom of the vial. Turn on the magnetic stir plate.
Increase the pressure in the pressure bomb by slowly turning the valve to the open position.
-
Fill ~5 mm of the tip with 3.5-μm BEH slurry. If desired, a lower pressure of helium (200–300 psi) can be used for this step. Packing the tip can be accomplished by opening the valve for 1–2 s, and then by slowly releasing the pressure. Run methanol through the column to push all packing material toward the top. If necessary, pack with more 3.5-μm BEH material. A light source (lamp, flashlight) placed beside the column can be used to visually monitor its packing.
TROUBLESHOOTING Prepare a slurry of 1.7-μm BEH packing material by transferring the packing material (~0.5 mg) to a glass vial containing a mini magnetic stir bar. Add 1 ml of acetone to the vial.
-
Fill the analytical column with 1.7-μm BEH packing material to a length of ~35 cm.
TROUBLESHOOTING -
Release the pressure by turning the valve to the closed position slowly to avoid column unpacking.
CRITICAL STEP Use a light source to check for any gaps in the packing material. If gaps are present, run methanol through the column to push the packing material upward. Dry the column for 10–15 min on the pressure bomb with air.
Box 2 | Optimizing the settings of the mass analyzer
The recently introduced Orbitrap Fusion combines an Orbitrap mass analyzer with a quadrupole mass filter, a collision cell and a dual-pressure linear ion trap. The unique geometry of the Fusion allows the processes of ion injection, precursor isolation, peptide fragmentation and detection to be parallelized, significantly increasing the MS/MS acquisition rate. The optimization of various instrument parameters has been discussed in previous publications4,5
In the data-dependent method described in Step 21, survey scans are performed at an MS resolving power of 60,000 (at 200 m/z), precursor peptides are isolated for fragmentation in the quadrupole at a width of 0.7 m/z, fragmented in the collision cell followed by mass analysis of the fragments in the ion trap. The instrument is operated in top speed mode with a cycle time of 5 s, meaning that MS/MS events are continuously performed on precursor ions for either 5 s or until no acceptable precursors remain. Between 15 and 80 min of the LC-MS/MS separation, an average of nine MS/MS scans are taken between MS scans; this number can vary considerably across the gradient, however, with up to 55 MS/MS events triggered from a single MS scan. Details on adjusting these parameters within the instrument method can be found in supplementary Figure 2. The scan sequence for these events is outlined in supplementary Figure 3. When operating in this mode, the instrument is capable of scanning rates >20 Hz. Of course, other combinations of isolation events, fragmentation methods and detection options are available, but these may affect the duty cycle of the instrument, and in turn the MS/MS acquisition rate
We found that optimizing MS1 AGC target, MS1 resolving power and MS2 maximum injection time had the greatest effect on increasing the number of identified peptides (Fig. 3). The MS1 AGC target defines the number of charges introduced to the Orbitrap for the survey scan. To test the effect of MS1 AGC, we analyzed several settings between 8e4 and 5e5, the highest AGC setting offered on the instrument. Increasing the number of charges introduced to the mass spectrometer uniformly increases the number of MS/MS scans, PSMs and unique identified peptides
An increase in the number of unique peptides is also afforded from selecting an MS1 resolving power of 60,000. Decreasing the resolution correlates linearly with decreased transient collection time; this allows more MS/MS scans to be taken at a resolving power of 15,000 than at other resolving powers with longer transients. However, these extra scans do not translate to more unique peptide identifications. Although the number of MS/MS scans is comparable at resolving powers of 30,000, 60,000 and 120,000, the number of unique peptides was greatest at resolving powers of 60,000 and 120,000, presumably because the higher resolving power allows better resolution of peptidic features, which allows deeper sampling of the proteome. No further increase in unique peptides is observed above 120,000, as any benefits from increased resolving power are outweighed by longer transient time, which results in fewer MS/MS scans, PSMs and peptides
Increasing the MS/MS max inject time, or the maximum amount of time allowed for MS/MS analysis, predictably results in a decrease in the number of MS/MS scans. This decrease also translates to fewer unique peptide identifications at 45 and 60 ms MS/MS max inject times. As analysis in the ion trap requires 30–35 ms, lower max inject times ensure full parallelization of the mass spectrometer
Figure 2|.

Column fabrication. (a) To pull an emitter tip, first cut an appropriate length (35–40 cm) of 360 pm o.d. × 75 μm i.d. fused silica. Remove ~2 cm of polyimide coating 3–4 cm from one end of the fused silica. (b) The column is packed with 1.7-μm BEH C18 packing material according to the protocol. (c,d) Setup of the pressure bomb.
Figure 3|.
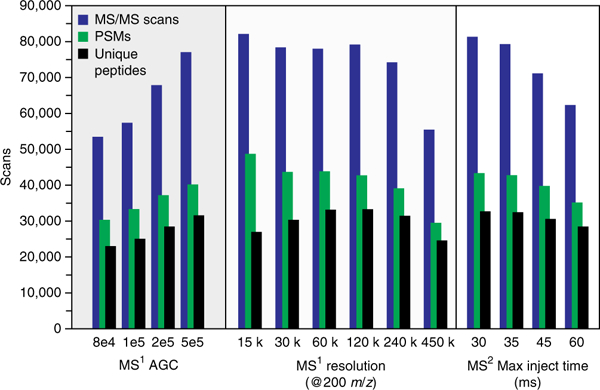
Effect of MS1 AGC target, resolution and MS2 max inject time on the number of MS/MS scans, PSMs and unique peptides.
Sample injection and separation method
For sample injection, first the pre-programmed volume of sample is placed in the sample loop. Second, trapping is performed at a flow rate of 0.325–0.375 μl/min in 100% solvent A for 13 min. The total trapping volume should be at least the sample injection volume plus 1 μl. During trapping, the flow path includes the sample loop, 25-μm capillary and column. To avoid sample loss, analytes are loaded directly onto the analytical column during trapping (e.g., no precolumn is used). Note that one must remove all salts before injection when the pre-column is omitted. During the separation, the percentage of solvent B is adjusted as described below, and 2 kV is applied directly to the stainless steel union. After the gradient, the column is washed with 70% solvent B for 5 min and then equilibrated in 100% solvent A for 15 min.
With this setup and a 75-μm i.d. bare-fused silica capillary column packed to 35 cm with 1.7-μm BEH packing material, a pressure of 7,000–9,000 psi is expected for 100% solvent A at a flow of 350 μl/min. The pressure will depend on a number of factors. Decreasing the diameter of the electrospray tip orifice, the diameter of the packing material or the column i.d. will cause increased back-pressure, as will increasing the column length.
The following RP chromatography gradient was used in our laboratory (see also the TROUBLESHOOTING section). We chose a step-wise gradient over a linear gradient because it resulted in more peptide identifications, presumably by providing better separation of the large group of hydrophilic peptides that elute early in the gradient at Step 23.
| Time interval (min) | Gradient (%B) | Flow rate (μl/min) |
|---|---|---|
| 0 | 0 | 350 |
| 0.1 | 4 | 350 |
| 32 | 12 | 350 |
| 60 | 22 | 350 |
| 70 | 30 | 350 |
| 75 | 70 | 350 |
| 80 | 70 | 350 |
| 100 | 0 | 350 |
PROCEDURE
-
1|
Vigorously resuspend a yeast pellet prepared using your chosen method (see Reagent Setup) with chilled lysis buffer. The pellet can first be pipetted up and down, followed by vortexing for ~1 min to ensure even resuspension. A target protein concentration of ~2 mg/ml is recommended.
-
2|
Add 2 ml of glass beads to each mixer mill jar. Glass beads can be measured in a 2-ml centrifuge tube and then transferred to the mixer mill jar.
-
3|
Add 2 ml of yeast lysate to each mixer mill jar.
-
4|
Fasten the jar into the mixer mill. Mill for 4 min at 30 Hz, followed by a 1-min rest. Repeat this step seven additional times.
-
5|
Transfer the lysate from the mixer mill jar to a 2-ml centrifuge tube.
 PAUSE POINT Yeast lysate should be kept at 4 °C; it can be stored for up to 1 week.
PAUSE POINT Yeast lysate should be kept at 4 °C; it can be stored for up to 1 week.-
6|
According to the manufacturer’s protocol, perform a BCA assay to determine the protein concentration.
Protein reduction and alkylation
-
7|
Prepare a 250 mM stock of DTT. Add the appropriate volume of 250 mM DTT to the yeast lysate to obtain a final concentration of 5 mM DTT. Incubate the sample for 45 min at 57 °C to reduce disulfide bonds.
CRITICAL STEP Do not perform the reduction step above a temperature of 60 °C or for much longer than 45 min, as urea can degrade into isocyanic acid, which reacts with the primary amines of the protein analytes. To test the extent of this reaction at 57 °C, we searched our data with carbamylation of lysines and N-terminal peptides set as variable modifications. Unique peptides (1.7%) were carbamylated, which is a very small portion of the data set. -
8|
Allow the yeast lysate to cool to room temperature. To alkylate cysteines, incubate the lysate with 15 mM IAA for 45 min in the dark at room temperature.
CRITICAL STEP IAA is light-sensitive. IAA stock solution must be freshly prepared and kept in the dark, and the alkylation reaction must be performed in the dark.
TROUBLESHOOTING -
9|
Incubate the lysate with 5 mM DTT for 15 min at ambient temperature to quench any remaining IAA.
Trypsin digestion
-
10|
Dilute the lysate with 50 mM Tris (pH 8) until the urea concentration is 1.5 M.
CRITICAL STEP Before adding trypsin, ensure that the pH of the lysate in 1.5 M urea is ~8. To determine the pH, pipette 0.2 μl of lysate onto a pH paper; match the color on the paper with the provided chart. -
11|
Add trypsin at a 1:50 enzyme to protein mass ratio and incubate the mixture for ~16 h at ambient temperature with gentle rocking.
-
12|
After 16 h, add a second bolus of trypsin at a 1:50 enzyme-to-protein ratio, and incubate the mixture on a rocker for 1–2 h.
Desalting (Sep-Pak)
-
13|
Desalt the peptides before analysis to remove urea and salts. Select a Sep-Pak cartridge size that corresponds to the amount of starting protein material. Use ~20 times more bulk material than protein sample. For example, for 4 mg of protein, use a 100-mg Sep-Pak. The steps below are for the use of either a 50-mg or a 100-mg Sep-Pak. For 500-mg Sep-Paks, increase all volumes listed by five.
-
14|
Add a minimum amount of 10% (vol/vol) TFA to reduce the sample to a pH <2. Confirm approximate pH with a pH paper. Pipette ~0.2 μl of the peptide solution onto the pH paper; adjust the pH of the peptide solution with 10% (vol/vol) TFA until the pH paper reading matches the desired pH. Centrifuge the sample at ambient temperature for 1 min at 8,000g to pellet any insoluble material.
TROUBLESHOOTING -
15|
Wash and condition the Sep-Pak cartridge by adding 3 ml of 100% (vol/vol) ACN, followed by 1 ml of 75% (vol/vol) ACN/0.1% (vol/vol) TFA and 1 ml of 50% (vol/vol) ACN/0.1% (vol/vol) TFA.
-
16|
Equilibrate the cartridge with 3 ml of 0.1% (vol/vol) TFA.
-
17|
Slowly load the supernatant of the acidified sample onto the Sep-Pak.
-
18|
Wash the sample with 3 ml of 0.1% (vol/vol) TFA.
-
19|
Move the Sep-Pak to a 2-ml microcentrifuge tube. Elute the sample with 0.6 ml of 50% (vol/vol) ACN/0.1% (vol/vol) TFA, followed by 0.6 ml of 75% (vol/vol) ACN/0.1% (vol/vol) TFA. Note that this step can be performed by gravity or under vacuum. If you are performing this step under vacuum, do not let the bulk material run dry; the flow rate should not exceed 1 ml/min for the washing and equilibration step and ~0.2 ml/min for the sample loading step.
-
20|
Freeze the eluate at –80 °C and concentrate it in a SpeedVac. When this step is complete, the sample should be a fluffy white powder.
PAUSE POINT Samples can be stored at –80 °C for several months in this form.
LC-MS/MS analysis
-
21|Create an instrument method with the following parameters:
Method parameter Value MS1 detector type Orbitrap MS1 resolution 60,000 MS1 AGC target 5 × 105 Precursor charge states 2–6 Dynamic exclusion Exclusion count 1 Exclusion duration 15–45 s Exclusion width ±10 p.p.m. Data dependent mode – top speed 3–5 s Precursor priority Most intense MS2 isolation mode Quadrupole Isolation window 0.7 m/z Activation type HCD Collision energy 30% MS2 detector type Ion trap MS2 scan rate Rapid MS2 AGC target 1 × 104 MS2 max inject time 25–35 ms
CRITICAL STEP If the results obtained using this procedure are not as expected or are otherwise suboptimal, it might be possible to solve this problem by optimizing the parameters shown in this table using the advice in Box 2 (See also Supplementary Figs. 2 and 3).
-
22|
Resuspend the samples to a concentration of 1 μg/μl in 0.2% (vol/vol) FA.
-
23|
Inject 1.4 μl of the sample onto the LC-MS/MS system.
Database searching
-
24|
In our laboratory, raw data are processed using Proteome Discoverer (version 1.4.0.288, Thermo Fisher Scientific), although other software suites are also available. All MS/MS spectra are searched with the SEQUEST search engine against a database of 6,632 yeast open reading frames (database downloaded from http://www.yeastgenome.com, February 3, 2011). Regardless of the software used, set the enzyme specificity to trypsin, with up to two missed cleavages permitted. Set carbamidomethylation of cysteines as a fixed modification and oxidation of methionines and protein N-terminal acetylation as variable modifications. Search the precursor and product ion mass tolerances at 20 p.p.m. and 0.35 Da, respectively.
-
TROUBLESHOOTING
-
25|
Validate the PSMs using Percolator (through Proteome Discoverer) on the basis of q values at a 1% FDR (ref. 19).
-
TROUBLESHOOTING Troubleshooting advice can be found in Table 1.
TABLE 1|.
Troubleshooting table.
| Steps | Problem | Possible reason | Solution |
|---|---|---|---|
| Box 1, steps 4, 6–9 | Liquid is not spraying from the analytical column | Column is not fully open | Examine the column under a microscope; if it is not fully open, etch the tip by dipping it in HF for 1–2 min (see Box 1 for details. HF is extremely dangerous; follow the precautions outlined in Box 1). The column should also be examined for any obstructions (dust, particles) that may prevent it from packing |
| Open end of the column is clogged | The end of the column can become damaged or clogged when inserting it through the ferrule, leading to the high-pressure bomb. After insertion, cut ~0.2 cm off the end of the column using a ceramic scoring wafer | ||
| Box 1, step 15 | Column is packing too quickly with 3.5-|im packing material | Slurry of 3.5-μm packing material is too concentrated | Make a more dilute solution of packing material by increasing the amount of ACN |
| Box 1, step 17 | Column is not packing | Turn helium off, and then back on, using the valve connected to the pressure bomb. This will sometimes cause the column to begin packing again | |
| Open end of the column is clogged | The end of the column can become clogged with packing material. Cut ~0.2 cm off the end of the column using a ceramic scoring wafer | ||
| Slurry of 1.7-μm packing material is too concentrated | Ensure that all of the packing material is able to go into solution when stirred; if not, increase the amount of ACN in the slurry (note that ACN will evaporate over time) | ||
| Open end of the column does not reach the packing material slurry | Ensure that the end of the column is fully submerged in the packing material slurry | ||
| Gaps in packing material | Poor flow of packing material; blockage | Increase the pressure of the helium | |
| Connect to UPLC and flow the 100% mobile phase B through the column at a flow rate of 0.3 μl/min for 15 min. Discard the column if the gap does not fill | |||
| Reversed-phase Chromatography (Equipment Setup) | High back-pressure during LC-MS/MS separation | Analytical column may be clogged | Replace the column |
| High-pressure union is clogged | Over time, the high-pressure union can become blocked with packing material or fused silica. Remove the union. When held up to the light, you should be able to see through it. If the union is blocked, wash it with ethanol to remove the debris. If necessary, replace the union | ||
| Analytical column is attached too tightly at the union | If the analytical column is attached too tightly, the end can become crushed and block the union. Remove the column and trim the end with a scoring wafer. To avoid this problem, after threading the column through the ferrule, trim the end with a ceramic scoring wafer. The column should protrude ~1 mm out of the ferrule. Tighten the analytical column as much as possible, without crushing the tubing. Once the analytical column is connected, gently pull on it. If it comes loose, use slightly more force when you are tightening the union | ||
| 25-μm line is clogged | Verify by removing the analytical column; if there is not a drastic decrease in pressure, replace the 25-μm line | ||
| High back-pressure during sample loading (>8,500 psi) | Impurities in the sample | Ensure that the sample was properly desalted before analysis | |
| Chromatography | Very low back-pressure | Column not properly attached | Verify whether the liquid is flowing from the tip; if not, make sure that the 25-μm line and the analytical column are properly connected at the union. If no liquid flow is observed from the 25-μm line, check its connection to the LC system |
| Broad peaks (>1 min) | Column was packed >1 cm with 3.5-μm packing material | Discard the column (see step 11 from Box 1) | |
| Gaps in packing material in column | Flow 100% solvent A through the column at 300–350 μl/min (with column heater). Inspect the column; if there are no more gaps and the packed column is of suitable length, trim the unpacked end from the column using a ceramic scoring wafer. If gaps persist, discard the column | ||
| Sample overloading | Reduce the amount of material injected on the column | ||
| Late-eluting sample | Column degradation | Inject 100–300 fmol of tryptic BSA peptides on the column; if peaks have shifted significantly, discard the column. Similarly, if total ion chromatogram (TIC) and base peak intensities are significantly lower than usual and the instrument is working as expected, discard the column | |
| Trapping sample for too long | Decrease the trapping time according to the instructions in the ‘Reversed-phase chromatography’ section (Equipment Setup) | ||
| Column is not packed all the way to the end | Inspect the column using a light source. If it is not packed to the end, remove the unpacked portion using a ceramic scoring wafer | ||
| 25-μm line too long | If possible, trim this line to <50 cm | ||
| Pumps are not delivering the correct amount of solvent | Measure the flow rate of pumps A and B; if they are not delivering the correct amount of solvent, the gradient may need to be adjusted to accommodate the actual flow rates. Contact the LC vendor for maintenance help | ||
| Run-to-run variation | Column is not fully equilibrated | Increase the time of the equilibration period at the end of the LC gradient | |
| Pumps are not delivering the correct amount of solvent | Measure the flow rate of pumps A and B; if they are not delivering the correct amount of solvent, the gradient may need to be adjusted to accommodate the actual flow rates. Contact the LC vendor for maintenance help | ||
| Column degradation | Inspect the chromatogram for wide peaks. This can also be tested by injecting 100–300 fmol of tryptic BSA peptides on the column. If peaks are wider than ~40 s, discard the column | ||
| 8 | Amidation reaction | Urea degradation during reduction (Step 7) | Perform the reduction step (Step 7) at a lower temperature (i.e., at ambient temperature) |
| 24 | Fewer identifications than expected; data-base search gives more semi-tryptic peptides than tryptic peptides | Active endogenous proteases | Collect yeast at an earlier time point. If this is not an option, try digesting with a higher enzyme: protein ratio for a shorter period of time (for example, 1:5 ratio for 4 h) (Steps 11,12) |
| 24 | Large number (>50%) missed cleavages | Increase the enzyme to protein ratio to 1:10 (Steps 11,12) | |
| 14 | Protein precipitates out of solution on acidification with TFA | Use a less concentrated solution of TFA; add TFA slowly, by checking the pH after each addition to avoid overacidification | |
| 24 | Fewer identifications than expected | Instrument is out of calibration | Follow the recommended calibration schedule provided in the calibration console. If MS mass error is >6 p.p.m., recalibrate |
| Fewer identifications than expected; decrease in TIC intensity over time | Quadrupole may require maintenance | Over time, the quadrupole and other parts of the instrument may require cleaning or maintenance. Contact your service engineer for instructions | |
TIMING
Box 1, column fabrication: 2 h
Yeast protein resuspension and cell lysis: Steps 1–3, 5 min; Steps 4 and 5, 45 min per round of lysis
Determination of protein concentration by BCA assay: Step 6, 40 min
Protein reduction and alkylation: Step 7, 50 min; Step 8, 35 min; Step 9, 15 min
Trypsin digestion: Step 10, 5 min; Step 11, 16 h; Step 12: 2 h
Peptide desalting: Step 13, 5 min; Step 14, 30 min; Step 15, 10 min; Step 16, 2 min; Step 17, 10 min; Step 18, 10 min; Steps 19 and 20, 3–5 h
LC-MS/MS analysis, including sample loading on column, LC-MS/MS gradient and column washing and equilibration: Steps 21–23, 2 h
Database searching: Steps 24 and 25, 2 h (searching speed may vary depending on the computational platform)
ANTICIPATED RESULTS
In developing this protocol, yeast was analyzed as described in the MATERIALS and PROCEDURE sections. To test the reproducibility between runs, we performed the experiment in technical quintuplicate. On average, 80,460 MS/MS scans were taken per run. After database searching, this translates to an average of 43,400 PSMs and 34,255 unique peptides. In other words, 54% of MS/MS scans were mapped to a peptide; of these identified peptides, 79% were unique. These peptide identifications yielded an average of 3,977 proteins at 1% FDR. The number of MS/MS scans, PSMs and unique peptides for all data sets are presented in Supplementary Tables 2 and 3.
Key to these high identification rates is maintaining a consistent number of identifications across the entire LC-MS/MS gradient. Figure 4a shows the cumulative number of peptide identifications as a function of the retention time for each of the five replicates.
Figure 4|.
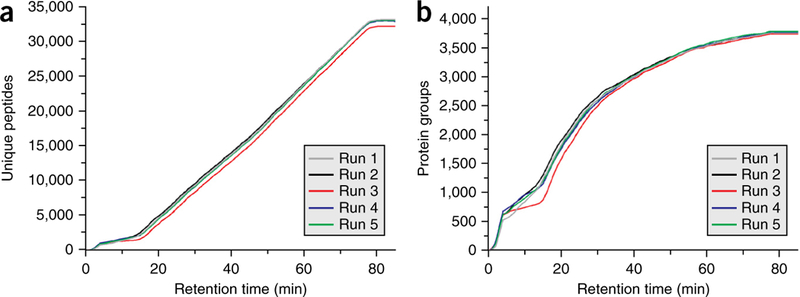
Yeast peptide and protein identifications for all replicates. (a) Plots the number of cumulative unique yeast peptide identifications for five technical replicates across the LC-MS/MS gradient. (b) Plots the corresponding proteins across the gradient. Cumulative peptide and protein identifications were determined using the Coon OMSSA Proteomic Analysis Software Suite20. PSMs passing a 1% FDR cutoff were exported to a text file and processed by a modified version of Protein Hoarder (version 2.4.1). These PSMs were iteratively processed in successive 1-min windows and grouped into proteins using the law of parsimony at a 1% FDR.
For all the replicates, the rate of identification is almost linear over the 70-min gradient and 10-min wash period. The average rate of unique peptide identifications per second is 8.3. Unique protein identifications for each run are plotted in Figure 4b. The consistency of identifications is further illustrated in Figure 5a, where the average number of unique peptides identified across the LC-MS/MS method is plotted for the five replicates. For all runs, we identify between 2,000 and 2,700 unique peptides per 5-min range from 15 min to 80 min. Expectedly, there is an initial spike in the number of protein identifications at the beginning of the gradient (Fig. 5b), with a steady decrease in new protein groups throughout the run. We also examined the effect of gradient length on the number of peptide and protein identifications. By using the same sample preparation and instrument parameters described above, yeast was analyzed over 30-, 45-, 120-, 180- and 240-min LC-MS/MS separations. Cumulative unique peptide identifications are plotted in Figure 6a. Similar rates of identification are achieved for the 30-, 45- and 70-min separations, as evidenced by the slope of the line. Predictably, longer gradients result in more peptide identifications. However, the number of unique peptides decreases substantially with increasing gradient length. For example, in the 180-min analysis, 48,970 unique peptides are identified, whereas 51,211 unique peptides were identified in 240 min, an increase of just 2,241 peptides in 60 min of analysis time. The number of proteins for each LC-MS/MS gradient is plotted in Figure 6b.
Figure 5|.
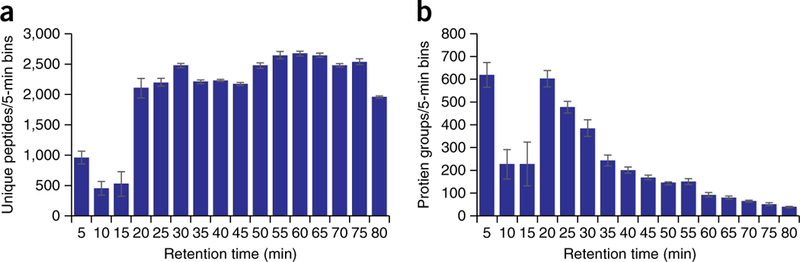
Unique peptides and proteins identified over the LC-MS/MS gradient. (a) Plots the number of unique yeast peptides identified in 5-min bins for five technical replicates across the LC-MS/MS gradient. (b) Plots the corresponding proteins across the gradient.
Figure 6|.
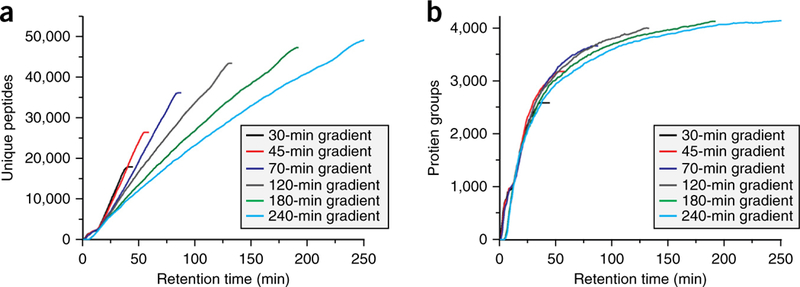
Effect of gradient length on peptide and protein identifications. (a) Plots the number of cumulative unique yeast identifications for LC-MS/MS gradients of 30, 45, 70, 120, 180 and 240 min. (b) Plots the corresponding protein identifications for each gradient.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to A. Merrill for yeast production. We thank A. Gasch for assistance with yeast growth. This work was supported by the US National Institutes of Health (R01 GM080148) and the National Science Foundation (0701846). A.L.R. gratefully acknowledges the support from a US National Institutes of Health–funded Genomic Sciences Training Program (5T32HG002760).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html
References
- 1.Tabb DL et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 9, 761–776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H, Sadygov RG & Yates JR III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Washburn MP, Wolters D & Yates JR III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Hebert AS et al. The one hour yeast proteome. Mol. Cell Proteomics 13, 339–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senko MW et al. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Goffeau A et al. Life with 6000 genes. Science 274 546 563–547 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Ghaemmaghami S et al. Global analysis of protein expression in yeast. Nature 425, 737–741 (2003). [DOI] [PubMed] [Google Scholar]
- 8.de Godoy LM et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Wu R et al. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell Proteomics 10, M111.009654 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb KJ, Xu T, Park SK & Yates JR III. Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. J. Proteome Res. 12, 2177–2184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagaraj N et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell Proteomics 11, M111.013722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulak NA, Pichler G, Paron I, Nagaraj N & Mann M Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Meye JG. & Komive EA. Charge state coalescence during electrospray ionization improves peptide identification by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 23, 1390–1399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahne H et al. DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods 10, 989–991 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Pirmoradian M et al. Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell Proteomics 12, 3330–3338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Richards A, Hebert A, Ulbrich A, Bailey D, Coughlin E, Westphall M & Coon J Preparation of yeast cells for proteomic analysis by LC-MS/MS. Protocol Exchange (2015) http://www.nature.com/doifinder/10.1038/protex.2015.030. [Google Scholar]
- 18.Treco DA & Winston F Growth and manipulation of yeast. Curr. Protoc. Mol. Biol. 82, 13.2.1–13.2.12 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ & Coon JJ COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.