

Abstract
Objective
Alzheimer’s disease (AD) is a severe neurodegenerative disorder and has become a global public health problem. Intensive research has been conducted for AD. But the pathophysiology of AD is still not elucidated. Disease comorbidity often associates diseases with overlapping patterns of genetic markers. This may inform a common etiology and suggest essential protein targets. US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) collects large-scale postmarketing surveillance data that provide a unique opportunity to investigate disease co-occurrence pattern. We aim to construct a heterogeneous network that integrates disease comorbidity network (DCN) from FAERS with protein–protein interaction (PPI) to prioritize the AD risk genes using network-based ranking algorithm.
Materials and Methods
We built a DCN based on indication data from FAERS using association rule mining. DCN was further integrated with PPI network. We used random walk with restart ranking algorithm to prioritize AD risk genes.
Results
We evaluated the performance of our approach using AD risk genes curated from genetic association studies. Our approach achieved an area under a receiver operating characteristic curve of 0.770. Top 500 ranked genes achieved 5.53-fold enrichment for known AD risk genes as compared to random expectation. Pathway enrichment analysis using top-ranked genes revealed that two novel pathways, ERBB and coagulation pathways, might be involved in AD pathogenesis.
Conclusion
We innovatively leveraged FAERS, a comprehensive data resource for FDA postmarket drug safety surveillance, for large-scale AD comorbidity mining. This exploratory study demonstrated the potential of disease-comorbidities mining from FAERS in AD genetics discovery.
Keywords: Alzheimer’s disease, FAERS, disease comorbidity network, protein–protein interaction, disease gene discovery
Introduction
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder characterized by the progressive loss of cholinergic neurons, leading to the onset of severe behavioral, motor, and cognitive impairments. An estimated 5.4 million Americans have AD. It is the sixth leading cause of death in the United States and the fifth leading cause of death in Americans age ≥ 65 years. Between 2000 and 2013, deaths from AD increased 71%.1 Though intensive research for AD has been conducted, the etiology of AD is still not elucidated.
Computational-based approaches have been widely used in disease gene discovery.2,3 Network-based disease algorithm utilizes disease relationship to prioritize candidate disease genes. The key for network-based disease gene discovery is to construct disease relationship. Disease manifestation and electronic medical record (EMR) have been used for this purpose. For example, we constructed a disease manifestation network (DMN) to predict novel genes for Parkinson’s disease.4 Bagley et al. discovered new genes for autoimmune disorder and neuropsychiatric disorder using EMR.5 Disease comorbidity often associates diseases with overlapping patterns of genetic markers5,6 and several comorbidity networks have also been built.7–9 Recently, a very interesting disease trajectory relationship were also established based on EMR data on 6.2 million patients.10 However, these networks are biased towards special population7 or single medical center9 and have not been used in disease gene discovery.
FDA Adverse Event Reporting System (FAERS) contains adverse event reports from manufacturers, consumers, and healthcare professionals for all marketed drug and therapeutic biologic products, which is a large-scale database that contains seven linked data files representing patient demographics, drugs, indications, outcomes, reactions, therapies, and reporting sources.11 FAERS data have been intensively used in drug safety issue studies. But the other possible usages have not been explored. We noticed that each case report in indication data contains information for all used drugs and diseases when drug adverse event occurs, which essentially reflects the co-occurring diseases in an individual. Based on this observation, we explore the possibility of FAERS in disease comorbidity study. Compared with EMR, indication data of FAERS have several advantages. First, all co-occurring diseases reported in FAERS are treated by drugs, which helps to reduce the disease noise. Second, large scale of FAERS makes data unbiased for specific diseases. Third, FAERS provides a unified reporting system in whole population level, which can avoid the potential bias of EMR toward specific population or discrepancy across health care systems.12,13
In this study, we used association rule mining to explore this large-scale data to construct a disease comorbidity network (DCN). One of the advantages of this method is that it can flexibly detect multiple disease comorbidities, which is common in clinic setting.14 DCN was further integrated with protein–protein interaction (PPI) network. We used network and functional analysis to reveal the novel genes and pathways for AD.
METHODS
Our overall methods are shown in Figure 1. First, we used association rule mining to construct a DCN from FAERS; second, we constructed a heterogeneous network by integration of DCN with PPI network; third, we used random walk with restart to prioritize AD risk genes and evaluated the performance of our methods using de novo prediction of validation gene set from AlzGene database; fourth, we used AD as the seed to prioritize the new AD risk genes; finally, we performed the pathway analysis using top-ranked genes to discover novel pathways that might be involved in AD pathogenesis.
Figure 1.

Overview of our method. ARM: association rule mining; DCN: disease comorbidity network; PPI: protein–protein interaction.
Data
FAERS data were downloaded from US Food and Drug Administration (FDA), which contains 17 305 542 case reports for indications from 2004 to 2017.11 Disease genetic data were extracted from Online Mendelian Inheritance in Man (OMIM). The OMIM catalog contains 15 462 disease–gene associations for 8832 genes and 6018 diseases/traits.15 Protein–protein interaction were obtained from STRING database, which contains 1 380 504 interactions for 17 860 genes.16 AlzGene database collects AD risk genes (679 genes) that were derived from comprehensive genetic association studies.17
Construction of disease comorbidity network
Data processing
Indication files in FAERS from 2014 to 2017 were used in this study to explore disease comorbidity patterns. After removing reports with unknown indications, data contain 6 480 372 case reports and represent 15 721 indications of drugs. Table 1 shows a sample indication data for one patient. We can see this patient was treated with 9 drugs for different diseases/symptoms.
Table 1.
Sample indication data for one patient
| Primary_id | Case_id | Drug_seq | Drug | Indication |
|---|---|---|---|---|
| 131970402 | 13197040 | 1 | Trifluridine | Adenocarcinoma of colon |
| 131970402 | 13197040 | 2 | Irinotecan | Adenocarcinoma of colon |
| 131970402 | 13197040 | 3 | Bevacizumab | Adenocarcinoma of colon |
| 131970402 | 13197040 | 4 | Fentanyl | Back pain |
| 131970402 | 13197040 | 5 | Acetaminophen | Back pain |
| 131970402 | 13197040 | 6 | Ondansetron hydrochloride | Prophylaxis of nausea and vomiting |
| 131970402 | 13197040 | 7 | Levothyroxine sodium | Hypothyroidism |
| 131970402 | 13197040 | 8 | Rivaroxaban | Deep vein thrombosis |
| 131970402 | 13197040 | 9 | Dexamethasone | Prophylaxis of nausea and vomiting |
Note: Primary_id is used to link other data in FAERS. Case_id indicates patient.
Indications in FAERS are represented as Medical Dictionary for Regulatory Activities (MedDRA) terms.18 In order to facilitate downstream analysis, we mapped indication terms into Unified Medical Language System (UMLS)19 using MetaMap (2016 V2 release).20 Considering these indications include not only diseases, but also treatment procedures, etc., we constrained the mapping to 12 semantic types that are categorized as disorders in UMLS, including Acquired Abnormality, Anatomical Abnormality, Cell or Molecular Dysfunction, Congenital Abnormality, Disease or Syndrome, Experimental Model of Disease, Finding, Injury or Poisoning, Mental or Behavioral Dysfunction, Neoplastic Process, Pathologic Function, and Sign or Symptom. Total 12 225 of 15 721 (77.76%) were mapped. The clean data set contains 6211 disorders and 5 784 501 case reports.
We then summarized the data on patient level, that is, each row represents co-occurring disorders in one patient. For example, the patient in Table 1 has multiple diseases, including adenocarcinoma of colon, back pain, prophylaxis of nausea and vomiting, hypothyroidism, and deep vein thrombosis, which will be constructed as one record in our data set.
Disease comorbidity pattern calculation
We applied Frequent Pattern-growth (FP-growth) algorithm (implemented in Weka)21,22 into this data to obtain disease co-occurrence patterns. FP-growth is a widely used association rule mining algorithm and the choice of support, and lift is a tradeoff between precision and recall. We experimented with different combinations of support and lift to evaluate the performance of comorbidity mining using manually curated disease comorbidities related to obesity, multiple sclerosis, and psoriasis. After experimentations, we used support >12 and lift >1 and generated 20 101 rules, which are lists of patterns between two sets of diseases, represented in the form , for example, {}.
Construct disease comorbidity network
We constructed an undirected and unweighted DCN based on these rules. Nodes in DCN included all diseases in the rules and edges were established between each pair of diseases in both sides. The DCN contains 1538 diseases and 21 321 edges.
Evaluation of performance for AD comorbidity
We considered neighbor nodes of AD as its comorbidities and obtained subcomorbidity network for AD. To test the performance of DCN, we manually curated comorbidities of AD from literature, then compared with comorbidities from DCN. Precision and recall were computed correspondingly.
Construction of a heterogeneous network by integration of disease comorbidity network and protein–protein interaction network
DCN was integrated with PPI by disease–gene association network from OMIM. Diseases in both DCN and OMIM were mapped to UMLS to enable the connection.
Prioritization of candidate genes for AD
We used random walk with restart to prioritize the AD candidate gene. We used AD as the seed and prioritized genes according to their scores, which represented the probability that each gene can be reached from the seed at steady state. Assuming is a seed vector, the updated score vector at step is defined:
| (1) |
where γ is the probability that the random walker restarts from the seeds at each step, and M is the transition matrix of the entire heterogeneous network, which contains two intranetwork transition matrices on the diagonal and two internetwork transition matrices on the off-diagonal defined below:
| (2) |
where and represent DCN and the genetic network, respectively. The value of γ was set to 0.5 according to de novo prediction result below and loop stopped when , indicating probability vector is stable.23
Evaluation of predicted genes for AD
To evaluate our methods, we obtained a validation gene set from AlzGene database. Currently, there are 679 genes in this database, which represented the largest AD risk gene set. We performed de novo prediction to test how well our approach ranks these genes. Specifically, we removed all edges between AD and its associated OMIM genes. Then, we used random walk with restart to prioritize the AD risk genes in gene network. We evaluated the performance of our algorithm from two aspects.
First, we split the whole ranked gene list into 36 bins with size of 500 genes and investigated the distribution of validation genes in each bin. We then calculated the fold enrichment of validation genes in the top 500 ranked genes. In order to calculate the statistical significance of enrichment, we randomized all 17 860 genes for 1000 times to generate random rankings. We then counted the number of AD risk genes in top 500 genes in each randomization to generate the background distribution. The P-value and fold enrichment of our ranking were calculated based on this distribution.
Second, we used different rank percentiles as thresholds to compute a receiver operating characteristic curve (ROC curve) and precision-recall curve. Given a percentile, for example 5%, we considered all genes that rank in top 5% are positive prediction (AD risk genes, denoted as ADgenes) and the other 95% genes are negative prediction (none-AD risk genes, denoted as nADgenes). , , , and were defined as following formulas, where AlzGene/nAlzGene are denoted as genes in/not in AlzGene database separately.
| (3) |
| (4) |
| (5) |
| (6) |
Once these values were calculated in each threshold, precision, recall, specificity, and sensitivity were computed following the standard definitions24 and ROC and precision-recall curve can be derived.
Comparison of DCN with randomized disease network
To further test the usefulness of DCN, we compared the performance of DCN in predicting AD risk genes with that of randomized disease network. To generate such networks, we kept all disease nodes and total number of edges unchanged but edges were randomly assigned between 2 nodes. We generated 1000 such networks. Then each network was integrated with protein–protein network, and random walk with restart was used to prioritize AD risk genes. We used 679 genes from AlzGene database as validation gene set to compute the Area Under the ROC curve (AUC). P-value of the AUC from real DCN was computed based on normal distribution of AUCs from 1000 randomized networks.
Functional analysis of candidate genes for AD
We used clusterProfiler (Version 3.4.4) (R package)25 to perform gene ontology analysis and gene set enrichment analysis to understand the functions of novel candidate genes we obtained from our methods.
RESULTS
Disease comorbidity network capture known comorbidities of Alzheimer’s disease
We extracted 20 101 comorbidity association rules from the indication data of FAERS across thirteen years. The comorbidity network based on these rules contains 1538 nodes and 21 312 edges. To obtain subcomorbidity network for AD, we considered all its neighbor nodes as comorbidities of AD. Figure 2A shows the extracted comorbidity network of AD. Total 98 comorbidities were found in our network, including five psychiatric disorders such as depression, anxiety disorder etc., and many nonpsychiatric disorders, such as hypertension, diabetes mellitus, type 2 etc.
Figure 2.

Comorbidity network of Alzheimer’s disease. (A) Diseases are represented as nodes and the size of each node is proportional to its degree. Node color represents disorder class (SOC in MedDRA) to which it belongs (yellow nodes indicate psychiatric disorders). Edges between nodes are represented as the co-occurrence of diseases. (B) Precision and recall for AD comorbidities from DCN.
To test the performance of our network, we compared comorbidities of AD from DCN with known comorbidities of AD from literature. Comorbidities of AD include psychiatric disorder such as depression, sleep disorder, bipolar disorder, and nonpsychiatric disorders, such as cardiovascular diseases (ischemia damage, hypertension, etc.), diabetes mellitus (type 2), hypercholesterolemia, hyperlipidemia, arthrosis, thyroid disease, osteoporosis, and glaucoma.26,27 Based on these reports, the precision and recall of AD comorbidities from our network are 66.3% and 91.7% separately. Considering some unknown comorbidities have not been identified, this result indicates that our network has good performance in capture disease comorbidities for AD.
DCN-based network rank algorithm prioritizes known AD associated genes
We used 679 AD associated genes from AlzGene database as validation gene set to evaluate our approach. All connections between AD and its associated genes reported in OMIM were removed and we used AD as the seed to prioritize all genes using random walk with restart. We’d like to emphasize that this de novo prediction high-lighten the contribution of DCN in disease gene discovery for AD. The top 500 genes in the ranking contain 93 validation genes, which is 5.53 folds enrichment comparing with random ranking () (Figure 3A). We also used ranking percentiles as threshold to compute the ROC (Figure 3B) and precision-recall curve (Figure 3C). Our approach achieved AUC of 0.770 and top-ranked genes showed high precision.
Figure 3.
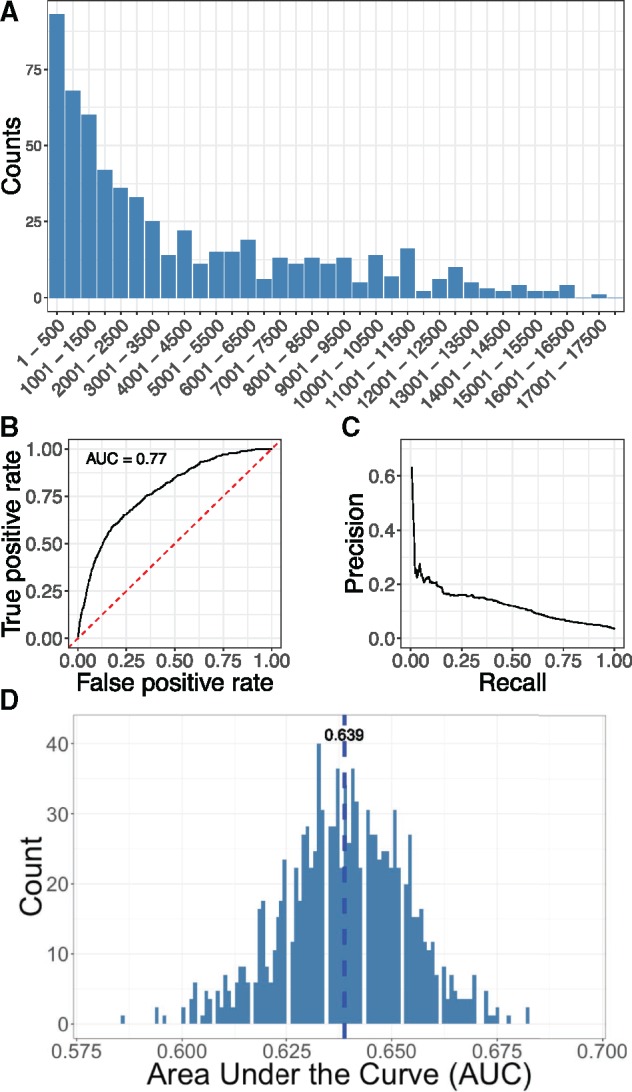
Evaluation of DCN-based AD risk gene prediction. (A) Distribution of validation gene set from AlzGene database in gene ranking. (B) ROC curve for de novo prediction of AD risk genes. (C) Precision-recall curve for de novo prediction of AD risk genes. (D) Distribution of AUCs generated from 1000 randomized disease networks.
To further demonstrate the usefulness of DCN, we generated 1000 randomized disease networks and used them to rank AD risk genes. Distribution of AUCs computed from these networks shows normal distribution with mean of 0.639 and variance of 0.0146 (Figure 3D). AUC (0.770) obtained from real DCN is significantly better than that from randomized networks ().
DCN-based network rank algorithm prioritizes new AD risk candidate genes
We used AD and AD associated genes reported in OMIM as seeds to rank new AD associated genes. Table 2 lists the top 20 ranked genes (see Supplementary Material for full ranked gene list).
Table 2.
Top 20 ranked new AD risk genes
| Rank | Gene_symbol | Gene_name | Location | Type |
|---|---|---|---|---|
| 1 | UBCa | Ubiquitin C | Cytoplasm | Enzyme |
| 2 | NOTCH1a | Notch 1 | Plasma Membrane | Transcription regulator |
| 3 | EGFRa | Epidermal growth factor receptor | Plasma Membrane | Kinase |
| 4 | ALB | Albumin | Extracellular Space | Transporter |
| 5 | APLP2a | Amyloid beta precursor like protein 2 | Cytoplasm | Other |
| 6 | APLP1a | Amyloid beta precursor like protein 1 | Extracellular Space | Other |
| 7 | CPa | Ceruloplasmin | Extracellular Space | Enzyme |
| 8 | PRDM10a | PR/SET domain 10 | Nucleus | Transcription regulator |
| 9 | APBA2a | Amyloid beta precursor protein binding family A member 2 | Cytoplasm | Transporter |
| 10 | NAE1a | NEDD8 activating enzyme E1 subunit 1 | Cytoplasm | Enzyme |
| 11 | NCSTN | Nicastrin | Plasma Membrane | Peptidase |
| 12 | SHC1a | SHC adaptor protein 1 | Cytoplasm | OTHER |
| 13 | KAT5a | Lysine acetyltransferase 5 | Nucleus | Transcription regulator |
| 14 | TSPOa | Translocator protein | Cytoplasm | Transmembrane receptor |
| 15 | BACE1 | Beta-secretase 1 | Cytoplasm | Peptidase |
| 16 | APBA3a | Amyloid beta precursor protein binding family A member 3 | Cytoplasm | Transporter |
| 17 | BLMH | Bleomycin hydrolase | Cytoplasm | Peptidase |
| 18 | GEN1a | GEN1, Holliday junction 5′ flap endonuclease | Cytoplasm | Enzyme |
| 19 | APBA1 | Amyloid beta precursor protein binding family A member 1 | Cytoplasm | Transporter |
| 20 | TP53 | Tumor protein p53 | Nucleus | Transcription regulator |
New AD risk genes that are not included in AlzGene database.
We can see 14 genes that are not included in AlzGene database have high rankings, such as UBC, PRDM10, EGFR, NOTCH1, APLP1, and APLP2 etc. The roles of most of these genes in the AD pathogenesis have been implicated or supported by recent studies. For instance, UBC is a major ubiquitin protein and it is reported that ubiquitin-proteasome system is impaired in AD patients28; Notch1 activity is significantly altered in the brain of AD patients29; EGFR gene plays a central role in neurometabolic aging and associates with AD.30,31 Hence, these highly ranked genes provide a start point for further experimental investigation of their roles in AD pathogenesis.
Pathway analysis of top-ranked novel AD candidate genes
To further investigate the function of the top-ranked AD risk genes, we performed gene ontology analysis using these genes. Figure 4A lists the top 10 enriched GO biological process terms.32 AD is characterized by disruption of calcium homeostasis, mitochondrial oxidative stress, impaired energy metabolism and abnormal glucose regulation, and ultimately neuronal cell death.33 Expectedly, several biological processes, such as cellular response to oxidative stress and neuron death are enriched in our analysis. Interestingly, we found a new pathway, ERBB signaling pathway, is also significantly enriched in our analysis. Indeed, Mei et al. reported that ERBB signaling pathway is involved in nervous system development and disruption of ERBB is associated with nervous disorders.34
Figure 4.
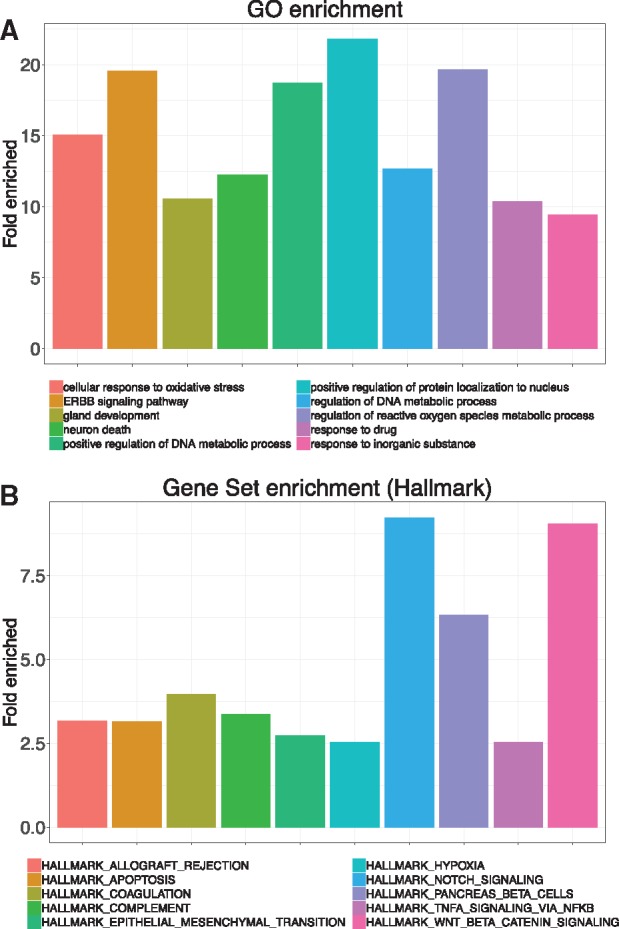
Functional analysis of top-ranked AD risk genes. (A) Top ten enriched biological process terms of gene ontology. (B) Top ten enriched Hallmark pathways of MSigDB using gene set enrichment.
We also performed gene set enrichment using Molecular Signatures Database (MSigDB) Hallmark pathways. MSigDB is a collection of annotated gene sets widely used in gene set enrichment analysis.35 There are 8 major gene set collections in MSigDB, and we used Hallmark gene set since it reduces noise and redundancy and provides a better delineated biological process.35Figure 4B lists the top 10 enriched Hallmark pathways. APOPTOSIS, NOTCH, TNFA, and HYPOXIA are well defined AD pathways.36–39 WNT, a recently identified AD pathway,40 is also ranked high in our analysis. Interestingly, we found that coagulation pathway is also significantly enriched (fold enrich = 3.97, P = .0002). A recent report detected the interactions of β-amyloid peptide with fibrinogen and coagulation factor XII,41 which provides preliminary evidence that coagulation system might be involved in AD pathogenesis.
CONCLUSIONS AND DISCUSSION
Alzheimer’s disease is complicated disease and its etiology is still not elucidated. Traditional in vitro- and in vivo-based experimental methods will continue to discover disease mechanisms, we propose a new framework to prioritize the AD risk genes by integration of DCN with PPI. We demonstrated that this framework can efficiently prioritize known AD risk genes, suggesting that the usefulness of our network in AD disease genetic analysis. We also predicted novel AD risk genes and pathways that have preliminary literature support. Further intensive experiment-based evidence needs to be performed to confirm our findings.
FAERS data have been considered as a largely uncurated and unstandardized database. A recent study reported that average 16 different names were given for each active drug ingredient and FAERS is biased towards serious or life-threatening outcomes.42 The data redundancy and bias may lead to wrong interpretation for drug-adverse event association.43 However, these problems don’t affect the investigation of disease co-occurrence pattern from indication data since we only focus on the co-occurring diseases in individual patients, which is reported as standard MedDRA terms.
One variability of DCN that is constructed using association rule mining is that we need to assign thresholds for support and lift. High thresholds will only identify very common comorbidities, which lead to poor recall for specific disease. On the contrary, low thresholds will identify very rare co-occurring diseases, which may not be real comorbidity disease and lead to poor precision. Therefore, these two values need to be carefully tuned to achieve a balance of precision and recall. However, two reasons make the evaluation difficult. One is that no comprehensive gold standard database for disease comorbidity is available. Another is that disease comorbidity is a dynamic concept that number of disease comorbidities for a specific disease changes over time. In this study, we manually curated disease comorbidities from literature or disease organizations for several diseases, including obesity, multiple sclerosis, and psoriasis. Then we used them as criteria to optimize the thresholds. Though it is not comprehensive, it is demonstrated that optimized DCN has good performance in terms of AD comorbidity as well as its risk gene discovery.
Systems approaches to study disease phenotypes can facilitate disease mechanism understanding. We in this study demonstrated that disease-comorbidity relationships mined from FAERS have potential in AD genetics prediction. In our future studies, we will integrate disease-comorbidity associations mined from FAERS with other disease phenotypic relationships (eg disease-manifestation) from other data resources (eg UMLS, biomedical literature), disease genetics and PPI for AD genetic discovery. We have recently used disease-manifestation relationships extracted from UMLS to construct a DMN network and have developed a combined phenome and genome-driven network approach for disease genetics prediction.44 We previously developed novel natural language processing techniques to extract large number of disease-phenotypic relationships from over 21 million published biomedical literature records and demonstrated the high potential of integrating the high-level disease-phenotypic relationships with lower-level genetic and genomic data in both disease genetics understanding and drug discovery.45–48
Modeling heterogeneous and complex relationships among tens of thousands biomedical entities extracted from different data resources (eg FAERS, biomedical literature) is a challenging task. Recently, we developed a novel a context-sensitive network (CSN) approach to model the complex, heterogeneous, and context-specific interactions among tens of thousands of biomedical entities, including diseases, disease phenotypes, drugs, drug phenotypes, and genes.49 Compared to existing biomedical networks where the relationships among entities are often modeled by pairwise similarity (similarity-based network or SBN), CSNs preserve the context information on how biomedical entities are connected. Our recent study showed that CSN-based approach for disease genetics prediction had significantly better performance than SBN-based approach.49 In future studies, we will use the CSN framework to model the context-specific (eg comorbidity, manifestation, risk/causal) relationships among diseases and other biomedical entities and integrate disease phenotypes with disease genetics and genomics data for disease genetics prediction and drug discovery.
Large-scale disease comorbidity relationships offer unique opportunities to understand shared genetic mechanisms underlying a disease and its comorbidities, for example, AD and its associated neuropsychiatric symptoms (eg anxiety, depression), AD, and type 2 diabetes. By integrating disease comorbidities and vast amounts of genetics, genomic and pathway data, we can understand how disease comorbidity occur, for example by directly sharing common disease genes or indirectly coregulated by high-level biological mechanisms such as cellular pathways.50
In summary, we demonstrated that we innovatively leveraged FAERS, a comprehensive data resource for FDA postmarket drug safety surveillance, for large-scale AD comorbidity mining. This early stage exploratory study demonstrated the potential of disease-comorbidities mining from FAERS in AD genetics discovery.
Data availability
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.3p9b4c2.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
Contributors
RX conceived the study. CZ performed the experiments and wrote the manuscript. Both authors have participated in study discussion and manuscript preparation. All authors read and approved the final manuscript.
Funding
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under the NIH Director’s New Innovator Award number DP2HD084068 (Xu), NIH National Institute of Aging (1 R01 AG057557-01, Xu), NIH National Institute of Aging (1 R01 AG061388-01, Xu), NIH National Institute of Aging (1 R56 AG062272-01, Xu), American Cancer Society Research Scholar Grant (RSG-16-049-01 - MPC, Xu), NIH Clinical and Translational Science Collaborative of Cleveland (1UL1TR002548-01, Konstan).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1. Alzheimer’s Association. 2016Alzheimer’s disease facts and figures. Alzheimers Dement 2016; 124: 459–509. [DOI] [PubMed] [Google Scholar]
- 2. Zhu C, Wu C, Aronow BJ, et al. Computational approaches for human disease gene prediction and ranking. Adv Exp Med Biol 2014; 799: 69–84. [DOI] [PubMed] [Google Scholar]
- 3. Opap K, Mulder N.. Recent advances in predicting gene-disease associations. F1000Res 2017; 6: 578.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen Y, Xu R.. Phenome-based gene discovery provides information about Parkinson’s disease drug targets. BMC Genomics 2016; 3117 Suppl 5: 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bagley SC, Sirota M, Chen R, et al. Constraints on biological mechanism from disease comorbidity using electronic medical records and database of genetic variants. PLoS Comput Biol 2016; 26124: e1004885.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rzhetsky A, Wajngurt D, Park N, et al. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA 2007; 10428: 11694–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park J, Lee DS, Christakis NA, Barabasi AL.. The impact of cellular networks on disease comorbidity. Mol Syst Biol 2009; 5: 262.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hidalgo CA, Blumm N, Barabási AL, et al. A dynamic network approach for the study of human phenotypes. PLoS Comput Biol 2009; 54: e1000353.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roque FS, Jensen PB, Schmock H, et al. Using electronic patient records to discover disease correlations and stratify patient cohorts. PLoS Comput Biol 2011; 78: e1002141.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jensen AB, Moseley PL, Oprea TI, et al. Temporal disease trajectories condensed from population-wide registry data covering 6.2 million patients. Nat Commun 2014; 5: 4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.FAERS: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm345338.htm. Accessed March 28, 2018.
- 12. Agniel D, Kohane IS, Weber GM.. Biases in electronic health record data due to processes within the healthcare system: retrospective observational study. BMJ 2018; 30361: k1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phelan M, Bhavsar NA, Goldstein BA.. Illustrating informed presence bias in electronic health records data: how patient interactions with a health system can impact inference. EGEMS 2017; 651: 22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van den Akker M, Buntinx F, Metsemakers JF, et al. Multimorbidity in general practice: prevalence, incidence, and determinants of co-occurring chronic and recurrent diseases. J Clin Epidemiol 1998; 515: 367–75. [DOI] [PubMed] [Google Scholar]
- 15. Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine. Baltimore, MD: Johns Hopkins University; 2018. https://omim.org/. [Google Scholar]
- 16. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43 (Database issue): D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007; 391: 17–23. [DOI] [PubMed] [Google Scholar]
- 18.MedDRA. https://www.meddra.org. Accessed September 22, 2017.
- 19.UMLS. https://www.nlm.nih.gov/research/umls.
- 20. MetaMap 2016v2 release. https://metamap.nlm.nih.gov. Accessed February 6, 2018.
- 21. Frank E, Hall MA, Witten IH.. The WEKA Workbench. Online Appendix for Data Mining: Practical Machine Learning Tools and Techniques. 4th ed. San Francisco, CA: Morgan Kaufmann; 2016. [Google Scholar]
- 22. Han J, Pei J, Yin Y. Mining frequent patterns without candidate generation. In: Proceedings of the 2000 ACM-SIGMID International Conference on Management of Data; New York, NY: ACM; 2000.
- 23. Köhler S, Bauer S, Horn D, et al. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008; 824: 949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis J, Goadrich M. The relationship between precision-recall and ROC curves In: ICML ’06 Proceedings of the 23rd international conference on Machine learning, June 25–29, 2006. Pittsburgh, PA: ACM; 2006: 233–240.
- 25. Yu G, Wang L, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012; 165: 284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcez ML, Falchetti AC, Mina F, et al. Alzheimer’s disease associated with psychiatric comorbidities. An Acad Bras Cienc 2015; 87 (2 Suppl): 1461–73. [DOI] [PubMed] [Google Scholar]
- 27. Duthie A, Chew D, Soiza RL.. Non-psychiatric comorbidity associated with Alzheimer’s disease. QJM 2011; 10411: 913–20. [DOI] [PubMed] [Google Scholar]
- 28. Tramutola A, Di Domenico F, Barone E, et al. It is all about (U)biquitin: role of altered ubiquitin-proteasome system and UCHL1 in Alzheimer disease. Oxid Med Cell Longev 2016; 2016: 1. doi: 10.1155/2016/2756068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Auber LA, Brai E, Marathe S, et al. Notch, a clinically relevant signaling pathway for Alzheimer’s Disease. Alzheimers Dement 2017; 137: 997. [Google Scholar]
- 30. Siddiqui S, Fang M, Ni B, et al. Central role of the EGF receptor in neurometabolic aging. Int J Endocrinol 2012; 2012: 739428.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen X, Wang C, Zhou S, et al. The impact of EGFR gene polymorphisms on the risk of Alzheimer’s disease in a Chinese Han population: a case-controlled study. Med Sci Monit 2018; 2024: 5035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gene ontology. http://www.geneontology.org. Accessed September 15, 2018.
- 33. Godoy JA, Rios JA, Zolezzi JM, et al. Signaling pathway cross talk in Alzheimer’s disease. Cell Commun Signal 2014; 2812: 23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mei L, Nave KA.. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014; 2831: 27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 2510243: 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Obulesu M, Lakshmi MJ.. Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 2014; 3912: 2301–12. [DOI] [PubMed] [Google Scholar]
- 37. Woo HN, Park JS, Gwon AR, et al. Alzheimer’s disease and Notch signaling. Biochem Biophys Res Commun 2009; 253904: 1093–7. [DOI] [PubMed] [Google Scholar]
- 38. Liu H, Qiu H, Xiao Q, et al. Chronic hypoxia-induced autophagy aggravates the neuropathology of Alzheimer’s disease through AMPK-mTOR signaling in the APPSwe/PS1dE9 mouse model. JAD 2015; 484: 1019–32. [DOI] [PubMed] [Google Scholar]
- 39. Cheng X, Shen Y, Li R.. Targeting TNF: a therapeutic strategy for Alzheimer’s disease. Drug Discov Today 2014; 1911: 1822–7. [DOI] [PubMed] [Google Scholar]
- 40. Inestrosa NC, Varela-Nallar L.. Wnt signaling in the nervous system and in Alzheimer’s disease. J Mol Cell Biol 2014; 61: 64–74. [DOI] [PubMed] [Google Scholar]
- 41. Ahn HJ, Chen ZL, Zamolodchikov D, et al. Interactions of β-amyloid peptide with fibrinogen and coagulation factor XII may contribute to Alzheimer’s disease. Curr Opin Hematol 2017; 245: 427–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maciejewski M, Lounkine E, Whitebread S, et al. Reverse translation of adverse event reports paves the way for de-risking preclinical off-targets. Elife Aug 8, 2017; doi: 10.7554/eLife.25818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McAdams M, Staffa J, Dal Pan G.. Estimating the extent of reporting to FDA: a case study of statin-associated rhabdomyolysis. Pharmacoepidem Drug Safe 2008; 173: 229–39. [DOI] [PubMed] [Google Scholar]
- 44. Chen Y, Zhang X, Zhang GQ, Xu R.. Comparative analysis of a novel disease phenotype network based on clinical manifestations. J Biomed Informatics 2015; 53: 113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu R, Li L, Wang Q.. Towards building a disease-phenotype knowledge base: extracting disease-manifestation relationship from literature. Bioinformatics 2013; 2917: 2186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu R, Li L, Wang Q.. dRiskKB: a large-scale disease-disease risk relationship knowledge base constructed from biomedical text. BMC Bioinformatics 2014; 151: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu R, Wang Q.. PhenoPredict: a disease phenome-wide drug repositioning approach towards schizophrenia drug discovery. J Biomed Informatics 2015; 56: 348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q, Xu R. Disease comorbidity-guided drug repositioning: a case study in schizophrenia. In: The 2018 Annual American Medical Informatics Association Symposium, Nov 2018. San Francisco, CA: AMIA. [PMC free article] [PubMed]
- 49. Chen Y, Xu R.. Context-sensitive network-based disease genetics prediction and its implications in drug discovery. Bioinformatics 2017; 1337: 1031–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ko Y, Cho M, Lee JS, Kim J.. Identification of disease comorbidity through hidden molecular mechanisms. Sci Rep 2016; 196: 39433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.3p9b4c2.