

Abstract
Cilia, organelles that move to execute functions like fertilization and signal to execute functions like photoreception and embryonic patterning, are comprised of a core of nine-fold doublet microtubules overlain by a membrane. Distinct types of cilia display distinct membrane morphologies, ranging from simple domed cylinders to the invaginations and membrane disks of photoreceptor outer segments. Critical for the ability of cilia to signal, both the protein and the lipid compositions of ciliary membranes are differentiated from those of other cellular membranes. This specialization presents a unique challenge for the cell as, unlike other membrane-associated organelles, the ciliary membrane is contiguous with the surrounding plasma membrane. This distinct ciliary membrane is generated in concert with the multiple membrane remodeling events that comprise the process of ciliogenesis. Once the cilium is formed, control of ciliary membrane composition relies on discrete molecular machines, including a barrier to membrane proteins entering the cilium at a specialized region of the base of the cilium called the transition zone and a trafficking adaptor that controls G protein-coupled receptor (GPCR) localization to the cilium called the BBSome. The ciliary membrane can be further remodeled by the removal of membrane proteins by the release of ciliary extracellular vesicles that may function in intercellular communication, removal of unneeded proteins or ciliary disassembly. Here, we review the structures and transport mechanisms that control ciliary membrane composition, and discuss how membrane specialization enables the cilium to function as the antenna of the cell.
Introduction
The cilium is a microtubule-based cellular projection that can be motile, sensory or both. Motile cilia and flagella beat to propel the cell or move the surrounding milieu. (Although different in their waveforms, flagella and cilia are used interchangeably throughout this review.) Immotile cilia, called primary cilia, function as antennae to communicate the external environment to the rest of the cell, although motile cilia can also have sensory functions. Emphasizing the generality of principles learned by studying the cilia of model organisms, the structure of cilia and, to at least some extent, mechanisms of ciliary signaling are conserved across the eukaryotic kingdoms, from unicellular organisms to animals. For example, all cilia are constructed atop mother centrioles, called basal bodies when associated with cilia. They have a skeleton, the ciliary axoneme, that is comprised of nine-fold microtubule doublets. And they are ensheathed by a membrane.
Cellular membranes are primarily comprised of amphipathic lipids. In aqueous solutions, the hydrophobic acyl groups of these amphipathic lipids associate with each other and the polar heads associate with water to generate lipid bilayers. All forms of life are made of cells surrounded by lipid bilayers, suggesting that one of the earliest steps in the evolution of life was to physically separate the cytoplasm from its surroundings with a plasma membrane [1,2]. As the last common eukaryotic ancestor is likely to have possessed a cilium, the evolution of the cilium was also probably an early step. Thus, the cilium and its membrane have had over a billion years over which to co-evolve and diversify into the highly specialized forms found in extant organisms.
Presumably, the ciliary membrane shares its biochemical origins with that of the plasma membrane: vesicles originating from the Golgi and recycling endosomes exocytose into the plasma membrane, or specialized proteins transfer lipids from the endoplasmic reticulum into the plasma membrane, and, subsequently, the ciliary membrane emerges from the plasma membrane through the process of ciliogenesis. As a consequence of the overlapping mechanisms of plasma and ciliary membrane biogenesis, proteins involved in some forms of polarized exocytosis, including RAB11, RAB8 and the exocyst, participate in ciliogenesis [3].
A common feature among all cilia regardless of their functions is that, in marked contrast to most other organelles, cilia are incompletely membrane-bounded. Instead, the ciliary membrane is contiguous with the plasma membrane, and the ciliary base is open to the cytosol. Access of cellular components to the cilium is occluded by the basal body and some specialized structures within the transition zone, a region of the axoneme near the ciliary base. Despite their contiguity and shared origins, the ciliary membrane has a distinct protein and lipid composition from that of the plasma membrane.
One important role for the ciliary membrane is that it provides the boundary across which select extracellular signals are communicated to the rest of the cell. For example, in the unicellular green algae Chlamydomonas reinhardtii, sexual reproduction is initiated by the flagella of the gametes of the two mating types adhering [4]. Adhesion between these flagella is enabled by the presence of complementary agglutinins specifically present in the flagellar membranes of the different mating types [5]. Agglutinin-mediated flagellar adhesion initiates intracellular signaling events leading to cellular fusion and sexual reproduction [6–9]. Thus, maintaining the distinct composition of the ciliary membrane is critical for the ability of cilia to transduce signals.
In vertebrates, morphogens of the Hedgehog (Hh) family require cilia to pattern tissues including the limb buds and the neural tube [10]. Hh ligands bind to their transmembrane receptor Patched (PTCH1), a receptor that localizes to and functions at cilia [11,12]. Subsequently, PTCH1 is shuttled out of the ciliary membrane, and concomitantly, the seven-pass transmembrane protein Smoothened (SMO) accumulates in the cilium to activate the downstream Hedgehog pathway [13]. Cilia are therefore required for and coordinate the signaling and trafficking events initiated by Hedgehog signals.
In this review, we focus on emerging studies of the ciliary membrane, how it is distinguished from other membranes, and how it participates in the specialized signaling functions of the cilium. We discuss the exquisite diversity of ciliary membrane morphologies, from that of the canonical primary cilium to the convoluted membrane of photoreceptor outer segments. We summarize current understanding of how several distinct molecular machines compromised in human ciliopathies coordinately control the composition of the ciliary membrane. Finally, we describe the extensive remodeling that occurs during cilia formation and the membrane remodeling that gives rise to ciliary extracellular vesicles.
Different types of cilia exhibit different membrane morphologies
Like the cells they adorn, ciliary membranes can adopt astoundingly different shapes. This diversity is perhaps nowhere on more prominent display than in C. elegans. In addition to the single cilia that are similar to those found on most mammalian cells, C. elegans neurons (e.g., ADF and ADL neurons) can possess pairs of cilia, at least superficially similar to the paired cilia found on mammalian tanycytes [14–17]. Even more impressively, other C. elegans neurons possess cilia with elaborate membrane extensions that have been compared to wings (AWA, AWB, AWC neurons), bags (BAG) or flaps (FLP).
In addition to tanycytes, other vertebrate cells can display cilia with diverse morphologies. As mentioned above, photoreceptor cells possess highly distended ciliary tips called outer segments. These outer segments are packed with Opsin-containing discs. While differences in the underlying ciliary axonemes are likely to contribute to these extreme morphologies, differences in the membrane surrounding the cilia are their most prominent features.
The specialization of ciliary membranes is also indicated by their distinct protein compositions. In Chlamydomonas species, different agglutinins within the plasma and ciliary membranes do not mix under most conditions, indicating that there are barriers between the plasma and ciliary membranes [18,19]. In mammals, diverse membrane-associated proteins, including the aforementioned Hedgehog signal transduction pathway components PTCH1 and SMO and the Polycystic Kidney Disease-associated proteins PKD1 and PKD2 can accumulate within the ciliary membrane [11,13,20,21]. Disruption of ciliary function can cause Hedgehog-associated developmental defects or kidney cysts [10,22,23], reflecting the intimate involvement of the ciliary environment in the function of these ciliary proteins. As there are no ribosomes, and therefore no translation, within the cilium, all ciliary proteins necessarily traffic there from elsewhere in the cell. Thus, targeted trafficking to and retention of membrane-associated proteins at the cilium are essential for the specialization of the ciliary membrane.
Outside of the cilium, membrane lipids actively participate in transmembrane protein trafficking and retention. For example, different Golgi membranes can have different thicknesses, and complementarity between the length of protein transmembrane domains and the thickness of the membrane can help sort proteins to subdomains of the Golgi apparatus [24,25]. In other membranes, localization can depend on additional physical characteristics of the membrane and transmembrane domains, such as charge and amphipathicity [26].
The physical characteristics of the ciliary membrane are largely unknown although the trypanosome flagellar membrane is highly enriched in dehydroergosterol, a sterol that vertebrates do not make [27], and the membrane of photoreceptor outer segments contains high levels of unsaturated lipids [28–30]. In the C. elegans AFD neuron, an unsaturated lipid-GFP fusion does not localize to the cilium [31], suggesting that cilia may exclude certain lipids, just as they exclude certain proteins.
Arguing from first principles, the high curvature of the ciliary membrane may enrich conical lipids such as phosphatidylethanolamine in the inner leaflet, and enrich inverted conical lipids such as lysophosphatidylcholine in the outer leaflet (Figure 1). Active regulation of the abundance of conical and inverted conical lipids in the ciliary membrane may generate the diverse morphologies seen across different types of cilia. What lipids are found within ciliary membranes, whether different types of cilia contain different lipids, whether lipids help determine which proteins localize to the cilium and how they participate in ciliary signaling will be interesting questions to answer.
Figure 1.
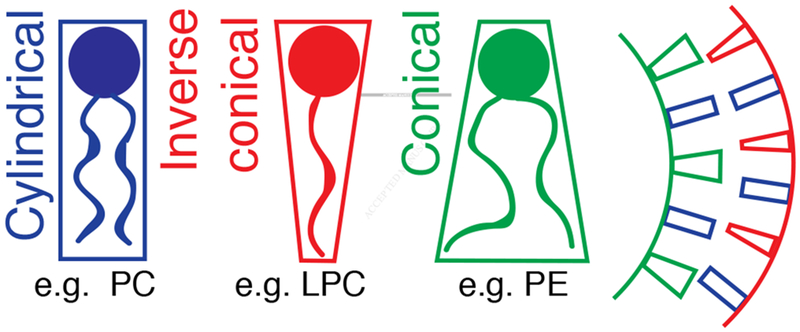
Lipids can determine membrane curvature
Lipids are categorized as cylindrical, conical or inverse conical according to their shape in the membrane. Phosphatidylcholine, PC. Lysophosphatidylcholine, LPC. Phosphatidylethanolamine, PE.
Different subdomains of the cilium have distinct membranes
The cilium consists of several functionally distinct subdomains associated with distinct subdomains of the ciliary membrane (Figure 2A). Surrounding the ciliary membrane of some cilia is the ciliary pocket, a membrane invagination that separates the ciliary membrane from the plasma membrane [32]. Like the ciliary membrane, the ciliary pocket is differently organized in different cell types. In fibroblasts, the ciliary pocket is a simple sheath around the cilium. In contrast, the ciliary pocket of frog outer segments, called the periciliary ridge complex, is a nine-fold symmetric series of ridges and grooves [33]. Newly-synthesized Opsin destined for the outer segments is first delivered to the periciliary ridge complex by vesicular transport, implicating ciliary pockets in trafficking of membrane-associated proteins to cilia [34].
Figure 2.
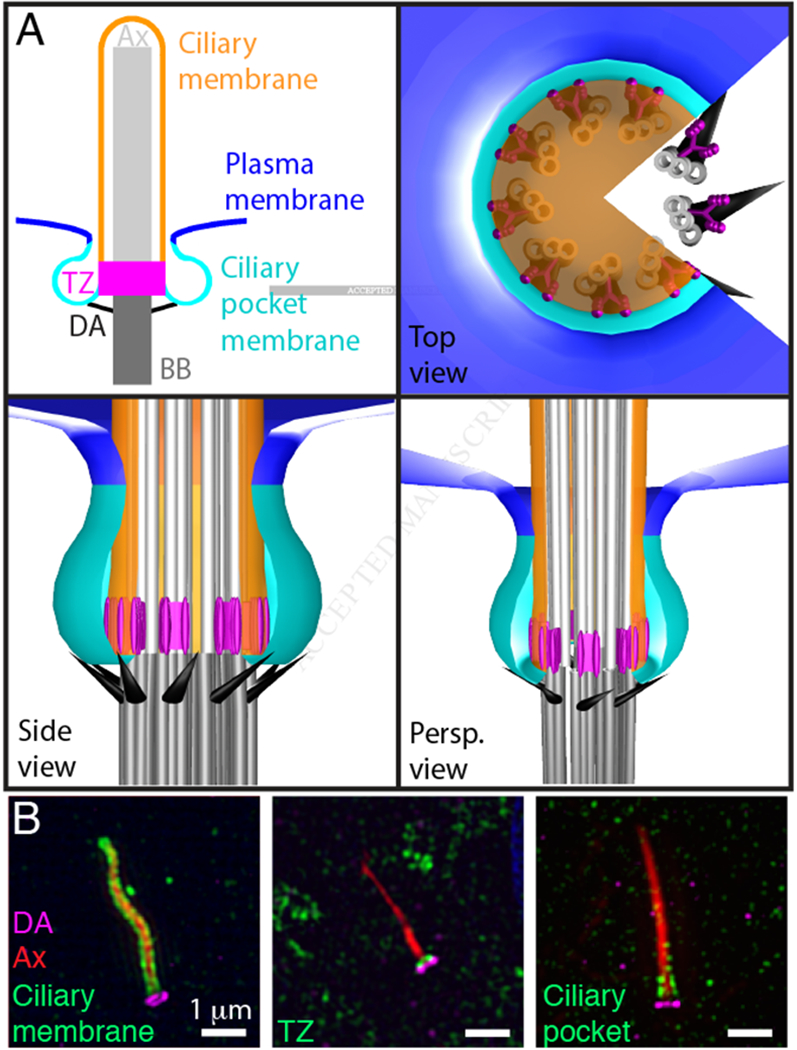
Compartmentalization of the ciliary membrane.
(A) The basal body (BB, dark grey) consists of nine-fold triplet microtubules. Doublet microtubules extend from the basal body to form the ciliary axoneme (Ax, light grey). Distal appendages (DA, black) project from the triplet microtubules at the distal end of the basal body. The transition zone (TZ, magenta) is a subdomain of the base of the cilium characterized by Y-shaped links between the ciliary membrane and doublet microtubules of the axoneme. The ciliary membrane (orange) ensheaths the axoneme. The ciliary pocket membrane (cyan) surrounds the ciliary membrane and separates it from the plasma membrane (blue). The plasma membrane, ciliary pocket, and the ciliary membrane are continuous. Panels depict a twodimensional model of the cilium (top left) or top, side, and perspective views (Persp.) of a three dimensional model.
(B) SIM detection of the distal appendages (CEP164, magenta), the axoneme (acetylated Tubulin, red) and either the ciliary membrane (ARL13B, green), the transition zone (TCTN2, green), or the ciliary pocket (EHD, green). Immunofluorescence staining and SIM were performed according to protocols described in [52].
Due to the absence of robust markers of the ciliary pocket, the boundary between the ciliary membrane and the ciliary pocket membrane has not been accurately defined. However, the boundary is presumably in the vicinity of the distal appendages, a ring of struts connecting the distal end of the basal body to the membrane at the base of the cilium. The nine-fold symmetric distal appendages project radially from the barrel of the basal body to contact the membrane [35]. Although several proteins that form the distal appendages have been identified, how the distal appendages attach to the membrane remains unknown [36,37].
In addition to serving as membrane attachment anchors, distal appendages are docking sites for intraflagellar transport (IFT) machinery [38]. IFT is the primary means by which active transport from the basal body to the ciliary tip and back again occurs [39]. The distal appendages may serve as organizing sites for IFT complexes entering and/or leaving cilia. Interestingly, the mature sensory cilia in C. elegans lack basal bodies and distal appendages, suggesting that membrane compartmentalization between the ciliary membrane and the periciliary membrane can be achieved independent of both structures [17,40,41].
Although lacking basal bodies, nematode cilia do have a specialized domain surrounding the base of their cilia called the periciliary membrane compartment (PCMC). Perhaps analogously, mammalian MDCK cells have a domain of ordered lipids surrounding the ciliary base in which Galectin-3 localizes [42]. Other mammalian cells have a membrane domain at the ciliary base rich in PI(4,5)P2 [43,44]. Freeze-fracture electron microscopy has identified a ring of intramembranous particles called the ciliary bracelet surrounding the ciliary base [45,46]. How the PCMC, the ordered lipid domain, the PI(4,5)P2-rich domain, and the ciliary bracelet correspond to the ciliary pocket is not clear, but it is possible that each of these independent works describes different aspects of the same region.
Immediately distal to the distal appendages is a subcompartment of the cilium called the transition zone [47]. The transition zone is named for the most proximal region in which the triplet microtubules that comprise the basal body have transitioned into the doublet microtubules of the ciliary axoneme. Ultrastructurally, the transition zone is characterized by Y-shaped densities that bridge the microtubule axoneme to the surrounding ciliary membrane and rings of intramembranous particles called the ciliary necklace [48]. Interestingly, identification of sterol complexes in the ciliary membranes of motile cilia reveals that the transition zone is sterol poor, suggesting that even subdomains of the ciliary membrane may possess different lipid compositions [49].
At the distal end of the cilium is the ciliary tip, yet another specialized subdomain of the cilium. Activation of C. reinhardtii mating causes the flagellar tips to grow rapidly, revealing that this subdomain can be dynamically remodeled [6]. Similarly, in mammals, proteins involved in signal transduction, such as SUFU and the GLI transcription factors involved in Hedgehog signaling, increase their localization to the ciliary tip in response to pathway activation [50,51].
For decades, the gold standard by which distinct ciliary compartments have been defined has been electron microscopy. The ability of electron microscopy to resolve fine, electron-dense, features in a cellular context is offset by the difficulty in identifying the localization of specific proteins. The advent of super-resolution fluorescence microscopy has made the detection of specific proteins within the cilium at resolutions of 20–100 nm much more tractable. Using conventional immunofluorescence staining techniques, it is now possible to resolve multiple, distinct ciliary compartments with structured illumination microscopy (SIM). For example, SIM can resolve the ciliary membrane, transition zone, and ciliary pocket from the distal appendages (Figure 2B). Excitingly, SIM can also resolve the ciliary membrane from the axoneme, structures that differ in diameter by only ~100 nm (Figure 2B, left). Another type of superresolution microscopy, stochastic optical reconstruction microscopy (STORM), can achieve even higher resolution (~20 nm) than SIM. The high resolution capabilities of STORM enable the detection of spatially distinct proteins within subdomains of cilia, such as the relative positions of distinct ciliopathy-associated complexes within the transition zone [52]. The complementary use of cellular electron microscopy and super-resolution fluorescence microscopy will enable further advancement in our understanding of how subdomains of the ciliary membrane differ from each other.
Ciliogenesis requires multiple steps of membrane remodelling
Ciliogenesis occurs in interphase. The two dominant models of ciliogenesis differ in the accompanying membrane remodeling events and in the cell types in which they occur (Figure 3). In the pathway that is thought to occur in polarized epithelial cells, the basal body docks directly to the plasma membrane [53]. Subsequently, the axoneme is built atop the basal body and the ciliary membrane expands to cover the nascent cilium. In the ciliogenesis pathway that is thought to occur in fibroblasts, the basal body distal appendages attach to small vesicles (the distal appendage vesicles, DAVs) [54]. The DAVs then fuse to form a single larger vesicle, the ciliary vesicle. Subsequently, the axoneme and the ciliary vesicle concomitantly extend to form the ciliary membrane, generating an internalized cilium. The outer surface of the large ciliary vesicle of an internalized cilium can then fuse with the plasma membrane to externalize the cilium.
Figure 3.
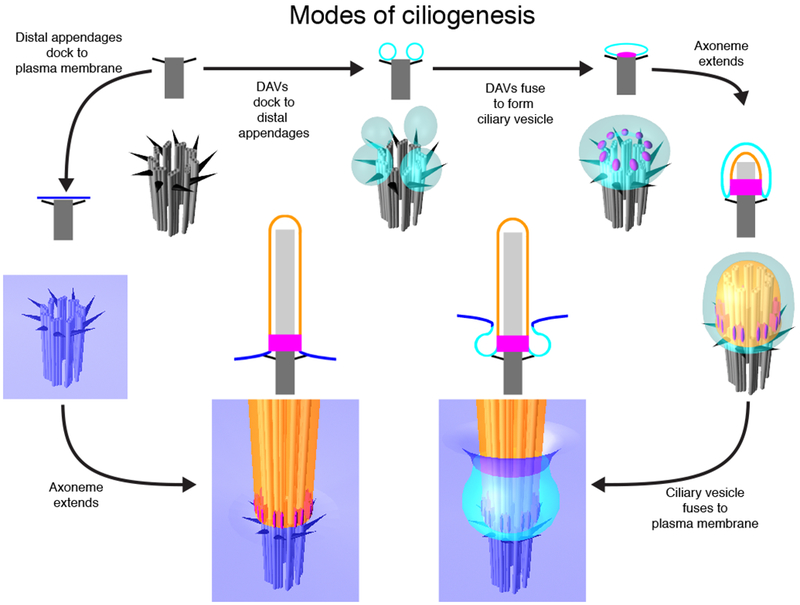
Modes of ciliogenesis
In polarized epithelial cells, the basal body (dark grey) docks to the plasma membrane (blue), and then the axoneme (light grey) and ciliary membrane (orange) extend to form the mature cilium. In fibroblasts and retinal pigmented epithelial cells, distal appendage vesicles (DAVs, cyan) dock to the distal appendages (black). As transition zone proteins (magenta) are recruited to the distal end of the basal body, DAVs fuse together to form a single ciliary vesicle. Subsequently, the ciliary vesicle and axoneme extend. The internal membrane surface of the ciliary vesicle gives rise to the ciliary membrane (blue). The external membrane surface of the extended ciliary vesicle fuses with the plasma membrane to form a partially externalized cilium. Illustration depicts two-dimensional models (top) or three-dimensional models (bottom) of ciliary features at each step of ciliogenesis.
Molecular determinants of membrane remodeling during ciliogenesis include membrane shaping proteins EHD1 and EHD3 [55]. Other proteins, including SNAP29, are required for the coalescence of the DAVs into the ciliary vesicle. Identifying additional molecular determinants of ciliary membrane remodeling is an active area of research.
Once the axoneme and ciliary membrane have extended, further membrane remodeling events are required in cilia with more complex membrane topographies, such as the outer segments of photoreceptor cells. Beyond outer segments, the molecular differences that underpin different modes of ciliogenesis and different membrane topographies are largely unknown.
After the completion of ciliogenesis, ciliary membranes may be reshaped by ciliary signaling. For example, cilia of a C. elegans neuron called AWB display an elongated morphology in the presence of nutrient-rich bacterial food. However, when fed an axenic, minimal medium, the morphology of the cilium changes to a fan-like shape, similar to the shape of cilia in nematodes mutant for chemosensory genes such as the guanylyl cyclase odr-1 and the cyclic nucleotide gated channel tax-4 [56]. Therefore, cilia membrane morphology can depend on the signaling state of the cilium.
Ciliary morphology is also determined by proteins that localize to the ciliary membrane or to the surrounding periciliary membrane. OSTA-1 is an organic solute transmembrane transporter in the PCMC of ciliated neurons of C. elegans that shapes sensory ciliary morphology and regulates trafficking of cargoes to the cilium [57]. In osta-1 mutants, the ciliary membrane displays altered branching and fanning, and select ciliary GPCRs are aberrantly enriched in the PCMC [57].
Further emphasizing the relationship between membrane trafficking and ciliary membrane morphology, endocytosis regulators affect the morphology of the ciliary membrane. For example, endocytosis proteins AP-2, Clathrin, RAB-5 and Dynamin all localize to the PCMC, and nematodes with partially disrupted AP-2 function display enlarged PCMC, expanded ciliary membranes, and altered localization of ciliary membrane proteins [58].
In addition to endocytosis effectors, regulatory proteins such as kinases help shape ciliary membrane morphology. Cyclin-dependent kinase 20 (Cdk20, also known as Ccrk) mutant mice display neuroepithelial cilia that are swollen and short [59]. Cdk20 mutant mouse embryonic fibroblasts have attenuated Hedgehog signaling, reflected by reduced ciliary SMO upon pathway activation [59]. The role of CDK20 in controlling ciliary length and morphology is highly conserved across diverse species, as mutants in LF2, a C. reinhardtii homolog of Cdk20, also display bulbous cilia [60].
CDK20 biochemically interacts with the putative Rab-GAP Broad-minded (BROMI), and like those of Cdk20 mutants, neuroepithelial cilia of Bromi mutant mice show a shortened, bulbous morphology [61]. In zebrafish, Bromi is required for the proper apposition of the axoneme and ciliary membrane, suggesting that BROMI, and potentially CDK20, coordinate assembly of the axoneme and expansion of the ciliary membrane during ciliogenesis [61].
Together, the identification of these diverse regulators of ciliary membrane morphology demonstrate that the formation of the cilium involves extensive membrane remodeling, and after the cilium is formed the composition of the ciliary membrane must be precisely controlled to support signaling.
The transition zone controls ciliary membrane protein composition
The transition zone, described above, is a specialized region of the ciliary base critical for controlling the composition of the ciliary membrane. The transition zone is comprised of several protein complexes, including the NPHP complex, the CEP290 complex and the MKS complex, that form the border between the ciliary and plasma membranes. The MKS complex, named after the association of many of its components with a severe ciliopathy called Meckel syndrome (MKS), is essential for ciliogenesis in some tissues such as the node and the neural tube [62–65]. In other tissues, such as the limb bud, the MKS complex is not required for ciliogenesis, but is required for developmental patterning [62–65]. In the absence of the MKS complex, some membrane-associated proteins, such as ARL13B, PKD2, GPR161 and SMO, fail to accumulate within the ciliary membrane [62–67].
In addition to promoting the ciliary localization of certain membrane-associated proteins, the transition zone also limits the entry of plasma membrane-associated proteins. For example, in the absence of MKS components, such as TCTN-1, C. elegans cilia can no longer exclude a normally non-ciliary membrane protein [67,68]. Similarly, knockdown of MKS complex components in mammalian cells allows normally non-ciliary constructs such as GFP-CEACAM1 to gain access to the ciliary membrane [63]. Thus, the MKS complex and, by extension, the transition zone are critical for delineating the ciliary membrane and the contiguous plasma membrane.
Although it remains unclear how the transition zone separates the components of these two domains, the transition zone is a structural tether between the membrane and the underlying microtubules. In C. reinhardtii, loss of the transition zone component CEP290 causes the membrane to lose its close apposition with the axonemal microtubules [69]. In wild type C. reinhardtii, the transition zone membrane is resistant to detergent extraction, a feature also dependent on CEP290 [69,70]. These results suggest that CEP290 imparts, directly or indirectly, distinct biophysical characteristics to the transition zone membrane.
Unlike Chlamydomonas, loss of the C. elegans ortholog of CEP290 is not sufficient to cause detachment of the transition zone membrane from the axoneme, but loss of both an MKS and NPHP complex is, indicating that the MKS and NPHP complexes have overlapping roles in the connecting the ciliary membrane to microtubules [68,71].
Proteins and dextrans below about 70 kD bypass the transition zone to gain entry to the cilium, suggesting that molecular weight is one determinant by which the transition zone discriminates molecules [72–76]. Similarly, the transition zone may control the composition of the ciliary membrane by acting as a barrier to the diffusion of membrane proteins. A critical component of this diffusion barrier may be the Septin cytoskeleton, which acts as a protein diffusion barrier in other contexts [77]. Knockdown of SEPT2 reveals that it is important for transition zone formation, indicating an intimate relationship between Septins and dedicated transition zone components [63].
In addition to functions as a diffusion barrier, recent super-resolution microscopy has revealed that the transition zone is a point at which many ciliary membrane-associated proteins accumulate, suggesting that this may be a waypoint at which proteins’ credentials are checked before being allowed entry or turned away (Figure 4) [52,78].
Figure 4.
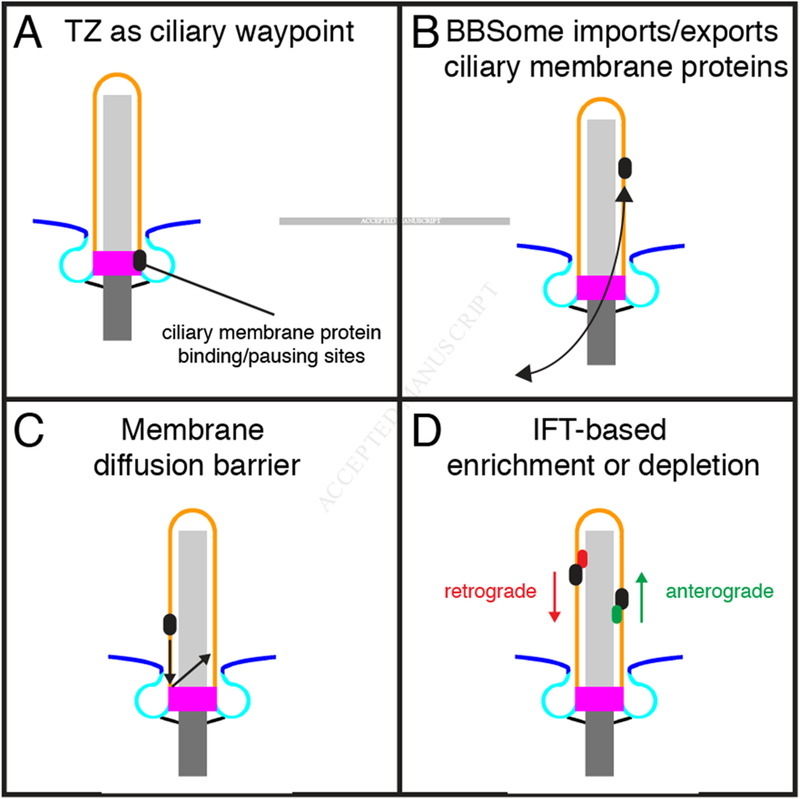
Models of compositional control for the ciliary membrane
A, The transition zone contains binding sites for select ciliary membrane proteins [52], and live imaging studies have shown that SMO pauses in the transition zone [78]. Thus, the transition zone may control the entry and exit of membrane-associated proteins from the cilium. B, The BBSome exports and imports ciliary membrane proteins such as GPCRs. C, A membrane diffusion barrier retains ciliary membrane proteins and excludes non-ciliary membrane proteins. D, Intraflagellar transport complexes enrich or deplete proteins in the cilium by coordinating with BBSome-facilitated trafficking across the transition zone.
Mutations in MKS components can cause, in addition to MKS, Joubert syndrome, a ciliopathy characterized by hypoplasia of the cerebellar vermis and brainstem abnormalities [79,80]. Cells from Joubert syndrome individuals show defects in the architecture of the transition zone and, most probably consequently, defects in the protein composition of the ciliary membrane [52]. We have proposed that at least some forms of Joubert syndrome are due to defective transition zones causing problems in ciliary signaling. How these defects in ciliary signaling disrupt the development of the cerebellum and brainstem remains to be elucidated. Changes in canonical and non-canonical Hedgehog signaling resulting in changes in patterning, cell proliferation and axon pathfinding may prove to cause the developmental changes underlying Joubert syndrome.
In addition to transmembrane proteins, peripheral membrane proteins that localize to the ciliary membrane must also surmount the transition zone. Ciliary prenylated and myristoylated proteins gain access to the cilium by the lipidated protein intraflagellar targeting (LIFT) system. LIFT requires proteins that bind and transport the lipidated cargo (e.g., PDE6D, UNC119), those that unload the cargo (e.g., small GTPase ARL13B), and those that direct the unloading specifically in the ciliary membrane (e.g., GTPase-activating protein RP2) [81–84].
The transition zone controls the composition of the ciliary membrane, but it does not act alone. For example, components of the transition zone NPHP complex in C. elegans, and the MKS complex in mammals genetically interact with components of a complex called the BBSome, revealing a tight functional coupling between the transition zone and a cargo adaptor complex to support ciliogenesis, ciliary trafficking and signaling [67].
The BBSome cooperates with the TZ to control ciliary membrane protein composition
Bardet-Biedl syndrome (BBS) is an inherited syndromic ciliopathy characterized by retinal degeneration, kidney cysts, polydactyly and obesity [85]. Intimately involved in the control of membrane-associated protein localization to cilia is the BBSome, a complex of eight proteins associated with Bardet-Biedl syndrome (BBS) [86–88].
A key role for the BBSome is the control of the ciliary localization of select GPCRs. To do so, the BBSome is recruited to membranes by the small GTPase ARL6 which, when binding GTP, interacts with the BBS1 component of the BBSome [89,90]. In addition to its interaction with membranes through ARL6, the BBSome interacts with phosphoinositides [89]. Apart from the membrane, the BBSome associates with the IFT machinery to localize select cilia-associated GPCRs to cilia [87,89,91–97]. For example, BBS2 and BBS4 are required for the localization of the GPCR SSTR3 to cilia of hippocampal neurons [94]. Moreover, ARL6 and BBS4 are required for the ciliary localization of a normally non-ciliary protein (CD8α) fused to the ciliary targeting sequence of the third intracellular loop of SSTR3, providing evidence of a discrete ciliary targeting sequence that functions as a zipcode for BBSome-mediated ciliary delivery [89].
Somewhat confusingly, the BBSome is also required for the ciliary export of at least some proteins, including some GPCRs [67,97–101]. In C. reinhardtii, the BBSome itself is exported from flagella in a way that depends on the IFT-A component IFT139, and the BBSome component BBS4 limits the flagellar accumulation of a limited number of membrane-associated proteins [99,100]. Without BBS4 activity, for example, C. reinhardtii flagella accumulate Phospholipase D with consequent increases in DAG, a product of Phospholipase D [99]. Thus, similar to the role of PDE6D in controlling the ciliary phosphoinositide composition through regulation of INPP5E localization (as described in the next section), the BBSome indirectly controls ciliary lipid composition through regulation of the localization of an enzyme that hydrolyzes ciliary membrane lipids.
Recently, SSTR3, a GPCR whose ciliary localization depends on BBS2 and BBS4 in the mouse hippocampus, was found to exit the IMCD3 cilium in a manner dependent on BBS2, BBS4 and another BBS-associated protein, ARL6 [94,102,103]. How might the BBSome promote the exit of SSTR3 from IMCD3 cilia, but promote the ciliary localization of SSTR3 in the hippocampus? Perhaps tagged SSTR3 expressed by IMCD3 cells and endogenous SSTR3 expressed by neurons traffic or ectocytose differently, or perhaps dynamic imaging reveals an involvement of BBSome proteins in ciliary exit that is missed by static snapshots.
Another possible explanation for the apparently contradictory findings that BBS proteins participate in trafficking GPCR to cilia and promoting their exit from cilia is that they do both, but under different conditions. A few GPCRs localize to cilia in an activity-dependent manner. For example, Dopamine receptor 1 (D1) localizes to cilia of neurons in the central nervous system, but rapidly exits cilia upon binding to a pharmacologic agonist through a mechanism that requires BBS4 [97]. Similarly, GPR161 exits cilia upon activation of the Hedgehog pathway in a way that requires the BBSome [67,104,105]. It is tempting to speculate that, in the absence of activation, the BBSome promotes the ciliary localization of diverse client GPCRs and that an activation-dependent event, such as binding to β-Arrestin-2, changes BBSome function to promote the ciliary exit of the very same GPCRs. Regardless, the phenotypes associated with BBS are presumably due to altered ciliary protein and lipid localization, disrupting multiple aspects of ciliary signaling.
Both the BBSome and the transition zone are complex macromolecular machines that function in the ciliary localization of membrane-associated proteins, but the transition zone is static whereas the BBSome is transported within the cilium by the IFT machinery. Knockdown of BBS1, 3 or 5 inhibits the ciliary localization of SMO [106] and knockout of BBS7 causes misaccumulation of SMO in cilia [107], but these effects on ciliary SMO activity may be minor as null mutations in mouse BBS-associated genes do not cause phenotypes associated with Hedgehog loss of function such as holoprosencephaly. Although absence of the BBSome does not dramatically disrupt ciliary localization of SMO, the presence of polydactyly in BBS-affected individuals and the effects of BBS-associated gene inhibition in zebrafish on fin patterning suggest that it can alter Hedgehog signaling [101,104,107–109]. Although loss of BBS-associated genes do not cause polydactyly in mice, they are expressed in the developing limb bud. Moreover, mutations in mouse Bbs1 or Bbs4 enhance the polydactyly seen in mutants affecting MKS complex components, demonstrating that they do have ancillary roles in mouse limb patterning and that the BBSome and the transition zone MKS complex genetically interact [67,108,110,111]. One scenario that may account for the disparate findings is if the transition zone actively recognizes BBSomes carrying inactive receptors to allow them to enter and BBSomes carrying activated receptors to allow them to exit [112]. Perhaps differential association with β-Arrestin-2 is one way in which activated receptors could be distinguished from inactive receptors [113]. Regardless, in addition to the membrane proteins that are localized specifically to the cilium by the transition zone and the BBSome, lipids themselves can be enriched in the cilium.
Phosphoinositide compartmentalization and consequences of aberrant composition
Phosphoinositides (PI) are phosphorylated lipids that confer distinct molecular identities to different cellular membranes [114]. For example, endosome and Golgi membranes possess PI(3)P and PI(4)P, respectively, whereas the plasma membrane possesses both PI(4)P and PI(4,5)P2 [115–118]. Differences in the types of phosphoinositides help compartmentalize signaling events within cells to different membranes. Crucial to maintaining the distinct phosphoinositide compositions of different membranes is their physical separation from each other. In contrast to organelles such as endosomes and Golgi, the cilium possesses a membrane contiguous with that of the plasma membrane. Despite the lack of a physical separation, the ciliary membrane nevertheless maintains a distinct phosphoinositide composition with high levels of PI(4)P and low levels of PI(4,5)P2, relative to the plasma membrane [43,44].
How does the ciliary membrane maintain a unique lipid composition? One part of the answer is ciliary localization of lipid biosynthetic enzymes. In C. elegans, a phosphoinositide 5-phosphatase called CIL-1 localizes in the region of cilia and promotes ciliary localization of PKD-2, the ortholog of Polycystic kidney disease 2 (PKD2) [119]. In mammals, three phosphoinositide 5-phosphatases can localize to cilia, prominent among which is INPP5E. INPP5E converts PI(4,5)P2 into PI(4)P and, in the absence of INPP5E, ciliary levels of PI(4)P decrease and PI(4,5)P2, increase, revealing that INPP5E is critical for generating the distinct phosphoinositide composition of cilia [43,44].
In addition, a phosphoinositide 5-phosphatase paralog of INPP5E, OCRL, can localize to primary cilia [120,121]. Mutations in OCRL is associated with Lowe syndrome, a multisystem disorder characterized by eye, nervous system and renal dysfunction [122]. The cilia of fibroblasts derived from Lowe syndrome patients have increased levels of PI(4,5)P2 and reduced levels of PI(4)P, suggesting that multiple phosphoinositide 5-phosphatases may have overlapping functions in controlling ciliary phosphoinositide composition.
For INPP5E and other phosphoinositide 5-phosphatases to generate the ciliary pool of PI(4)P, they must localize specifically to cilia. INPP5E is a peripheral membrane protein and, as discussed above for many other membrane-associated proteins, the ciliary localization of INPP5E depends on the transition zone MKS complex. The MKS complex localizes the small GTPase ARL13B to the cilium [62], and in conjunction with with PDE6D, ARL13B is itself critical for the ciliary localization of INPP5E [123–125]. Thus, it is likely that the transition zone MKS complex works through ARL13B to localize INPP5E to the cilium to generate the sharp boundary in phosphoinositide composition.
In wild type cells, the border between the ciliary PI(4)P domain and the PI(4,5)P2 domain is sharp, and how the sharpness of this border is maintained remains a question of interest. In addition to its role in directing the ciliary localization of INPP5E, there are two lines of evidence that raise the possibility that the transition zone may control the PI(4)P/PI(4,5)P2 border directly. First, several transition zone components contain a C2 domain, or the related B9 domain, both of which can bind phospholipids [64,126]. Second, many other transition zone components are transmembrane proteins that may generate an “anchored protein picket.” Models of anchored transmembrane proteins suggest that collectively they can restrict the diffusion of phospholipids through steric hindrance and hydrodynamic slowing, forming picket fences to partition membrane lipids [127]. For example, in the axon initial segment, anchored, densely packed transmembrane proteins limit the free diffusion of phospholipids [128].
Although tempting to speculate that the transition zone may restrict the movement of lipids into or out of the cilium, thereby drawing the PI(4)P/PI(4,5)P2 border, the C2 domain of the transition zone protein RPGRIP1 does not bind lipids [129]. These data raise the possibility that transition zone C2 domains instead function to facilitate protein-protein interactions. Adding to the complexity, beyond the transition zone affecting the distribution of phosphoinositides, INPP5E and phosphoinositides may reciprocally influence the composition of the transition zone [130]. Once established within cilia, phosphoinositides help control the distribution of ciliary proteins. The ciliary localization of TULP3, a Tubby-family protein that binds PI(4,5)P2 [131], increases as ciliary PI(4,5)P2 increases and PI(4)P decreases, suggesting that ciliary TULP3 levels are restrained by wild type ciliary phosphoinositide composition [43,44]. In a PI(4,5)P2-dependent fashion, TULP3 binds to the IFT-A complex and helps deliver diverse membrane-associated proteins to cilia, including GPCRs and PKD2 [131–133]. However, when TULP3 encounters PI(4)P in cilia, it may disassociate from the membrane and release its cargo within the ciliary membrane [44,133]. Loss of function mutations affecting TULP3, components of the IFT-A complex, or the ciliary GPCR GPR161 cause misactivation of Hedgehog signaling [132,134–139]. As loss of INPP5E attenuates Hedgehog signaling, it is likely that INPP5E impinges on signaling by limiting ciliary PI(4,5)P2 levels, thereby limiting ciliary TULP3, IFT-A and GPR161 [43,44]. Thus, a protein complex at the ciliary base (the MKS complex) controls the ciliary localization of an enzyme (INPP5E) which determines the lipid composition of the ciliary membrane (phosphoinositides) to control the ciliary localization of a lipid sensor (TULP3) to regulate the levels of a protein that controls the signaling output of cilia (GPR161).
It is likely that other Tubby family members function in manners analogous to TULP3 as cell type-specific sensors of ciliary phosphoinositides. In support of this possibility, mutation of either the founding member of the family, TUBBY, or its paralog, TULP1, are associated with obesity and retinal degeneration, phenotypes also observed in ciliopathies [140–142]. TUBBY is expressed predominantly by the retina and brain, and, like TULP3, it is involved in localizing GPCRs to cilia [133,143]. It is possible that photoreceptor phosphoinositides are read out by TUBBY and TULP1 to promote the delivery of Rhodopsin to the outer segment, defects in which cause photoreceptor loss, and read out by TUBBY to promote the delivery of MC4R to the paraventricular nucleus cilium [144], defects in which cause obesity.
TUBBY family proteins are present in diverse ciliated organisms, including Chlamydomonas, Tetrahymena and Paramecia. These data suggest that TUBBY family proteins, and by extension, phosphoinositides have phylogenetically ancient roles in ciliary biology. Whether phosphoinositides help direct ciliary protein trafficking in protists, or whether they might play other roles such as allosteric regulators of ciliary protein function, determinants of ciliary membrane viscosity, or contributors to membrane electrostatics, has yet to be established. Other ciliary proteins, including a component of the BBSome, can bind phosphoinositides, raising the possibility that proteins other than TUBBY family members are involved in interpreting ciliary phospholipids [86,89].
In further support of the possibility that ciliary phosphoinositides are critical for ciliary signaling, INPP5E is important for oncogenic Hedgehog signaling [145]. Misactivation of the Hedgehog pathway in the cerebellar external granule layer causes medulloblastoma, the most common pediatric brain cancer [146–148]. Medulloblastoma cells can be ciliated, and conditional deletion of Inpp5e in a mouse genetic model of Hedgehog-associated medulloblastoma promotes cilia loss and slows tumor progression [145,149]. Reduced INPP5E expression in human medulloblastoma patients is similarly associated with improved survival [145], but it is not known whether these differences are due to a direct effect on ciliary signaling.
Similar to genes encoding several transition zone components, mutations in human INPP5E are a cause of Joubert syndrome, and knockout of mouse Inpp5e results in phenotypes characteristic of ciliopathies, including cystic kidneys and polydactyly [130,150,151]. It will be interesting to assess whether, beyond phosphoinositides, additional ciliary lipids participate in ciliary signaling and the pathogenesis of ciliopathies.
With respect to the relationship between ciliary Hedgehog signaling and ciliary membrane lipid composition, cholesterol and cholesterol-derivatives such as oxysterols bind to Smoothened to modulate the Hedgehog transcriptional program [152–157]. Indeed, some of the developmental manifestations of SHH mutation are phenocopied by mutation of the cholesterol biosynthetic enzyme DHCR7 in Smith-Lemli-Opitz syndrome (SLOS), most notably holoprosencephaly. The most abundant oxysterol in the brain of a mouse genetic model of SLOS can inhibit Hedgehog signaling, and C. reinhardtii cilia contain an ergosterol which inhibits Hedgehog signal transduction by binding to Smoothened [158,159]. These data tantalizingly suggest that ciliary lipids may be the long-sought ligands that activate Smoothened downstream of Patched! Although it has yet to be determined if cilia contain cholesterol derivatives that stimulate the Hedgehog pathway, it is notable that the cerebrospinal fluid of medulloblastoma patients is enriched in an oxysterol that can activate Smoothened [160,161].
Ciliary-derived vesicles participate in diverse cellular functions.
Extracellular vesicles (ECVs) have emerged as a mechanism by which cells can communicate over long distances. Apart from the rest of the cell, cilia can be a source of ECVs with specialized critical functions. For example, in C. reinhardtii, ECVs released from flagella contain proteolytic enzymes that degrade the mother cell wall to release daughter cells (Figure 5) [162].
Figure 5.
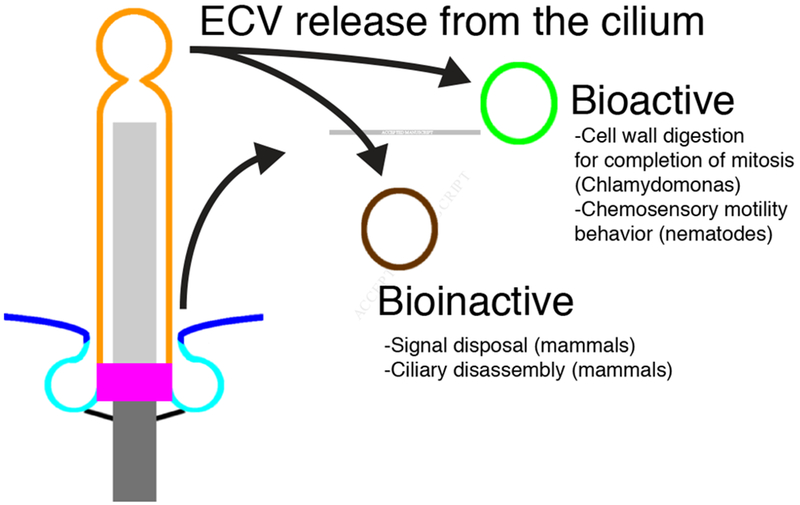
Ciliary-derived extracellular vesicles
Vesicles derived from either the ciliary membrane (orange) or the periciliary membrane (cyan) may fall into at least two distinct categories. Bioactive vesicles have a role in cell-to-cell communication in C. elegans or in cell wall digestion for the completion of mitosis in C. reinhardtii. Bioinactive vesicles have a role in signal disposal or ciliary disassembly in mammals.
ECVs are also associated with animal cilia. In C. elegans, ECVs containing LOV-1 and PKD-2, the orthologs of PKD1 and PKD2, are released from sensory neurons [163]. These vesicles, most probably derived from the PCMC or ciliary membranes of select cilia, accumulate in the extracellular space surrounding cilia. Furthermore, a genetic screen for mutants that fail to localize PKD-2 to the cilium identified a key role for CIL-7, an invertebrate myristoylated protein, in ECV biogenesis [164]. Without CIL-7, ECVs accumulate in the extracellular space surrounding the ciliary base and the PCMC and fail to be released into the environment [164]. ECVs induce male tail chasing behavior, suggesting a role of the ECVs in intercellular communication [163]. Consistently, loss of CIL-7 also results in a defect in male mating behavior [164]. As the C. reinhardtii, flagellar membrane glycoproteins are shed from the flagellar base [165], the ciliary base or the PCMC may to be an evolutionarily conserved source of ciliary ECVs.
In the vertebrate retina, the tip of each outer segment sheds ECVs derived from about six discs per day, removing Opsins with oxidized retinols [166]. These cilium-derived ECVs are taken up by the neighboring retinal pigmented epithelium where the retinol is reduced and transported back to the photoreceptor to regenerate function Opsins [166]. At the base of the outer segment, membrane discs are formed by membrane evagination and are retained within the outer segment and restrained from being shed by the disc-specific protein Peripherin [167,168]. The simultaneous processes of controlled vesicular shedding at the tip and membrane disc formation at the base allows continuous renewal of the outer segments. Defects in many steps of outer segment biogenesis and renewal result in retinal degeneration.
The source of ECVs in other mammalian tissues are more mysterious, but ECVs purified from urine contain many ciliary proteins, including PKD1 and PKD2, suggesting that nephron cilia may be a source of ECVs [169]. In cultured cells, these urinary ECVs interact with primary cilia of kidney epithelial cells, raising the possibility that they may be communicating to cilia [169]. In another context, primary cilia of neuroepithelial cells release ECVs containing Prominin-1 into neural tube fluid [170]. Whether these ECVs are involved in recycling degraded cellular components, as in the photoreceptor, or whether they, as in C. reinhardtii and C. elegans, are involved in triggering biological events, is not yet clear.
Similar to photoreceptor cells, cultured mammalian cells generate ECVs from their ciliary tips [103,171]. As Actin filaments are not prominent features in the cilium, it is a surprise that Actin-dependent events are deployed within the cilium to release ciliary ECVs from the tip. These ECVs are released in a signal-dependent manner and contain transmembrane signaling molecules such as the anorexigenic GPCR NPY2R [103]. Perhaps these ECVs are a way by which cilia discard ciliary membrane and associated proteins to terminate signaling. In this way, these cilia-derived ECVs may function analogously to retinal ECVs as garbage bins critical for maintaining ciliary homeostasis.
Parallel live imaging studies have shown that intraciliary PI(4,5)P2 and F-actin trigger ECV release from the ciliary tip to promote ciliary disassembly [171]. Thus, cilia-derived ECVs may also prepare for ciliary disassembly, an event that must occur before the initiation of mitosis so that the basal body can relocalize to the spindle pole. A non-mutually exclusive role for ECV release may therefore be to rapidly discard ciliary membrane and proteins to facilitate ciliary disassembly in preparation for mitosis [171].
Thus, ciliary ECVs may fall into at least two categories (Figure 5). Bioactive ciliary ECVs can perform cellular functions like degrading the C. reinhardtii cell wall or coordinating C. elegans male mating behavior, whereas bioinactive ciliary ECVs can help discard unneeded material from the cilium.
Conclusion and future directions of ciliary membrane investigation
As an important interface with the rest of the world, the ciliary membrane is a fascinating example of how membrane specialization confers critical functions, allowing the cilium to function as the antenna for the cell. Essential for the signaling functions of cilia is precise control of ciliary membrane compartmentalization, composition and morphology. Different types of cilia exhibit diverse membrane morphologies, and the morphology and remodeling of ciliary membranes can be dynamically modified by signaling activity.
Ciliary signaling depends not just on proteins. Indeed, cilia have distinct membrane lipid compositions that work together with protein complexes, such as the BBSome, to create the complexity required for signaling diverse inputs to the cell body. For example, phosphoinositides enriched in the ciliary membrane promote the deposition of specific membrane signaling proteins in the cilium.
Highlighting the importance of both lipids and proteins, many ciliopathies are characterized by disruptions in the ciliary localization of both, with concomitant effects on ciliary signaling. Recent studies of vesicles derived from the ciliary membrane have shed light on novel functions of ECVs, and suggest that many secrets of how the ciliary membrane functions remain to be uncovered. These cilia-derived ECVs may play function in intercellular signaling, serve as a ciliary waste disposal system, or both.
Finding the answers to open questions about how the ciliary cilium functions will rely on disparate approaches, from genetic perturbations in model organisms to biochemical reconstitution of minimal systems. What lipids are found within ciliary membrane can be determined by biochemical fractionation and lipid mass spectrometry. How proteins and protein complexes shape membranes can be determined using biochemical reconstitution and transmission electron microscopy. Which proteins localize to the cilium or subcompartments within the cilium can be assessed using proximity-labeling proteomics and fluorescence microscopy, especially super-resolution microscopy. How ciliary proteins participate in ciliary signaling required for development and organismal behavior can be determined using genetically tractable model organisms. These diverse strategies will be key to the future work of uncovering the molecular mechanisms by which ciliary membranes form and function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Keating CD. Aqueous phase separation as a possible route to compartmentalization of biological molecules. Acc Chem Res 2012;45:2114–24. doi: 10.1021/ar200294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Szostak JW. An optimal degree of physical and chemical heterogeneity for the origin of life? Philos T rans R Soc Lond, B, Biol Sci 2011. ;366:2894–901. doi: 10.1098/rstb.2011.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pan J, Snell WJ. Signal transduction during fertilization in the unicellular green alga, Chlamydomonas. Curr Opin Microbiol 2000;3:596–602. [DOI] [PubMed] [Google Scholar]
- [5].Ferris PJ, Waffenschmidt S, Umen JG, Lin H, Lee J-H, Ishida K, et al. Plus and minus sexual agglutinins from Chlamydomonas reinhardtii. The Plant Cell 2005;17:597–615. doi: 10.1105/tpc.104.028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mesland DA, Hoffman JL, Caligor E, Goodenough UW. Flagellar tip activation stimulated by membrane adhesions in Chlamydomonas gametes. J Cell Biol 1980;84:599–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Snell WJ, Buchanan M, Clausell A. Lidocaine reversibly inhibits fertilization in Chlamydomonas: a possible role for calcium in sexual signalling. J Cell Biol 1982;94:607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bloodgood RA, Levin EN. Transient increase in calcium efflux accompanies fertilization in Chlamydomonas. J Cell Biol 1983;97:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pijst HLA, van Driel R, Janssens PMW, Musgrave A, van den Ende H. Cyclic AMP is involved in sexual reproduction of Chlamydomonas eugametos. FEBS Lett 1984;174:132–6. doi: 10.1016/0014-5793(84)81091-1. [DOI] [Google Scholar]
- [10].Huangfu D, Liu A, Rakeman AS, Murcia Ns, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003;426:83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- [11].Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007;317:372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- [12].Kim J, Hsia EYC, Brigui A, Plessis A, Beachy PA, Zheng X. The role of ciliary trafficking in Hedgehog receptor signaling. Sci Signal 2015;8:ra55–5. doi: 10.1126/scisignal.aaa5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature 2005;437:1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- [14].Mirzadeh Z, Kusne Y, Duran-Moreno M, Cabrales E, Gil-Perotin S, Ortiz C, et al. Bi- and uniciliated ependymal cells define continuous floor-plate-derived tanycytic territories. Nat Commun 2017;8:13759. doi: 10.1038/ncomms13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ward S, Thomson N, White JG, Brenner S. Electron microscopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans.?2UU. J Comp Neurol 1975;160:313–37. doi: 10.1002/cne.901600305. [DOI] [PubMed] [Google Scholar]
- [16].Ware RW, Clark D, Crossland K, Russell RL. The nerve ring of the nematode Caenorhabditis elegans: Sensory input and motor output. Journal of Comparative Neurology 1975;162:71–110. doi: 10.1002/cne.901620106. [DOI] [Google Scholar]
- [17].Perkins LA, Hedgecock EM, Thomson JN, Culotti JG. Mutant sensory cilia in the nematode Caenorhabditis elegans. Developmental Biology 1986;117:456–87. [DOI] [PubMed] [Google Scholar]
- [18].Musgrave A, de Wildt P, van Etten I, Pijst H, Scholma C, Kooyman R, et al. Evidence for a functional membrane barrier in the transition zone between the flagellum and cell body of Chlamydomonas eugametos gametes. Planta 1986;167:544–53. doi: 10.1007/BF00391231. [DOI] [PubMed] [Google Scholar]
- [19].Hunnicutt GR, Kosfiszer MG, Snell WJ. Cell body and flagellar agglutinins in Chlamydomonas reinhardtii: the cell body plasma membrane is a reservoir for agglutinins whose migration to the flagella is regulated by a functional barrier. J Cell Biol 1990;111:1605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 2002;13:2508–16. [DOI] [PubMed] [Google Scholar]
- [21].Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol 2002;12:r378–80. [DOI] [PubMed] [Google Scholar]
- [22].Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 2000; 151:709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LSB, Somlo S, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 2003;100:5286–91. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science 1993;261:1280–1. [DOI] [PubMed] [Google Scholar]
- [25].Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 2010;142:158–69. doi: 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fueller J, Egorov MV, Walther KA, Sabet O, Mallah J, Grabenbauer M, et al. Subcellular Partitioning of Protein Tyrosine Phosphatase 1B to the Endoplasmic Reticulum and Mitochondria Depends Sensitively on the Composition of Its Tail Anchor. PLoS ONE 2015;10:e0139429. doi: 10.1371/journal.pone.0139429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tyler KM, Fridberg A, Toriello KM, Olson CL, Cieslak JA, Hazlett TL, et al. Flagellar membrane localization via association with lipid rafts. J Cell Sci 2009;122:859–66. doi: 10.1242/jcs.037721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Poo M, Cone RA. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature 1974;247:438–41. [DOI] [PubMed] [Google Scholar]
- [29].Aveldñno MI, Bazan NG. Molecular species of phosphatidylcholine, -ethanolamine, -serine, and -inositol in microsomal and photoreceptor membranes of bovine retina. J Lipid Res 1983;24:620–7. [PubMed] [Google Scholar]
- [30].Boesze-Battaglia K, Schimmel R. Cell membrane lipid composition and distribution: implications for cell function and lessons learned from photoreceptors and platelets. J Exp Biol 1997;200:2927–36. [DOI] [PubMed] [Google Scholar]
- [31].Nguyen PAT, Liou W, Hall DH, Leroux MR. Ciliopathy proteins establish a bipartite signaling compartment in a C. elegans thermosensory neuron. J Cell Sci 2014;127:5317–30. doi: 10.1242/jcs.157610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, et al. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci 2010;123:1785–95. doi: 10.1242/jcs.059519. [DOI] [PubMed] [Google Scholar]
- [33].Peters KR, Palade GE, Schneider BG, Papermaster DS. Fine structure of a periciliary ridge complex of frog retinal rod cells revealed by ultrahigh resolution scanning electron microscopy. J Cell Biol 1983;96:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Papermaster DS, Schneider BG, Besharse JC. Vesicular transport of newly synthesized opsin from the Golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in Xenopus retinas. Invest Ophthalmol Vis Sci 1985;26:1386–404. [PubMed] [Google Scholar]
- [35].Wilsman NJ, Farnum CE, Reed-Aksamit DK. Incidence and morphology of equine and murine chondrocytic cilia. Anat Rec 1980;197:355–61. doi: 10.1002/ar.1091970309. [DOI] [PubMed] [Google Scholar]
- [36].Graser S, Stierhof Y-D, Lavoie SB, Gassner OS, Lamla S, Le Clech M, et al. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 2007; 179:321–30. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lau L, Lee YL, Sahl SJ, Stearns T, Moerner WE. STED microscopy with optimized labeling density reveals 9-fold arrangement of a centriole protein. Biophys J 2012;102:2926–35. doi: 10.1016/j.bpj.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol 2001;11:1586–90. [DOI] [PubMed] [Google Scholar]
- [39].Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol 1995;131:1517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Doroquez DB, Berciu C, Anderson JR, Sengupta P, Nicastro D. A high-resolution morphological and ultrastructural map of anterior sensory cilia and glia in Caenorhabditis elegans. eLife 2014;3:e01948. doi: 10.7554/eLife.01948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Serwas D, Su TY, Roessler M, Wang S, Dammermann A. Centrioles initiate cilia assembly but are dispensable for maturation and maintenance in C. elegans. J Cell Biol 2017;216:1659–71. doi: 10.1083/jcb.201610070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WLC, Simons K. FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc Natl Acad Sci USA 2006;103:18556–61. doi: 10.1073/pnas.0608291103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chávez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev Cell 2015;34:338–50. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- [44].Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G, Abedin M, Schurmans S, et al. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev Cell 2015;34:400–9. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Melkonian M The functional analysis of the flagellar apparatus in green algae. Symp Soc Exp Biol 1982;35:589–606. [PubMed] [Google Scholar]
- [46].Weiss RL, Goodenough DA, Goodenough UW. Membrane particle arrays associated with the basal body and with contractile vacuole secretion in Chlamydomonas. J Cell Biol 1977;72:133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Garcia-Gonzalo FR, Reiter JF. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb Perspect Biol 2016:a028134. doi: 10.1101/cshperspect.a028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. J Cell Biol 1972;53:494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Montesano R Inhomogeneous distribution of filipin-sterol complexes in the ciliary membrane of rat tracheal epithelium. Am J Anat 1979;156:139–45. doi: 10.1002/aja.1001560115. [DOI] [PubMed] [Google Scholar]
- [50].Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chen Y, Yue S, Xie L, Pu X-H, Jin T, Cheng SY. Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3beta regulates its stability and localization in the primary cilium. J Biol Chem 2011;286:13502–11. doi: 10.1074/jbc.M110.217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shi X, Garcia G, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, et al. Superresolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol 2017;57:842–1188. doi: 10.1038/ncb3599. [DOI] [PubMed] [Google Scholar]
- [53].Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci 1968;3:207–30. [DOI] [PubMed] [Google Scholar]
- [54].SOROKIN S Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol 1962;15:363–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol 2015;17:228–40. doi: 10.1038/ncb3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mukhopadhyay S, Lu Y, Shaham S, Sengupta P. Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev Cell 2008;14:762–74. doi: 10.1016/j.devcel.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Olivier-Mason A, Wojtyniak M, Bowie RV, Nechipurenko IV, Blacque OE, Sengupta P. Transmembrane protein OSTA-1 shapes sensory cilia morphology via regulation of intracellular membrane trafficking in C. elegans. Development 2013;140:1560–72. doi: 10.1242/dev.086249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kaplan OI, Doroquez DB, Cevik S, Bowie RV, Clarke L, Sanders AAWM, et al. Endocytosis genes facilitate protein and membrane transport in C. elegans sensory cilia. Curr Biol 2012;22:451–60. doi: 10.1016/j.cub.2012.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Snouffer A, Brown D, Lee H, Walsh J, Lupu F, Norman R, et al. Cell Cycle-Related Kinase (CCRK) regulates ciliogenesis and Hedgehog signaling in mice. PLoS Genet 2017;13:e1006912. doi: 10.1371/journal.pgen.1006912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tam L-W, Wilson NF, Lefebvre Pa. A CDK-related kinase regulates the length and assembly of flagella in Chlamydomonas. J Cell Biol 2007;176:819–29. doi: 10.1083/jcb.200610022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell 2010;18:237–47. doi: 10.1016/j.devcel.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 2011. ;43:776–84. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- [64].Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SGM, Corbit KC, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Roberson EC, Dowdle WE, Ozanturk A, Garcia-Gonzalo FR, Li C, Halbritter J, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol 2015;209:129–42. doi: 10.1083/jcb.201411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto eA, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011;145:513–28. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yee LE, Garcia-Gonzalo FR, Bowie RV, Li C, Kennedy JK, Ashrafi K, et al. Conserved Genetic Interactions between Ciliopathy Complexes Cooperatively Support Ciliogenesis and Ciliary Signaling. PLoS Genet 2015;11 :e1005627. doi: 10.1371/journal.pgen.1005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 2011;192:1023–41. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Craige B, Tsao C-C, Diener DR, Hou Y, Lechtreck K-F, Rosenbaum JL, et al. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol 2010;190:927–40. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kamiya R, Witman GB. Submicromolar levels of calcium control the balance of beating between the two flagella in demembranated models of Chlamydomonas. J Cell Biol 1984;98:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Schouteden C, Serwas D, Palfy M, Dammermann A. The ciliary transition zone functions in cell adhesion but is dispensable for axoneme assembly in C. elegans. J Cell Biol 2015;210:35–44. doi: 10.1083/jcb.201501013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Awata J, Takada S, Standley C, Lechtreck KF, Bellve KD, Pazour GJ, et al. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci 2014;127:4714–27. doi: 10.1242/jcs.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, Nachury MV. An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. J Cell Biol 2013;203:129–47. doi: 10.1083/jcb.201212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lin Y-C, Niewiadomski P, Lin B, Nakamura H, Phua SC, Jiao J, et al. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat Chem Biol 2013;9:437–43. doi: 10.1038/nchembio.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Najafi M, Maza NA, Calvert PD. Steric volume exclusion sets soluble protein concentrations in photoreceptor sensory cilia. Proc Natl Acad Sci USA 2012;109:203–8. doi: 10.1073/pnas.1115109109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Calvert PD, Schiesser WE, Pugh EN. Diffusion of a soluble protein, photoactivatable GFP, through a sensory cilium. J Gen Physiol 2010;135:173–96. doi: 10.1085/jgp.200910322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 2010;329:436–9. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Milenkovic L, Weiss LE, Yoon J, Roth TL, Su YS, Sahl SJ, et al. Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched! Proc Natl Acad Sci USA 2015;112:8320–5. doi: 10.1073/pnas.1510094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].De Haene Acta Neurol Belg A, 1955. Agénésie partielle du vermis du cervelet à caractère familial. n.d. [PubMed]
- [80].Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969;19:813–25. [DOI] [PubMed] [Google Scholar]
- [81].Wright KJ, Baye LM, Olivier-Mason A, Mukhopadhyay S, Sang L, Kwong M, et al. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev 2011. ;25:2347–60. doi: 10.1101/gad.173443.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Fansa EK, Kösling SK, Zent E, Wittinghofer A, Ismail S. PDE6δ-mediated sorting of INPP5E into the cilium is determined by cargo-carrier affinity. Nat Commun 2016;7:11366. doi: 10.1038/ncomms11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gotthardt K, Lokaj M, Koerner C, Falk N, Giessl A, Wittinghofer A. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. eLife 2015;4:213. doi: 10.7554/eLife.11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jaiswal M, Fansa EK, Kösling SK, Mejuch T, Waldmann H, Wittinghofer A. Novel Biochemical and Structural Insights into the Interaction of Myristoylated Cargo with Unc119 Protein and Their Release by Arl2/3. J Biol Chem 2016;291:20766–78. doi: 10.1074/jbc.M116.741827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- [86].Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007;129:1201–13. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- [87].Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell 2008; 15:854–65. doi: 10.1016/j.devcel.2008.11.001. [DOI] [PubMed] [Google Scholar]
- [88].Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J Med Genet 2014;51:132–6. doi: 10.1136/jmedgenet-2013-101785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, et al. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010;141:1208–19. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mourão A, Nager AR, Nachury MV, Lorentzen E. Structural basis for membrane targeting of the BBSome by ARL6. Nat Struct Mol Biol 2014;21:1035–41. doi: 10.1038/nsmb.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, et al. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 2004;18:1630–42. doi: 10.1101/gad.1194004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ou G, Blacque oE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature 2005;436:583–7. doi: 10.1038/nature03818. [DOI] [PubMed] [Google Scholar]
- [93].Ou G, Koga M, Blacque OE, Murayama T, Ohshima Y, Schafer JC, et al. Sensory ciliogenesis in Caenorhabditis elegans: assignment of IFT components into distinct modules based on transport and phenotypic profiles. Mol Biol Cell 2007;18:1554–69. doi: 10.1091/mbc.E06-09-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA 2008;105:4242–6. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K. Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell 2008;19:1540–7. doi: 10.1091/mbc.E07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA 2004; 101:16588–93. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, Mykytyn K. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci 2011;68:2951–60. doi: 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Xu Q, Zhang Y, Wei Q, Huang Y, Li Y, Ling K, et al. BBS4 and BBS5 show functional redundancy in the BBSome to regulate the degradative sorting of ciliary sensory receptors. Sci Rep 2015;5:11855. doi: 10.1038/srep11855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lechtreck KF, Brown JM, Sampaio JL, Craft JM, Shevchenko A, Evans JE, et al. Cycling of the signaling protein phospholipase D through cilia requires the BBSome only for the export phase. J Cell Biol 2013;201:249–61. doi: 10.1083/jcb.201207139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lechtreck K-F, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, et al. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol 2009;187:1117–32. doi: 10.1083/jcb.200909183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Zhang Q, Nishimura D, Seo S, Vogel T, Morgan DA, Searby C, et al. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci USA 2011;108:20678–83. doi: 10.1073/pnas.1113220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McIntyre JC, Hege MM, Berbari NF. Trafficking of ciliary G protein-coupled receptors. Methods Cell Biol 2016;132:35–54. doi: 10.1016/bs.mcb.2015.11.009. [DOI] [PubMed] [Google Scholar]
- [103].Nager AR, Goldstein JS, Herranz-Pérez V, Portran D, Ye F, Garcia-Verdugo JM, et al. An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell 2016. doi: 10.1016/j.cell.2016.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Eguether T, San Agustin JT, Keady BT, Jonassen JA, Liang Y, Francis R, et al. IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell 2014;31:279–90. doi: 10.1016/j.devcel.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Liew GM, Ye F, Nager AR, Murphy JP, Lee JS, Aguiar M, et al. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell 2014;31:265–78. doi: 10.1016/j.devcel.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, et al. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet 2011. ;7:e1002358. doi: 10.1371/journal.pgen.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet 2012;21:1945–53. doi: 10.1093/hmg/dds004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Goetz SC, Bangs F, Barrington CL, Katsanis N, Anderson KV. The Meckel syndrome-associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling. PLoS ONE 2017;12:e0173399. doi: 10.1371/journal.pone.0173399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Tayeh MK, Yen H-J, Beck JS, Searby CC, Westfall TA, Griesbach H, et al. Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet 2008;17:1956–67. doi: 10.1093/hmg/ddn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhang Y, Seo S, Bhattarai S, Bugge K, Searby CC, Zhang Q, et al. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum Mol Genet 2014;23:40–51. doi: 10.1093/hmg/ddt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Barbelanne M, Hossain D, Chan DP, Peranen J, Tsang WY. Nephrocystin proteins NPHP5 and Cep290 regulate BBSome integrity, ciliary trafficking and cargo delivery. Hum Mol Genet 2015;24:2185–200. doi: 10.1093/hmg/ddu738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ye F, Nager AR, Nachury MV. BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. bioRxiv 2017:180620. doi: 10.1101/180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Pal K, Hwang S-H, Somatilaka B, Badgandi H, Jackson PK, DeFea K, et al. Smoothened determines β-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J Cell Biol 2016;212:861–75. doi: 10.1083/jcb.201506132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Balla T Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 2013;93:1019–137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006;443:651–7. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- [116].Roth MG. Phosphoinositides in constitutive membrane traffic. Physiol Rev 2004;84:699–730. doi: 10.1152/physrev.00033.2003. [DOI] [PubMed] [Google Scholar]
- [117].Hammond GRV, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, et al. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012;337:727–30. doi: 10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Hammond GRV, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol 2014;205:113–26. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Bae Y-K, Kim E, L’hernault SW, Barr MM. The CIL-1 PI 5-phosphatase localizes TRP Polycystins to cilia and activates sperm in C. elegans. Curr Biol 2009;19:1599–607. doi: 10.1016/j.cub.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, et al. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 1992;358:239–42. doi: 10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- [121].Prosseda PP, Luo N, Wang B, Alvarado JA, Hu Y, Sun Y. Loss of OCRL increases ciliary PI(4,5)P2 in Lowe oculocerebrorenal syndrome. J Cell Sci 2017;130:3447–54. doi: 10.1242/jcs.200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].De Matteis Ma, Staiano L, Emma F, Devuyst O The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat Rev Nephrol 2017;13:455–70. doi: 10.1038/nrneph.2017.83. [DOI] [PubMed] [Google Scholar]
- [123].Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat 2014;35:137–46. doi: 10.1002/humu.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci USA 2012;109:19691–6. doi: 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kösling SK, Fansa EK, Maffini S, Wittinghofer A. Mechanism and dynamics of INPP5E transport into and inside the ciliary compartment. Biol Chem 2017;0. doi: 10.1515/hsz-2017-0226. [DOI] [PubMed] [Google Scholar]
- [126].Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol 2012;197:697–709. doi: 10.1083/jcb.201111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J Cell Biol 2002;157:1071–81. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Nakada C, Ritchie K, Oba Y, Nakamura M, Hotta Y, Iino R, et al. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol 2003;5:626–32. doi: 10.1038/ncb1009. [DOI] [PubMed] [Google Scholar]
- [129].Remans K, Burger M, Vetter IR, Wittinghofer A. C2 domains as protein-protein interaction modules in the ciliary transition zone. Cell Rep 2014;8:1–9. doi: 10.1016/j.celrep.2014.05.049. [DOI] [PubMed] [Google Scholar]
- [130].Dyson JM, Conduit SE, Feeney SJ, Hakim S, DiTommaso T, Fulcher AJ, et al. INPP5E regulates phosphoinositide-dependent cilia transition zone function. J Cell Biol 2017;216:247–63. doi: 10.1083/jcb.201511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, et al. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev 2010;24:2180–93. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 2013;152:210–23. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- [133].Badgandi HB, Hwang S-H, Shimada IS, Loriot E, Mukhopadhyay S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J Cell Biol 2017;216:743–60. doi: 10.1083/jcb.201607095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Norman RX, Ko HW, Huang V, Eun CM, Abler LL, Zhang Z, et al. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum Mol Genet 2009;18:1740–54. doi: 10.1093/hmg/ddp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Patterson VL, Damrau C, Paudyal A, Reeve B, Grimes DT, Stewart ME, et al. Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Hum Mol Genet 2009;18:1719–39. doi: 10.1093/hmg/ddp075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Cameron DA, Pennimpede T, Petkovich M. Tulp3 is a critical repressor of mouse hedgehog signaling. Dev Dyn 2009;238:1140–9. doi: 10.1002/dvdy.21926. [DOI] [PubMed] [Google Scholar]
- [137].Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci USA 2011;108:1456–61. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet 2008;40:403–10. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Liem KF, Ashe A, He M, Satir P, Moran J, Beier D, et al. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol 2012;197:789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Noben-Trauth K, Naggert JK, North MA, Nishina PM. A candidate gene for the mouse mutation tubby. Nature 1996;380:534–8. doi: 10.1038/380534a0. [DOI] [PubMed] [Google Scholar]
- [141].Kleyn PW, Fan W, Kovats SG, Lee JJ, Pulido JC, Wu Y, et al. Identification and characterization of the mouse obesity gene tubby: a member of a novel gene family. Cell 1996;85:281–90. [DOI] [PubMed] [Google Scholar]
- [142].Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet 1998;18:174–6. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- [143].Sun X, Haley J, Bulgakov OV, Cai X, McGinnis J, Li T. Tubby is required for trafficking G protein-coupled receptors to neuronal cilia. Cilia 2012; 1:21. doi: 10.1186/2046-2530-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Siljee JE, Wang Y, Bernard AA, Ersoy BA, Zhang S, Marley A, et al. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat Genet 2018;50:180–5. doi: 10.1038/s41588-017-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Conduit SE, Ramaswamy V, Remke M, Watkins DN, Wainwright BJ, Taylor MD, et al. A compartmentalized phosphoinositide signaling axis at cilia is regulated by INPP5E to maintain cilia and promote Sonic Hedgehog medulloblastoma. Oncogene 2017;36:5969–84. doi: 10.1038/onc.2017.208. [DOI] [PubMed] [Google Scholar]
- [146].Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2015;17 Suppl 4:iv1–iv62. doi: 10.1093/neuonc/nov189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997;277:1109–13. [DOI] [PubMed] [Google Scholar]
- [148].Vorechovsky I, Tingby O, Hartman M, Strömberg B, Nister M, Collins VP, et al. Somatic mutations in the human homologue of Drosophila patched in primitive neuroectodermal tumours. Oncogene 1997;15:361–6. doi: 10.1038/sj.onc.1201340. [DOI] [PubMed] [Google Scholar]
- [149].Han Y-G, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 2009; 15:1062–5. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 2009;41:1027–31. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- [151].Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, et al. Mutations in INPp5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 2009;41:1032–6. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, et al. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell 2013;26:346–57. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol 2013;9:557–64. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, et al. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. eLife 2013;2:e01340. doi: 10.7554/eLife.01340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Byrne EFX, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016;535:517–22. doi: 10.1038/nature18934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Kelley RL, Roessler E, Hennekam RC, Feldman GL, Kosaki K, Jones MC, et al. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet 1996;66:478–84. doi: 10.1002/(SICI)1096-8628(19961230)66:4<478::AID-AJMG22>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- [157].Roux C, Wolf C, Mulliez N, Gaoua W, Cormier V, Chevy F, et al. Role of cholesterol in embryonic development. Am J Clin Nutr 2000;71:1270S–9S. [DOI] [PubMed] [Google Scholar]
- [158].Sever N, Mann rK, Xu L, Snell WJ, Hernandez-Lara CI, Porter NA, et al. Endogenous B-ring oxysterols inhibit the Hedgehog component Smoothened in a manner distinct from cyclopamine or side-chain oxysterols. Proc Natl Acad Sci USA 2016; 113:5904–9. doi: 10.1073/pnas.1604984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Xu L, Mirnics K, Bowman AB, Liu W, Da J, Porter NA, et al. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol Dis 2012;45:923–9. doi: 10.1016/j.nbd.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 2016;5:1055. doi: 10.7554/eLife.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bernini F, Sangiovanni G, Pezzotta S, Galli G, Fumagalli R, Paoletti P. Selected ion monitoring technique for the evaluation of sterols in cerebrospinal fluid: a new approach to desmosterol test for central nervous system tumors. Journal of NeuroOncology 1986;4:31–5. [DOI] [PubMed] [Google Scholar]
- [162].Wood cR, Huang K, Diener DR, Rosenbaum JL. The cilium secretes bioactive ectosomes. Curr Biol 2013;23:906–11. doi: 10.1016/j.cub.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Wang J, Silva M, Haas LA, Morsci NS, Nguyen KcQ, Hall DH, et al. C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr Biol 2014;24:519–25. doi: 10.1016/j.cub.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Maguire JE, Silva M, Nguyen KCQ, Hellen E, Kern AD, Hall DH, et al. Myristoylated CIL-7 regulates ciliary extracellular vesicle biogenesis. Mol Biol Cell 2015;26:2823–32. doi: 10.1091/mbc.E15-01-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Bloodgood RA, Woodward MP, Salomonsky NL. Redistribution and shedding of flagellar membrane glycoproteins visualized using an anti-carbohydrate monoclonal antibody and concanavalin A. J Cell Biol 1986;102:1797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol 1969;42:392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Salinas RY, Pearring JN, Ding J-D, Spencer WJ, Hao Y, Arshavsky VY. Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J Cell Biol 2017;216:1489–99. doi: 10.1083/jcb.201608081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Ding J-D, Salinas RY, Arshavsky VY. Discs of mammalian rod photoreceptors form through the membrane evagination mechanism. J Cell Biol 2015;211:495–502. doi: 10.1083/jcb.201508093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol 2009;20:278–88. doi: 10.1681/ASN.2008060564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Dubreuil V, Marzesco A-M, Corbeil D, Huttner WB, Wilsch-Brauninger M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol 2007;176:483–95. doi: 10.1083/jcb.200608137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Phua SC, Chiba S, Suzuki M, Su E, Roberson EC, Pusapati GV, et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell 2017;168:264–279. e15. doi: 10.1016/j.cell.2016.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]