

Abstract
Synthetic high density lipoprotein nanoparticles (sHDL) capable of mobilizing excess cholesterol from atherosclerotic arteries and delivering it to the liver for elimination have been shown to reduce plaque burden in patients. Unfortunately, sHDLs have a narrow therapeutic index and relative to the endogenous HDL shorter circulation half-life. Surface modification with polyethylene glycol (PEG) was investigated for its potential to extend sHDL circulation in vivo. Various amounts (2.5, 5 and 10%) and different chain lengths (2 and 5 kDa) of PEG-modified lipids were incorporated in sHDL’s lipid membrane. Incorporating PEG did not reduce the ability of sHDL to facilitate cholesterol efflux, nor did it inhibit cholesterol uptake by the liver cells. By either adding more PEG or using PEG of longer chain lengths the circulation half-life was extended. Addition of PEG also increased the area under the curve for the phospholipid component of sHDL (p < 0.05), but not for the apolipoprotein A-I peptide component of sHDL — suggesting sHDL is remodeled by endogenous lipoproteins in vivo. The extended phospholipid circulation led to a higher mobilization of plasma free cholesterol, a biomarker for facilitation of reverse cholesterol transport. The area under the cholesterol mobilization increased about 2 to 4-fold (p < 0.05), with greater increases observed for longer PEG chains and higher molar percentages of incorporated pegylated lipids. Mobilized cholesterol was associated primarily with the HDL fraction, led to a transient increases in VLDL cholesterol, and returned to baseline 24 h post-dose. Overall, pegylation of sHDL leads to beneficial changes in sHDL particle pharmacokinetic and pharmacodynamic behaviors.
Keywords: Apolipoproteins, Cholesterol, HDL, Lipoproteins/Metabolism, Pharmacokinetics, Apolipoprotein A-I mimetic peptides, Lipoprotein remodeling, PK-PD modeling
Graphical Abstract
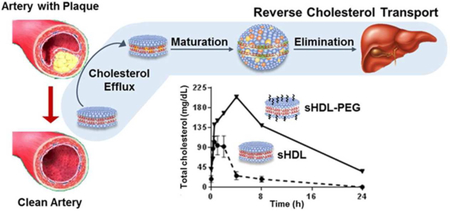
1. Introduction
Cardiovascular disease (CVD) is the leading cause of death in the United States, responsible for 17% of national health expenditures1. High-density lipoprotein cholesterol (HDL-C) levels are known to inversely correlate with the risk of CVD2. HDL is natural nanoparticle composed primarily of apolipoprotein A-I (ApoA-I) and lipids. One of the key functions of HDL is to maintain cholesterol homeostasis through reverse cholesterol transport (RCT), whereby HDL effluxes free cholesterol from peripheral tissues through ATP-binding cassette transporters (ABCA1 and ABCG1) and then delivers it to liver for excretion by the scavenger receptor B type I (SR-BI) receptor3. Thus, the idea of administrating synthetic HDL (sHDL) to facilitate RCT and promote the regression of atherosclerotic plaque has gained significant interest4-7. Several sHDL therapeutic products have been developed that consist of either ApoA-I protein purified from human plasma, recombinant ApoA-I, or ApoA-I mimetic peptides8-9. The protein component is usually combined with phospholipids to form sHDL nanoparticles, which are administered intravenously following a cardiovascular event. It was hypothesized that the acute administration of several sHDL infusions could lead to a rapid reduction in plaque volume and prevent the likelihood of subsequent cardiovascular events7, 10. A number of sHDL products have been tested in Phase I and II clinical trials and were able to increase HDL-C levels, reduce inflammation and decrease atherosclerotic plaque volume9.
Despite initial clinical success, the translation of sHDL nanomedicine into an FDA approved therapy has been difficult. This class of drugs has a unique pharmacokinetic-pharmacodynamic relationship. Administration of sHDL causes rapid mobilization of cholesterol, which is subsequently delivered to the liver for elimination11. The amount of mobilized cholesterol is often several folds higher than pre-dose plasma cholesterol levels, leading to saturation of hepatic receptors and pathways for cholesterol metabolism12. Thus, administration of sHDL often results in drug-related toxicity exhibited by transient elevation of liver transaminases (ALT and AST), triglycerides, low and very low-density lipoprotein cholesterol levels (LDL-C and VLDL-C). In addition, the half-life of endogenous ApoA-I protein in humans is only 3.3 days13. It is believed that the protein participates in multiple cycles of RCT by forming HDL, effluxing cholesterol, delivering it to the liver, and returning to the peripheral tissues to efflux more cholesterol11. In contrast, administration of sHDL results in a protein or peptide half-life of only 8-12 hours14-15.
Due to the rapid elimination of sHDL, the duration of time following administration when sHDL plasma levels remain above the level required to obtain therapeutic effect is quite short. This rapid elimination necessitates either frequent dosing, which is inconvenient for patients especially for an intravenous product, or administration of relatively high doses, which can lead to off-target toxicity. In addition, following administration in vivo, sHDL nanoparticles rapidly interact with endogenous lipoproteins by exchanging lipid and protein components. This is a natural phenomenon in lipoprotein metabolism, known as remodeling, that leads to the disassembly of administered sHDL particles, and results in differences in elimination kinetics of ApoA-I and lipid components of sHDL16. This remodeling, in turn, affects both the efficacy and safety of administered sHDL. The main hypothesis of this study is that by modifying the sHDL surface with polyethylene glycol, it may be possible to slow down sHDL remodeling by endogenous lipoproteins, thereby reducing sHDL nanoparticle elimination by reticuloendothelial system and increasing its circulation half-life in vivo.
Polyethylene glycol (PEG) has been widely used to coat or pegylate nanoparticle surfaces to archive “stealth-like” characteristics17. Pegylated drug delivery systems have been shown to be more physically stable in vitro and have longer circulation half-lives in vivo, resulting from the ability of PEG to escape uptake by the reticuloendothelial system18. Pegylation has been shown to be clinically safe and currently used in several products approved by the Food and Drug Administration19. It has been reported that by conjugating PEG to ApoA-I protein, protein in vivo circulation was prolonged and anti-inflammatory activity in a murine model of atherosclerosis was improved20. The PEG density on the nanoparticle surfaces, PEG molecular weight, and branch structure are all known to affect nanoparticle circulation and efficacy21, but this has not been systematically investigated for sHDL. In this study, we incorporated PEG-modified phospholipids in sHDL nanoparticles and examined how the degree of surface modification and PEG chain length affected sHDL pharmacokinetics and pharmacodynamics in vivo. We investigated whether the addition of PEG would increase in vivo stability of sHDL and increase sHDL circulation time without compromising the nanoparticle’s ability to facilitate RCT.
To test our hypothesis, we decided to pegylate the surface of a model peptide-based sHDL, ETC-642. ETC-642 was previously examined in single- and multiple-dose clinical studies in dyslipidemic patients15, 22. It was shown that infusion of ETC-642 increased HDL-C levels at doses as low as 3 mg/kg and was well tolerated by patients at doses up to 30 mg/kg22. However, the plasma circulation half-life of 22A-peptide in humans was only ~12 h, which is significantly shorter than the reported half-life of endogenous ApoA-I protein, 68 h23-24. In this study, the abilities of pegylated sHDL to promote cellular cholesterol efflux and facilitate cholesterol uptake in vitro were examined. The remodeling of pegylated sHDL by endogenous lipoproteins was also examined in vitro, following incubation with human serum, as well as in vivo following administration to Sprague-Dawley (SD) rats. The pharmacokinetic properties of both unmodified and pegylated sHDL nanoparticles in addition to their abilities to mobilize, esterify and eliminate cholesterol were also examined in SD rats. Because of the enhanced cholesterol mobilization caused by prolonged circulation of sHDL, improvements in sHDL pharmacokinetic parameters may be translated to reduction of dose and frequency of sHDL dosing and, thus, improvement of safety profile for this drug class.
2. Experimental Section
2.1. Materials
ApoA-I mimetic peptides 22A (PVLDLFRELLNELLEALKQKLK) and 5A (DWLKAFYDKVAEKLKEAFPDWAKAAYDKAAEKAKEAA) were synthesized by Genscript (Piscataway, NJ). The peptide purities were determined to be over 95% by reserve phase HPLC. 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), N-(Carbonyl-methoxypolyethyleneglycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG-2k) and N-(Carbonyl-methoxypolyethyleneglycol 5000) −1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG-5k) were purchased from NOF Corporation (White Plains, NY). DSPE-PEG-FITC amd Bodipy-labeled cholesterol were purchased from Avanti Polar Lipids, Inc (Alabaster, AL). All other materials were obtained from commercial sources.
2.2. Preparation of sHDL particles
All investigated particles were prepared using a lyophilization method. Briefly, 22A peptide, DPPC, DSPE-PEG2000 (PEG-2k) or DPSE-PEG5000 (PEG-5k) were dissolved in acetic acid at a molar ratio of 1:7.15 for peptide to total lipids. The molar percentage of DSPE-PEG2000 in total lipids was 0, 2.5%, 5% and 10% for sHDL, sHDL-PEG-2k (2.5%), sHDL-PEG-2k (5%), and sHDL-PEG-2k (10%), respectively. Similarly, HDL-PEG-5k (5%) contained DSPE-PEG5000 at 5% of total lipids. After freeze-drying for over 24 hours, PBS (pH = 7.4) was added to hydrate to a final 22A peptide concentration of 10 mg/mL. The mixture was vortexed briefly, heated to 50°C for 10 min and cooled to 25°C for 10 min, and this cycle repeated three times. sHDL particle concentrations are expressed in terms of 22A peptide concentration.
sHDL and sHDL-PEG particles loaded with bodipy-labeled cholesterol were prepared by a freeze-drying method that dissolved labelled cholesterol in glacial acidic acid together with all other components. The final concentration of bodipy-cholesterol after hydration of lyophilized powder was 30 μg/mL.
2.3. Characterization of HDL and HDL-PEG particles
The purity and particle size of sHDL and sHDL-PEG was analyzed by gel permeation chromatography (GPC), with UV detection wavelength of 220 nm, using a Tosoh TSK gel G3000SWx 7.8mm × 30cm column (Tosoh Bioscience, King of Prussia, PA) on Waters Breeze Dual Pump system. Samples were diluted to 1 mg/ml and 50 μL injection volume was used. The samples were eluted with PBS (pH 7.4). Particle size distribution was also determined by dynamic light scattering (DLS), using a Zetasizer Nano ZSP, Malvern Instruments (Westborough, MA). The samples were diluted to 1 mg/ml prior to analysis.
2.4. Cholesterol efflux evaluation
Baby hamster kidney (BHK)-mock and BHK cells transfected with ABCA1 or SR-BI receptors were grown in Dulbecco’s modified eagle medium with 10% FBS and 1% pen strep glutamine. 105 cells were then seeded into 24-well plates and grown for 24 hours. Cells were washed with PBS (pH 7.4) once at room temperature and labeled with cholesterol for 24 hours in DMEM/BSA/antibiotic media (0.5 mL) containing 1 μCi of [3H] cholesterol/mL of media. Subsequently, labeled cells were washed with PBS (pH 7.4) twice to remove [3H] cholesterol that was not taken up by cells. Tritium-labeled cholesterol was effluxed from cells for 18 hours, using various lipid complex acceptors (5 μM) in duplicate. Finally, all cell media (0.5 mL) was removed into separate Eppendorf tubes and centrifuged at 10,000 rpm for 5 min. Remaining cells were lysed with 0.1%SDS/0.1M NaOH solution for 2 hours. Radioactive counts of media and cell fractions were measured separately, using a Perkin Elmer liquid scintillation counter. Percent cholesterol effluxed from cells was calculated by dividing media counts by the total sum of media and cell counts and then multiplying this number by 100%.
2.5. Liver cell uptake of cholesterol in sHDL
The uptake of bodipy-labeled cholesterol loaded in either sHDL or sHDL-PEG by HepG2 cells was quantified by confocal laser scanning microscopy and flow cytometry. Briefly, liver hepatocellular carcinoma (HepG2) cells were cultured in Dulbecco’s Modified Eagle Medium with 10% FBS and 1% Pen Strep Glutamine and maintained in an incubator at 37°C and 5% CO2. On day 0, 104-105 cells were seeded in a MatTek 35 mm petri dish in DMEM. The next day, media was aspirated and cells were washed with PBS. Fresh media containing sHDL and sHDL-PEG loaded with bodipy-labeled cholesterol was added at a final 22A concentration of 100 μg/ml. After incubation at 37 °C for 2 hours, cells were washed twice with PBS followed by fixation with 4% paraformaldehyde in PBS for 15 min at room temperature. 1% Triton-X solution was then added to the dish for 15 min and washed with PBS twice. Finally, the cell nuclei were stained with DAPI. The nuclei and bodipy fluorescence images were acquired on a Nikon A-1 Spectral Confocal microscope system (Nikon Corporation, Tokyo), with an excitation wavelength of 495 nm for bodipy. Quantification of cellular fluorescent signal was performed using a cell sorter (Beckman Coulter FC500 5-colour analyzer) at an excitation wavelength of 495 nm.
2.6. Particle remodeling in serum
To study the remodeling of sHDL by endogenous lipoproteins, sHDL or sHDL-PEG-2K (5%) particles were incubated with human serum at 1 mg/mL 22A concentration at 37 °C for 1 h with shaking at 300 rpm. To track the exchange of DSPE-PEG 2000 between sHDL and endogenous lipoproteins, sHDL particles were prepared with fluorescent FITC-DSPE-PEG-2K, which has excitation and emission spectrum peak wavelengths of approximately 495 nm and 519 nm respectively. The cholesterol distribution among different lipoproteins in samples after incubation was analyzed using Waters HPLC system equipped with a Superose 6, 10/300 GL column (GE Healthcare, Piscataway, NJ) and on-line detection of cholesterol was achieved via post-column enzymatic reactions25. The post column reaction was done using a 5 mL reaction coil at 37°C by mixing separated lipoprotein and enzymatic reagent whose flow rate was 0.2 mL/min. The UV absorbance of the product was detected at 490 nm. The enzymatic reagent solution was composed of 100 mM phosphate buffer (pH 7.0), 4 M sodium chloride, 0.2% triton X-100, 10 mM sodium cholate, 2.5 mM 4-aminoantipyrine, 7.5 mM 2-hydroxy-3,5 dichlorobenzene, 0.0625 U/ml cholesterol oxidase, 1.25 U/ml peroxidase, 1.25 U/ml lipase and 0.1 U/ml cholesterol ester hydrolase. To examine the distribution of FITC-DSPE-PEG-2K among endogenous lipoproteins, the HPLC fractions of HDL, LDL and VLDL were collected after separation, and fluorescence for each fraction was determined, using a Synergy NEO HTS Multi-Mode Microplate Reader (Bio-Tek).
To determine the effect of sHDL induced remodeling of endogenous HDL subclasses, lipoproteins after incubation were separated by one-dimensional native polyacrylamide gel electrophoresis (PAGE) based on particle size and visualized by western blot for ApoA-I protein. Samples were subject to electrophoresis in 10-well Tris-Borate-EDTA gradient (3-25%) acrylamide native gels (Jule, Inc., Milford, CT). For each line, 5 μl of human serum after incubation with PBS, 22A peptide or 22A-sHDL was mixed with 5 μl of 2X TBE sample buffer and 6 μl of the resulting mixtures were loaded per line. Gels were run at 200 V until the sample dye was 2.5 cm away from the bottom of the gel. Proteins were visualized by western blot by transfer onto PVDF membrane and incubation overnight with anti-human apoA-I-HRP conjugated antibody (Meridian Life Science, Memphis, TN). Bands were visualized using SuperSignal™ West Pico Chemiluminescence Substrate (Thermo Fisher) and images were acquired on a FluorChem M Imager (Protein Simple, San Jose, CA). Image J was used for spot densitometry.
2.7. Pharmacokinetic and pharmacodynamic study in rats
The sHDL and sHDL-PEG formulations were prepared at a final 22A concentration of 20 mg/mL in PBS. Sixteen male Sprague-Dawley rats were randomly assigned to four groups: sHDL, sHDL-PEG-2k (5%), sHDL-PEG-2k (10%) and sHDL-PEG-5k (5%), containing four rats each. All rats were fasted overnight before dosing and received different sHDL formulations at the dose of 50 mg/kg based on 22A peptide concentration via tail vein injection. Blood samples of approximately 0.3 mL were collected from the jugular vein in heparinized BD centrifuge tubes (BD, Franklin Lakes, NJ) at pre-dose and 0.25, 0.5, 1, 2, 4, 8, 24 and 48 hours after dosing. Serum samples were separated immediately by centrifugation at 10,000 rpm for 10 minutes at 4°C and stored at −20°C until further analysis.
2.8. Measurement of 22A peptide serum concentration by LC-MS and pharmacokinetic analysis
To determine 22A peptide concentration in serum, 10 μL of 5A peptide (3 mg/mL) was added to 10 μL of each serum sample as an internal standard (IS). The sample was then mixed with 100 μL of methanol containing 0.1% acetic acid and vortexed for 30 seconds. The mixture was centrifuged at 10,000 rpm for 10 minutes at 4°C and supernatant was collected for LC-MS analysis. Samples were injected into the Agilent 6520 Accurate-Mass Q-TOF LC/MS system equipped with a dual electrospray ionization source (Dual-ESI) (Agilent Technologies, CA). An Agilent 300SB-C18 column (2.1 mm × 50 mm, 3.5 μm) was used for separation and the UV detector was set to a wavelength of 220 nm. The flow rate was 0.3 mL per minute, with the initial mobile phase composed of 90% solvent A (water containing 0.1% v/v formic acid) and 10% solvent B (methanol containing 0.1% v/v formic acid). A linear gradient to 40% solvent A and 60% solvent B at 3.5 min, followed by an additional linear gradient to 5% solvent A and 95% solvent B at 8 min was used. Mass spectra were acquired in negative ion mode with the mass range set at m/z 100-3200. The conditions used for the ESI source included a capillary voltage of 3500 V, a drying gas temperature of 332°C, a drying gas flow of 5 L/min, and a nebulizer pressure of 45 psi as well as a fragmentor voltage of 225 V. MassHunter Workstation software (Agilent Technologies, CA) was used for data acquisition and processing. The extracted ion chromatogram (EIC) of 22A was exported from the total ion chromatogram (TIC) by monitoring the key fragment of 22A at m/z 656.6. Since 22A(−)Lys (21A) is the main metabolite of 22A peptide after administration, we also determined 22A(−)Lys levels. The EIC of 22A(−)Lys metabolite and IS 5A was extracted at m/z 832.5 and m/z 844.4, respectively. The total integral area of 22A peak and 22A(−)Lys metabolite peak was used to calculate concentration of 22A. A pharmacokinetic non-compartmental analysis (NCA) was also performed to derive basic pharmacokinetic parameters of peptide using Phoenix© WinNonlin® Version 7.3 (Pharsight Corporation, Mountain View, CA, USA), including maximum serum concentration (Cmax), area under the serum concentration-time curve (AUC), elimination rate constant (K10), elimination half-life (T1/2), total clearance (CL) and volume of distribution (Vss) for each rat using Phoenix© WinNonlin® Version 6.3 (Pharsight Corporation, Mountain View, CA, USA). Coefficient of variation was calculated for each parameter.
2.9. Quantification of serum phospholipids and cholesterol
The levels of serum phospholipids (PL), total cholesterol (TC), and unesterified or free cholesterol (FC) were determined by enzymatic analysis, using commercially available kits (Wako Chemicals, Richmond, VA). Cholesterol ester levels (CE) were calculated as the difference between TC and FC levels at each time point. Briefly, serum samples were diluted with PBS (pH 7.4) for TC and FC detection, or with Milli Q water for PL detection. Defined amounts of standards or diluted samples were transferred to 96-well plates (50 μL, 60 μL and 20 μL for TC, FC and PL analyses, respectively), and assay reagents were added per manufacturer’s instructions. The plates were gently shaken using an orbital shaker and incubated at 37 °C for 5 min. The UV absorbance at 600 nm was measured by a Synergy NEO HTS Multi-Mode Microplate Reader (Bio-Tek). A pharmacokinetic non-compartmental analysis (NCA) was also performed to derive basic pharmacokinetic parameters of phospholipids using Phoenix© WinNonlin® Version 7.3 (Pharsight Corporation, Mountain View, CA, USA). The pharmacodynamic effect in each mouse was determined as the area under the total effect curve (AUEC) from dosing time point to 48 hours after dosing, which were calculated by trapezoidal rule. Secondary pharmacodynamic endpoints (maximal effect [Emax] and time to Emax [Tmax, E]) were also analyzed to compare pharmacodynamic effects. The coefficient of variation was calculated for each parameter.
2.10. Cholesterol distribution among lipoproteins
To study the effect of sHDL or sHDL-PEG on lipoprotein profiles in vivo, cholesterol distribution between lipoprotein fractions was determined for rat serum collected at various post-dose time intervals. Briefly, serum lipoproteins were separated by size using a Waters HPLC system equipped with a Superose 6, 10/300 GL column (GE Healthcare, Piscataway, NJ) and cholesterol distribution between VLDL, LDL, and HDL lipoprotein fractions was determined by post-column enzymatic reactions. Rat serum prior to dosing and 2, 4, 12 and 24 h post-injection was analyzed. Serum aliquots (50 μl) were injected and eluted with 154 mM sodium chloride/0.02% sodium azide solution at 0.8 ml/min. The post column reaction was used to determine cholesterol concentration as described in section 2.6.
2.11. Statistical Analysis
The significance of difference between data points, calculated Km and Vmax of cholesterol efflux, pharmacokinetic (Cmax, AUC, T1/2, Kl0, CL, Vss) and pharmacodynamic parameters (Emax, AUEC) were determined by one-way ANOVA analysis, with sHDL group as the control. Data are expressed as mean value with CV% of independent experiments. Statistical significance was established at p < 0.05, p < 0.01, p < 0.001 and p < 0.0001.
3. Results
3.1. Preparation and Characterization of PEG Modified sHDL Nanoparticles
The sHDL selected for PEG modification was composed of 22A ApoA-I mimetic peptide and DPPC at 1:2 w/w (1:7.15 mol/mol) ratio of peptide:lipid. This composition was very similar to the composition of ETC-642, a peptide-based sHDL that has been tested in both single- and multiple-dose Phase I clinical trials in dyslipidemic patients15, 22. The molar ratio between peptide and phospholipid components of sHDL dictates both the size and purity of the resulting sHDL. For our study, a 1:2 weight ratio resulted in formation of homogeneous preβ-like HDL nanodiscs with average diameter of 8 - 10 nm26. Therefore, the ratio between peptide and lipid was kept constant while a portion of DPPC was substituted for DPSE-PEG to achieve surface pegylation of sHDL. Two different molecular weights of PEG were used (2 kDa and 5 kDa) and DSPE-PEG were substituted for DPPC at either 2.5, 5 or 10 molar percent. One unmodified and four pegylated sHDL nanoparticle formulations were prepared and their compositions are summarized in Table 1.
Table 1.
The characterization summary of different 22A-sHDL particles
| sHDL Formulations (molar ratio) | RTa (min) | Particle size (nm) | PDIb |
|---|---|---|---|
| 22A: DPPC (1:7.15) | 7.89 | 9.21 ± 0.29 | 0.36 ± 0.09 |
| 22A: DPPC: DSPE-PEG2k (1:6.97:0.18) | 7.78 | 10.44 ± 0.16 | 0.15 ± 0.06 |
| 22A: DPPC: DSPE-PEG2k (1:6.79:0.36) | 7.44 | 11.38 ± 0.22 | 0.24 ± 0.06 |
| 22A: DPPC: DSPE-PEG2k (1:6.43:0.72) | 6.92 | 13.06 ± 0.03 | 0.23 ± 0.06 |
| 22A: DPPC: DSPE-PEG5k (1:6.79:0.36) | 6.45 | 14.91 ± 0.02 | 0.26 ± 0.02 |
RT: retention time
TDI: polydispersity index.
The size distributions of sHDL and sHDL-PEG particles were determined by dynamic light scattering (DLS) (Table 1, Figure 1A). The average diameter of non-pegylated sHDL was 9.2 ± 0.3 nm, similar to other reported sHDL sizes26. The average particle size of sHDL increased gradually with increasing amounts of surface modifications, from 10.4 ± 0.2, 11.4 ± 0.2 to 13.1 ± 0.1 nm with addition of 2.5, 5 and 10% of PEG-2K, respectively. The size further increased to 14.9 ± 0.1 nm upon surface modification with the longer PEG-5K polymer. The homogeneity of particle size distribution was preserved, as evidenced by the low polydispersity index. Further characterization of sHDL nanoparticle size and purity was performed by gel permeation chromatography (GPC) (Figure 1B). In agreement with our DLS findings, the GPC retention time decreased from 7.89 min to 6.45 min with pegylation, indicating the formation of larger particles.
Figure 1.

Particle size distribution of different 22A-sHDL particles determined by dynamic light scattering (Panel A) and gel permeation chromatography (Panel B).
3.2. Impact of PEG modification of sHDL surface on cholesterol efflux capacity
The ability of PEG modified sHDL to facilitate cholesterol efflux was evaluated, using BHK cells stably transfected with either human ABCA1, ABCG1 or SR-BI transporters. Efflux from non-transfected or mock-transfected cells was measured for comparison. The cells were first loaded with 3H- cholesterol and then incubated with sHDL, sHDL-PEG2k (5%), sHDL-PEG2k (10%) or sHDL-PEG5k (5%) for 18 hours at concentrations of 0, 1.25, 2.5, 5, 10 or 20 μM 22A to obtain dose-response curves (Figure 2). The percent of cholesterol effluxed into the medium by sHDL was determined by counting the radioactivity in the medium and combined radioactivity of medium and lysed cells.
Figure 2.

Cholesterol Efflux of cholesterol from BHK cells transfected with ABCA1 (Panel A), ABCG1 (Panel B), SR-BI (Panel C), BHK-mock cell (Panel D) by sHDL particles.
The ABCA1 transporter is one of the major efflux mechanism for removing cholesterol from macrophages in the atheroma, and primarily interacts with the lipid-free and or lipid-poor forms of ApoA-I protein. In contrast, the ABCG1 transporter interacts with larger, more mature HDL to efflux cholesterol. The SR-BI transporter is found in the calveolar regions of macrophage and endothelial cell membranes and is responsible for mediating bi-directional cholesterol efflux and anti-inflammatory signaling induced by HDL. All four formulations showed significant ability to efflux cholesterol passively from the cellular membrane of BHK mock cells (Figure 2A). Induction of ABCA1, ABCG1 and SR-BI transporters slightly increased cholesterol efflux. The amount of effluxed cholesterol reached saturation at 5-10 μM of sHDL in all four cell types and for all sHDL formulations. In summary, pegylation of sHDL does not appear to shield the functional interaction between sHDL and cholesterol transporters.
3.3. Impact sHDL pegylation on cholesterol uptake by liver cells
After cholesterol is effluxed from peripheral tissues by sHDL, it is taken up by hepatocyte receptors, metabolized and excreted into the bile10. To evaluate cholesterol uptake by hepatic cells, sHDL particles were loaded with bodipy-labeled cholesterol and incubated with HepG2 cells. Following a two-hour incubation, cellular uptake of bodipy-cholesterol from both unmodified and pegylated sHDL was visualized by confocal imaging and quantified by flow cytometry (Figure 3). The bottom row shows the channel overlay, with nuclei in blue and bodipy-cholesterol in green. The fluorescence of bodipy appears to distribute throughout cellular membranes and cytosol for all groups. When bodipy uptake by HepG2 cells was quantified by flow cytometry and compared between different formulations, there were no significant differences observed between groups. Thus, HDL nanoparticle pegylation at up to 10% with PEG-2k and up to 5 % with PEG-5K does not prevent cellular uptake of cholesterol by hepatocytes.
Figure 3.

Cell uptake of bodipy-labelled cholesterol by HepG2 cells at 2 hours after incubation of sHDL particles (Panel A). The fluorescent intensity was quantified by flow cytometry (Panel B).
3.4. Effect of sHDL pegylalion on lipoprotein remodeling in vitro
Following in vivo administration, sHDL are known to undergo significant remodeling in blood, a process in which both lipid and protein components of sHDL nanoparticle exchange with lipid and protein components of endogenous lipoproteins27. The rate of this exchange and how composition of sHDL affects this rate is not well characterized. To examine if pegylation of lipid component of sHDL affects particle remodeling, the sHDL nanoparticles were incubated in serum for 1 h at 37°C. By incorporation of fluorescently labelled DSPE-PEG2k-FITC in sHDL and monitoring fluorescence, we confirmed that pegylated lipid eluted with the HDL fraction before and after incubation with serum (Figure 4A). We used post-column reaction to measure levels of cholesterol in VLDL, LDL and HDL fractions before and after serum incubations with various sHDLs (Figure 4B). After one hour incubation with non-pegylated sHDL, VLDL cholesterol levels appear to decrease and cholesterol distributes to smaller HDL particles likely due to partitioning to cholesterol-free sHDL. When sHDL-PEG-2K (5%) were mixed with serum, cholesterol redistribution toward HDL appears to be reduced.
Figure 4.

The cholesterol distribution between VLDL, LDL and HDL lipoprotein fractions before and at 1 hour after incubation of sHDL and FITC labelled sHDL-PEG2k (5%) with human serum at 37 °C (Panel A). The distribution of FITC-labelled PEG2k component in sHDL-PEG2k (5%) among lipoproteins after incubation was shown in Panel B. The ApoA-I distribution between various subclasses of HDL after 1 hr incubation was determined by 1-D native page electrophoresis and visualized by western blot using anti-apoA-I antibody (Panel C). Lane 1 was the control serum, lane 2, 3, 4, 5 ,6 represented free 22A, sHDL, sHDL-PEG 2k (5%), sHDL-PEG2k (10%), sHDL-PEG5k (5%) at peptide concentration of 0.3 mg/ml. Lane 7, 8, 9 represented sHDL, sHDL-PEG 2k (5%), sHDL-PEG2k (10%) at peptide concentration of 1.0 mg/ml.
Potential differences in how serum incubations with pegylated and non-pegylated sHDL affect redistribution of endogenous apoA-I between various HDL sub-classes were also examined by 1-D native page electrophoresis. Different free 22A peptide and various sHDLs formulation were added at either 0, 0.3, and 1.0 mg/ml peptide, incubated for 30 min, separated by 1-D native page electrophoresis and visualized by western blot for human ApoA-I (Figure 4C). Compared to control serum (lane 1), addition of all sHDL formulations resulted in the appearance of a lipid-free band of ApoA-I (30 kDa) and in some instances a small pre-β HDL particle or ApoA-I dimer around 60 kDa. For serum incubations with pegylated HDL, increase of ApoA-I signal in larger α-HDL was seen by the decreased intensity between 250 and 720 kDa. The increase in pre-β HDL levels are considered to be a beneficial effect of sHDL infusions as this sub-class of smaller HDL is thought to be responsible for cholesterol efflux from macrophages by the ABCA1 transporter28. Thus, serum incubation with non-pegylated sHDL leads to more favorable endogenous HDL remodeling. However, formation of larger alpha-HDL, in case of pegylated sHDL incubations, could be just an artifact of increased hydrodynamic diameter of lipoprotein due to presence of PEG on the surface.
3.5. Pharmacokinetics of 22A peptide and phospholipids
The pharmacokinetics of the peptide and phospholipid components of sHDL nanoparticles were examined following IV bolus administration in SD rats (Figure 5). Nanoparticles with different degrees of pegylation were injected at 50 mg/kg dose of 22A peptide. The peptide concentration in serum was detected by LC-MS analysis. The total choline containing phospholipids in serum were measured by plate assay and the pre-dose levels of lipids were subtracted. The serum concentration kinetics data for peptide and lipid components of sHDL were fitted using a non-compartment analysis (NCA) by WinNonlin software (Version 7.3). Pharmacokinetic parameters derived included maximum serum concentration (Cmax), area under the serum concentration-time curve (AUC), elimination rate constant (K10), elimination half-life (T1/2), total clearance (CL) and volume of distribution at steady state (Vss).
Figure 5.

Pharmacokinetics of 22A peptide (Panel A) and phospholipids (Panel B) after intravenous bolus of various sHDL to Sprague-Dawley rats at 22A peptide dose of 50 mg/kg corresponding to 100 mg/kg dose of phospholipids. Serum peptide concentrations were determined by LC-MS and total choline containing phospholipids was measured by a commercial choline oxidase assay. (*) denotes statistical significant differences of total phospholipids changes for each group compared with sHDL group, with p-values less than 0.0001.
Similar 22A peptide PK profiles were observed among all treatment groups (Figure 5), indicating that PEG modification had no effect on peptide elimination following sHDL administration in vivo. The elimination rate constants obtained from NCA analysis showed no significant difference among all groups and small inter-subject variability for each group. The calculated area under the curve (AUC) values were comparable in all groups as well (Table 2). Different from 22A peptide, the PK behavior of sHDL’s phospholipids was significantly altered by PEG addition. Phospholipids in sHDL-PEG5k (5%) had a higher peak concentration Cmax (578 mg/dL) relative to the Cmax of unmodified sHDL (384 mg/dL) (p < 0.05). Higher surface density of PEG-2000 led to a longer lipid serum half-life in vivo and a higher AUC (Figure 5 and Table 3). The AUC increased gradually as more PEG was added, from 2071 mg*h/dL for non-pegylated sHDL to 3042 and 4473 mg*h/dL for 5 and 10 mol% PEG-2k, respectively. Similarly, the addition of longer PEG chains at the same surface density of polymer led to a longer half-life and a larger AUC, with half-lives of 8.6 h for sHDL-PEG2k (5%) and 12.5 h for sHDL-PEG5k (5%) (NS) and AUC of 4473 mg*h/dL for sHDL-PEG2k (5%) and 6108 mg*h/dL for sHDL-PEG5k (5%) (p < 0.01).
Table 2.
Pharmacokinetic parameters (% CV) of 22A peptide after 50 mg/kg doses of sHDL, sHDL-PEG2k (5%), sHDL-PEG2k (10%) and sHDL-PEG5k (5%) treatments.
| Parameters | Groups |
|||
|---|---|---|---|---|
| sHDL | sHDL-PEG2k (5%) | sHDL-PEG2k (10%) | sHDL-PEG5k (5%) | |
| Cma a (mg/dL) | 633.5 (22.4) | 677.1 (15.2)ns | 612.3 (27.5)ns | 712.0 (15.2)ns |
| AUCb (mg*h/dL) | 4185 (19.4) | 4150 (22.6)ns | 5885 (8.50)* | 5163 (10.4)ns |
| K10c (h−1) | 0.1521 (7.94) | 0.1693 (21.4)ns | 0.1071 (33.7)ns | 0.1377 (6.46)ns |
| T1/2 d (h) | 4.588 (8.58) | 4.25 (17.4)ns | 7.14 (28.8)ns | 5.05 (6.34)ns |
| CLe (dL/h) | 0.0034 (21.4) | 0.0032 (19.9)ns | 0.0022 (8.40)* | 0.0025 (9.18)ns |
| Vss f (dL) | 0.0221 (21.1) | 0.0188 (9.84)ns | 0.0216 (9.6)ns | 0.0179 (13.3)ns |
Cmax: the maximum plasma concentration of peptide
AUC: the area under the curve in plot of concentration of peptide against time
K10: elimination rate constant
T1/2: the half-life of elimination
CL: total clearance for peptide
Vss: volume of distribution for peptide at steady state. Data was shown as mean with CV%. Significance:
p<0.05
p<0.01
p<0.001
p<0.0001
: no significant difference compared with sHDL group.
Table 3.
Pharmacokinetic parameters (% CV) of total phospholipids after 50 mg/kg doses of sHDL, sHDL-PEG2k (5%), sHDL-PEG2k (10%) and sHDL-PEG5k (5%) treatments.
| Parameters | Groups |
|||
|---|---|---|---|---|
| sHDL | sHDL-PEG2k (5%) | sHDL-PEG2k (10%) | sHDL-PEG5k (5%) | |
| Cmaxa (mg/dL) | 383.9 (28.0) | 438.1 (9.5)ns | 477.0 (27.3)ns | 578.0 (4.8)* |
| Tmaxb (h) | 0.38 (38.5) | 0.88 (28.6)* | 1.6 (46.2)* | 1.0 (0)*** |
| AUCc (mg*h/dL) | 2071 (21.7) | 3042 (26.2)ns | 4473 (11.8)*** | 6108 (18.5)*** |
| K10d (h-1) | 0.17 (34.5) | 0.11 (54.9)ns | 0.072 (19.7)* | 0.065 (37.9)* |
| T1/2e (h) | 4.59 (43.3) | 8.62 (46.9)ns | 9.96 (18.2)** | 12.5 (38.2)* |
| CLf (dL/h) | 0.014 (23.6) | 0.0088 (20.5)* | 0.0057 (10.7)** | 0.0044 (30.4)** |
| VSSg (dL) | 0.083 (19.7) | 0.10 (37.2)ns | 0.084 (28.0)ns | 0.074 (27.4)ns |
Cmax: the maximum plasma concentration of phospholipids
Tmax: the time to observe Cmax
AUC: the area under the curve in plot of concentration of phospholipids against time
K10: elimination rate constant
T1/2: the half-life of elimination
CL: total clearance for phospholipids
VSS: volume of distribution for phospholipids. Data was shown as mean with CV%.
p<0.05
p<0.01
p<0.001
p<0.0001
: no significant difference compared with sHDL group.
The dichotomy of how pegylation did not affect pharmacokinetics of ApoA-I peptide component of sHDL but significantly altered elimination of phospholipid component was surprising. It became clear that pegylation of sHDL by insertion of pegylated lipid does not provide stealth properties to the entire nanoparticle, but rather just to the phospholipid bilayer component. This also points to the fact that following in vivo administration, sHDL undergoes extensive remodeling. The cholesterol-free lipid bilayers readily accept cholesterol; the ApoA-I peptide components exchange with endogenous apolipoproteins; and the phospholipid themselves likely exchange with endogenous lipoprotein lipids.
3.6. The impact of sHDL pegylation on in vivo pharmacodynamics
To examine the potential therapeutic effect of sHDL pegylation on cholesterol metabolism in vivo, the pharmacodynamics biomarkers were measured following administration of nanoparticles to normal SD rats by IV infusion at a dose of 50 mg/kg 22A peptide. sHDL has been reported to facilitate cholesterol efflux from peripheral tissues through RCT, and the level of cholesterol in serum can be used to reflect the transient efflux of cholesterol induced by administration of sHDL. The levels of plasma total cholesterol (TC, Figure 6A and B) and free cholesterol (FC, Figure 6C and D) were determined directly, while esterified cholesterol level was calculated by subtracting free cholesterol level from total cholesterol level at each time point (CE, Figure 6C). Pharmacodynamics parameters were calculated and summarized in Table 4. The typical pharmacological response following sHDL infusion is a rapid mobilization of free cholesterol into the plasma compartment, followed by a rise in cholesterol ester due to esterification by lecithin-cholesterol acyltransferase (LCAT) and subsequent elimination of cholesterol by the liver. These expected effects were observed for sHDL infusions, with a maximum FC mobilization of 75 mg/dL at 0.5 h followed by a peak in CE (46 mg/dL) at 4 h, and elimination of all mobilized cholesterol by 24 h post-dose. In line with the improvements in phospholipid PK, the pegylated sHDL exhibited greater cholesterol mobilization and longer pharmacodynamic effect relative to non-pegylated sHDLs. Both the higher degree of surface modification by PEG and longer PEG chain lengths resulted in higher maximum effect levels for TC, FC, and CE increases. For example, the maximum mobilized FC levels increased from 75 mg/dL for non-pegylated sHDL, to 110, 125 and 137 mg/dL for sHDL-PEG-2K (5%), sHDL-PEG-2K (10%) and sHDL-PEG-5K (5%). The area under the effect curve also increased for all pegylated formulations: from 422 mg*h/dL (sHDL) to 1241 mg*h/dL (a 1.9-fold increase) for sHDL-PEG-2K (10%) and to 1588 mg*h/dL (a 2.8-fold increase) for sHDL-PEG-5K (5%). Hence, the longer lipid circulation time for pegylated sHDL resulted in greater levels of mobilized cholesterol and a longer duration of the effect.
Figure 6.

Pharmacodynamic assessment after IV administration of sHDL particles from the pharmacokinetic study. The level of total cholesterol (Panel A), free cholesterol (Panel B) and cholesterol ester (Panel C) in rat serum were determined by commercially available kits. (*) denotes statistical significant differences of TC, FC or EC changes for each group compared with sHDL group, with p-values less than 0.0001.
Table 4.
Pharmacodynamic parameters (% CV) of total phospholipids after 50 mg/kg doses of sHDL, sHDL-PEG2k (5%), sHDL-PEG2k (10%) and sHDL-PEG5k (5%) treatments.
| Parameters | sHDL | sHDL-PEG2k (5%) | sHDL-PEG2k (10%) | sHDL-PEG5k (5%) | |
|---|---|---|---|---|---|
| TC | Tmax,Ea (h) | 0.94 (82.6) | 2.0 (70.7)ns | 2.0 (0)* | 3.3 (46.2)* |
| Emaxb (mg/dL) | 114.9 (25.4) | 177.8 (8.1)** | 181.4 (17.4)* | 212.5 (7.7)** | |
| AUECc (mg*h/dL) | 562.8 (25.9) | 1660 (37.2)* | 2031 (13.1)**** | 3171 (25.8)*** | |
| FC | Tmax,E (h) | 0.56 (55.9) | 0.88 (28.6)ns | 2.0 (0)**** | 1.5 (38.5)* |
| Emax (mg/dL) | 74.6 (26.8) | 109.6 (7.0)* | 125.1 (31.6)ns | 136.9 (12.0)** | |
| AUEC (mg*h/dL) | 421.7 (36.5) | 740.7 (31.5)ns | 1241 (35.6)* | 1588 (26.1)** | |
| CE | Tmax,E (h) | 1.2 (79.5) | 4.3 (67.6)ns | 5.0 (40.0)* | 7.0 (28.6)** |
| Emax (mg/dL) | 46.4 (43.0) | 76.2 (14.6)* | 73.5 (39.8)ns | 89.9 (33.5)ns | |
| AUEC (mg*h/dL) | 372.9 (21.4) | 869.6 (51.8)ns | 1034 (41.3)* | 1562 (32.1)** |
Tmax: time at which the Emax is observed.
Emax: the maximum plasma concentration of different cholesterol species.
AUEC: the area under the effect curve. Data was shown as mean with CV%.
p<0.05
p<0.01
p<0.001
p<0.0001
: no significant difference compared with sHDL group.
3.7. Distribution of mobilized cholesterol and lipoprotein remodeling in vivo
In order investigate in greater detail the mechanism of cholesterol mobilization and elimination following administration of pegylated and non-pegylated sHDL particles, we determined the relative distribution of mobilized cholesterol in the HDL, LDL and VLDL fractions by HPLC. The positions of HDL, LDL and VLDL sized particles containing cholesterol are labeled on Figure 7. The infusion of sHDL caused a rapid mobilization of cholesterol in the HDL fraction, with the maximum increase at 0.5 h post-dose (blue line, Figure 7A). The increase in HDL-C was accompanied by small increase in VLDL-C, and cholesterol returned to baseline levels by 24 h post-dose. The cholesterol profile changes were different when pegylated sHDLs were administered. Because pegylated sHDLs are slightly larger in size, the mobilized cholesterol appears to elute between HDL and LDL fractions at 0.5, 2 and 4 h post administration (Figure 7B-D). The increase in VLDL-C appears to be higher for all pegylated sHDLs relative to sHDL, especially for sHDL-PEG-2K (10%). It is possible that DSPE-PEG caused inhibition of lipoprotein lipase or stimulation of liver lipogenesis, which both lead to the transient increase in VLDL-C. However, for all formulations cholesterol increases were transient. The cholesterol level and its relative lipoprotein distribution returned to pre-dose levels, indicating completion of the RCT process triggered by administered sHDL.
Figure 7.

The cholesterol distribution between VLDL, LDL and HDL lipoprotein fractions at different time points after IV injection of sHDL (Panel A), sHDL-PEG2k (5%) (Panel B), sHDL-PEG2k (10%) (Panel C) and sHDL-PEG5k (5%) (Panel D) to SD rats.
4. Discussion
In this study, we successfully prepared pegylated sHDL with varying PEG surface densities of 5 and 10 mol%, as well as two different molecular weights of PEG, 2 kDa and 5 kDa. Incorporation of PEG resulted in a small increase in the sHDL hydrodynamic diameter, but the overall size homogeneity was preserved. Owing to the relatively low degree of PEG surface modifications, pegylated sHDL retained their ability to efflux cholesterol by passive uptake and through ABCA1, ABCG1 and SR-BI transporters and facilitate uptake of cholesterol by HEPG2 cells. Despite high similarity in vitro, the performance of pegylated sHDL differed when nanoparticles were examined normal rats. Administration of pegylated sHDL led to greater extent of cholesterol mobilization and longer duration cholesterol elevation relative to non-pegylated sHDL (Figure 6). However, the pharmacokinetic effect appeared to be different from cholesterol mobilization. While some increases phospholipid circulation times and AUCs were observed after administration of pegylated sHDL, the ApoA-I peptide PK parameters remained unchanged (Figure 5). Overall, 22A peptide appears to be eliminated more rapidly than sHDL’s lipid components. This indicates that peptide and lipid components of administered sHDL exchange in vivo with contents of endogenous lipoproteins and are eliminated independently. Some degree of remodeling was observed in vitro following incubation of serum with sHDL, with 22A peptide displacing endogenous ApoA-I proteins from various sub-fractions of HDL (Figure 4).
Our intention was to improve the duration of sHDL pharmacologic effect by increasing stability of the entire sHDL nanoparticle in blood and, thus, increasing circulation time. While extensive pegylation of nanoparticles (20% of lipid) is known to improve their stealth properties29, we were concerned that 20% pegylation would interfere with sHDL’s ability to interact with cholesterol efflux transporters and liver scavenger receptors, leading us to choose modest pegylation levels for our sHDL preparations (5 and 10%). We have previously shown a modest reduction in sHDL internalization by SR-BI receptors expressed by colon carcinoma cells using fluorescently labelled sHDL containing only 5 mol% DSPE-PEG-2K relative to non-pegylated sHDL30. When in vivo circulation times of fluorescently non-pegylated sHDL and peg-modified sHDL (5 mol% DPPG-PEG2K) were compared by measuring fluorescence intensity by Zhang et al., no differences were observed31. Careful examination of how the degree of nanoparticle pegylation influence PK was performed by Liu et al29. When lipid-calcium phosphate nanoparticles of 30 nm in diameter were modified with DPPE-PEG2K at 5 mol%, no changes in PK or biodistribution were observed. In contrast, modification with 20 mol% lead to a significant improvement in plasma nanoparticle stability and a longer circulation time. Hence, using a higher molar content of PEG on the sHDL surface may lead to a more significant reduction in sHDL remodeling by lipoproteins in vivo and further improvement in PK/PD profiles relative to those observed in the current study.
In this study, the PK parameters of lipid component of sHDL were improved by pegylation, but the half-life of 22A peptide remained unchanged. This result confirms that sHDL particle is not eliminated in vivo in its intact form. It is likely that elimination rates of sHDL’s protein and lipid components depend on their exchange with endogenous lipoproteins. The signs of this remodeling were confirmed by ex-vivo incubation of sHDL with human plasma (Figures 4), with 22A peptide displacing ApoA-I protein on endogenous HDL, as is evident by the appearance of lipid-free ApoA-I band in western blot (Figure 4). Previously, we observed longer circulation times for sHDL lipid components relative to 22A peptide, as well as extensive displacement of ApoA-I protein from endogenous HDL by 22A peptide26. Some differences in elimination rates of protein and phospholipid components were also noticed in clinical studies of sHDL products. Indeed, faster elimination of ApoA-I (T1/2 ~ 10 h) was observed relative to phospholipid (T1/2 ~ 46 h) following administration of 45 mg/kg of CER-001, ApoA-I-sphingomyelin nanoparticles32. The elimination of ApoA-I and lipid components of CSL-112, ApoA-I-soybean phosphatidylcholine nanoparticles, followed an opposite trend to CER-001 with a longer protein half-life of 36.4 h relative to phospholipid half-life of 18.0 h16. There are known differences in in vivo metabolism of DPPC used in this study to prepare sHDL, soybeanPC (CSL-112) and sphingomyelin (CER-001)33. In addition, full-length ApoA-I has about 10 amphipahtich α-helical segments, resulting in stronger binding to phospholipid relative to the much smaller 22A peptide. Thus, it is possible that ApoA-I-based sHDLs are more stable and less susceptible to the remodeling in vivo relative to ApoA-I-peptide-based sHDL.
Various strategies have been used to increase in vivo half-life of the ApoA-I component of sHDL. In one study, a trimer of the ApoA-1 protein was produced by recombinant expression34. An improvement in residence PK profiles was observed35-36, but clinical development of trimeric ApoA-I was halted likely due to high manufacturing cost and immunogenicity concerns. Using a similar concept, crosslinked dimers, trimers and star-like multimers of ApoA-I mimetic peptides were synthesized to improve the in vivo stability, circulation time and efficacy of peptide-based sHDL37. Having multiple α-helixes to bind phospholipids resulted in longer in vivo circulation time and lower plasma cholesterol levels in murine model of atherosclerosis, but a reduction in atheroma volume was not reported36. A different chemical approach, termed hydrocarbon stapling, was employed by Sviridov et al. to stabilize peptide α-helices and improve cholesterol efflux, but the impact of stapling on RCT was not fully characterized38. Finally, the ApoA-I protein of sHDL was selectively pegylated by attaching a single 20K PEG chain to protein N-terminal by Murphy et al.20 Pegylation of ApoA-I did not reduce its ability to efflux cholesterol, but did improve protein circulation in vivo and led to the suppression of bone marrow myeloid progenitor cell proliferation in hypercholesterolemic mice. Pegylation also appeared to improve anti-atherosclerotic properties of HDL as observed by the reduction in atheroma necrotic core area and lipid content of the aortic arch relative to placebo. However, differences between pegylated and unmodified HDL were not significant. This indicates that the composition of pegylated sHDL needs to be further optimized for their plasma stability and pharmacokinetic properties prior to formal efficacy studies.
The focus of this study was to understand how pegylation affects the RCT pathway facilitated by HDL. The impact of pegylation on cholesterol efflux in vitro and elevation of cholesterol levels in vivo were examined as biomarkers of RCT. However, the impact of pegylation on the anti-inflammatory and anti-thrombotic properties of sHDL has not yet been examined. In addition, the reduction in atheroma volume following HDL administration in animal models of atherosclerosis is the ultimate measure of sHDL’s efficacy in vivo. We plan to perform these studies following development of a complete understanding of the impact of pegylation on the pharmacokinetics and in vivo stability of sHDL, in addition to selecting the optimal surface-modified sHDL formulation.
The results of our study have broader impact of the field of sHDL design and nanoparticle drug delivery. This study underlines the importance of sHDL remodeling by endogenous lipoproteins in vivo in regards to sHDL’s PK and pharmacological effect. Hence, strategies for minimizing sHDL in vivo remodeling, such as crosslinking ApoA-I or lipid components, could potentially improve sHDL PK properties and efficacy. Similarly, HDL-like nanoparticles used for imaging and drug delivery purposes are likely to remodel in vivo, which could lead to rapid cargo release9, 31, 39-40. In the recent years, the formation of protein corona on the surface of nanoparticles used for drug delivery and imaging had emerged as a critical issue for in vivo efficacy of nanomedicines41-43. Endogenous lipoprotein components were found to be major components of the protein corona44. It has been reported that endogenous lipoproteins also rapidly remodel liposomal and micellar nanocarriers and alter cargo release45-46. Thus, understanding the forces driving lipoprotein remodeling in vivo and developing strategies to minimize it could lead to the design of more stable and efficacious sHDL based nanomedicines.
Supplementary Material
5. Acknowledgements
This research was funded in part by AHA 13SDG17230049, Michigan College of Pharmacy Upjohn Award, NIH R01 GM113832 and R21 NS091555. Emily E. Morin was partially supported by fellowships from the Cellular Biotechnology Training Program (T32 GM008353) and Translational Cardiovascular Research and Entrepreneurship Training Grant (T32 HL125242). Maria Fawaz was partially supported by a fellowship from the Pharmacological Sciences Training Program (T32 GM07767).
Abbreviations:
- ApoA-I
apoliprotein A-I
- IV
intravenous
- 22A/ESP24218
apoA-I mimetic peptide consisting of 22 amino acids–PVLDLFRELLNELLEALKQKLK
- 5A
apoA-I mimetic peptide consisting of 37 amino acids–DWLKAFYDKVAEKLKEAFPDWAKAAYDKAAEKAKEAA
- 22A-sHDL
phospholipid reconstituted HDL based on 22A
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DSPE-PEG
N-(carbonyl-methoxypolyethyleneglycol)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine
- DSPE-PEG-2k
DSPE-PEG with a 2000 Da molecular weight of PEG
- DSPE-PEG-5k
DSPE-PEG with a 5000 Da molecular weight of PEG
- HDL-C
HDL cholesterol
- PK
Pharmacokinetic
- PD
pharmacodynamics
- GPC
gel permeation chromatography
- DLS
dynamic light scattering
- IS
internal standard
- EIC
extracted ion chromatogram
- TIC
total ion chromatogram
- PL
phospholipids
- TC
total cholesterol
- FC
unesterified or free cholesterol
- CE
cholesterol ester
- RCT
reverse cholesterol transport
6. References
- 1.Heidenreich PA; Trogdon JG; Khavjou OA; Butler J; Dracup K; Ezekowitz MD; Finkelstein EA; Hong Y; Johnston SC; Khera A; Lloyd-Jones DM; Nelson SA; Nichol G; Orenstein D; Wilson PW; Woo YJ, Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 2011, 123 (8), 933–44. [DOI] [PubMed] [Google Scholar]
- 2.Rosenson RS; Brewer HB Jr.; Ansell BJ; Barter P; Chapman MJ; Heinecke JW; Kontush A; Tall AR; Webb NR, Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Remaley AT; Amar M; Sviridov D, HDL-Replacement Therapy: Mechanism of Action, Types of Agents and Potential Clinical Indications. Expert Rev Cardiovasc Ther 2008, 6 (9), 1203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwata A; Miura S; Zhang B; Imaizumi S; Uehara Y; Shiomi M; Saku K, Antiatherogenic Effects of Newly Developed Apolipoprotein a-I Mimetic Peptide/Phospholipid Complexes against Aortic Plaque Burden in Watanabe-Heritable Hyperlipidemic Rabbits. Atherosclerosis 2011, 218 (2), 300–7. [DOI] [PubMed] [Google Scholar]
- 5.Hafiane A; Genest J, HDL, Atherosclerosis, and Emerging Therapies. Cholesterol 2013, 2013, 891403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krause BR; Remaley AT, Reconstituted HDL for the Acute Treatment of Acute Coronary Syndrome. Curr Opin Lipidol 2013, 24 (6), 480–6. [DOI] [PubMed] [Google Scholar]
- 7.Darabi M; Guillas-Baudouin I; Le Goff W; Chapman MJ; Kontush A, Therapeutic Applications of Reconstituted HDL: When Structure Meets Function. Pharmacol Ther 2016, 157, 28–42. [DOI] [PubMed] [Google Scholar]
- 8.Li D; Gordon S; Schwendeman A; Remaley AT, Apolipoprotein Mimetic Peptides for Stimulating Cholesterol Efflux In Apolipoprotein Mimetics in the Management of Human Disease, Anantharamaiah GM; Goldberg D, Eds. Springer International Publishing: Cham, 2015; pp 29–42. [Google Scholar]
- 9.Kuai R; Li D; Chen YE; Moon JJ; Schwendeman A, High-Density Lipoproteins: Nature's Multifunctional Nanoparticles. ACS Nano 2016, 10 (3), 3015–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linsel-Nitschke P; Tall AR, HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov 2005, 4 (3), 193–205. [DOI] [PubMed] [Google Scholar]
- 11.Kingwell BA; Chapman MJ; Kontush A; Miller NE, HDL-Targeted Therapies: Progress, Failures and Future. Nat Rev Drug Discov 2014, 13 (6), 445–64. [DOI] [PubMed] [Google Scholar]
- 12.Charlton-Menys V; Durrington PN, Human Cholesterol Metabolism and Therapeutic Molecules. Exp Physiol 2008, 93 (1), 27–42. [DOI] [PubMed] [Google Scholar]
- 13.Hovorka R; Nanjee MN; Cooke CJ; Miller IP; Olszewski WL; Miller NE, Mass Kinetics of Apolipoprotein a-I in Interstitial Fluid after Administration of Intravenous Apolipoprotein a-I/Lecithin Discs in Humans. J Lipid Res 2006, 47 (5), 975–81. [DOI] [PubMed] [Google Scholar]
- 14.Michael Gibson C; Korjian S; Tricoci P; Daaboul Y; Yee M; Jain P; Alexander JH; Steg PG; Lincoff AM; Kastelein JJ; Mehran R; D'Andrea DM; Deckelbaum LI; Merkely B; Zarebinski M; Ophuis TO; Harrington RA, Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, After Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134 (24), 1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan M; Lalwani N; Drake S; Crockatt J; Dasseux J, Single-Dose Intravenous Infusion of ETC-642, a 22-Mer ApoA-I Analogue and Phospholipids Complex, Elevates HDL-C in Atherosclerosis Patients. Circulation 2003, 108 (17), 563–564. [Google Scholar]
- 16.Easton R; Gille A; D'Andrea D; Davis R; Wright SD; Shear C, A Multiple Ascending Dose Study of CSL112, an Infused Formulation of ApoA-I. J Clin Pharmacol 2014, 54 (3), 301–10. [DOI] [PubMed] [Google Scholar]
- 17.Vllasaliu D; Fowler R; Stolnik S, PEGylated Nanomedicines: Recent Progress and Remaining Concerns. Expert Opin Drug Deliv 2014, 11 (1), 139–54. [DOI] [PubMed] [Google Scholar]
- 18.Zundorf I; Dingermann T, PEGylation--a Well-Proven Strategy for the Improvement of Recombinant Drugs. Pharmazie 2014, 69 (5), 323–6. [PubMed] [Google Scholar]
- 19.Weissig V; Pettinger TK; Murdock N, Nanopharmaceuticals (Part 1): Products on the Market. Int J Nanomedicine 2014, 9, 4357–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy AJ; Funt S; Gorman D; Tall AR; Wang N, Pegylation of High-Density Lipoprotein Decreases Plasma Clearance and Enhances Antiatherogenic Activity. Circ Res 2013, 113 (1), e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM, PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv Drug Deliv Rev 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miles J; Khan M; Painchaud C; Lalwani N; Drake S; Dasseux J, Single-Dose Tolerability, Pharmacokinetics, and Cholesterol Mobilization in HDL-C Fraction Following Intravenous Administration of ETC-642, a 22-mer ApoA-I Analogue and Phospholipids Complex, in Atherosclerosis Patients. Arteriosclerosis Thrombosis and Vascular Biology 2004, 24 (5), E19–E19. [Google Scholar]
- 23.Calkin AC; Drew BG; Ono A; Duffy SJ; Gordon MV; Schoenwaelder SM; Sviridov D; Cooper ME; Kingwell BA; Jackson SP, Reconstituted High-Density Lipoprotein Attenuates Platelet Function in Individuals with Type 2 Diabetes Mellitus by Promoting Cholesterol Efflux. Circulation 2009, 120 (21), 2095–104. [DOI] [PubMed] [Google Scholar]
- 24.Patel S; Drew BG; Nakhla S; Duffy SJ; Murphy AJ; Barter PJ; Rye KA; Chin-Dusting J; Hoang A; Sviridov D; Celermajer DS; Kingwell BA, Reconstituted High-Density Lipoprotein Increases Plasma High-Density Lipoprotein Anti-Inflammatory Properties and Cholesterol Efflux Capacity in Patients with Type 2 Diabetes. J Am Coll Cardiol 2009, 53 (11), 962–71. [DOI] [PubMed] [Google Scholar]
- 25.Garber DW; Kulkarni KR; Anantharamaiah GM, A Sensitive and Convenient Method for Lipoprotein Profile Analysis of Individual Mouse Plasma Samples. J Lipid Res 2000, 41 (6), 1020–6. [PubMed] [Google Scholar]
- 26.Tang J; Li D; Drake L; Yuan W; Deshaine S; Morin EE; Ackermann R; Olsen K; Smith DE; Schwendeman A, Influence of Route of Administration and Lipidation of Apolipoprotein A-I Peptide on Pharmacokinetics and Cholesterol Mobilization. J Lipid Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Didichenko SA; Navdaev AV; Cukier AMO; Gille A; Schuetz P; Spycher MO; Therond P; Chapman MJ; Kontush A; Wright SD, Enhanced HDL Functionality in Small HDL Species Produced Upon Remodeling of HDL by Reconstituted HDL, CSL112: Effects on Cholesterol Efflux, Anti-Inflammatory and Antioxidative Activity. Circ Res 2016, 119 (6), 751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinecke JW, Small HDL Promotes Cholesterol Efflux by the ABCA1 Pathway in Macrophages: Implications for Therapies Targeted to HDL. Circ Res 2015, 116 (7), 1101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y; Hu Y; Huang L, Influence of Polyethylene Glycol Density and Surface Lipid on Pharmacokinetics and Biodistribution of Lipid-Calcium-Phosphate Nanoparticles. Biomaterials 2014, 35 (9), 3027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang J; Kuai R; Yuan W; Drake L; Moon JJ; Schwendeman A, Effect of Size and Pegylation of Liposomes and Peptide-Based Synthetic Lipoproteins on Tumor Targeting In AAPS 2016, Denver, CO, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z; Chen J; Ding L; Jin H; Lovell JF; Corbin IR; Cao W; Lo PC; Yang M; Tsao MS; Luo Q; Zheng G, HDL-Mimicking Peptide-Lipid Nanoparticles with Improved Tumor Targeting. Small 2010, 6 (3), 430–7. [DOI] [PubMed] [Google Scholar]
- 32.Keyserling CH; Hunt TL; Klepp HM; Scott RA; Barbaras R; Schwendeman A; Lalwani N; Dasseux J-L, Abstract 15525: CER-001, a Synthetic HDL-Mimetic, Safely Mobilizes Cholesterol in Healthy Dyslipidemic Volunteers. Circulation 2011, 124 (Suppl 21), A15525–A15525. [Google Scholar]
- 33.Agassandian M; Mallampalli RK, Surfactant Phospholipid Metabolism. Biochim Biophys Acta 2013, 1831 (3), 612–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graversen JH; Laurberg JM; Andersen MH; Falk E; Nieland J; Christensen J; Etzerodt M; Thogersen HC; Moestrup SK, Trimerization of Apolipoprotein A-I Retards Plasma Clearance and Preserves Antiatherosclerotic Properties. J Cardiovasc Pharmacol 2008, 51 (2), 170–7. [DOI] [PubMed] [Google Scholar]
- 35.Murphy AJ; Hoang A; Aprico A; Sviridov D; Chin-Dusting J, Anti-Inflammatory Functions of Apolipoprotein A-I and High-Density Lipoprotein Are Preserved in Trimeric Apolipoprotein A-I. J Pharmacol Exp Ther 2013, 344 (1), 41–9. [DOI] [PubMed] [Google Scholar]
- 36.Ohnsorg PM; Mary JL; Rohrer L; Pech M; Fingerle J; von Eckardstein A, Trimerized Apolipoprotein A-I (TripA) Forms Lipoproteins, Activates Lecithin:Cholesterol Acyltransferase, Elicits Lipid Efflux, and Is Transported Through Aortic endothelial Cells. Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids 2011, 1811 (12), 1115–1123. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y; Imura T; Leman LJ; Curtiss LK; Maryanoff BE; Ghadiri MR, Mimicry of High-Density Lipoprotein: Functional Peptide-Lipid Nanoparticles Based on Multivalent Peptide Constructs. J Am Chem Soc 2013, 135 (36), 13414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sviridov DO; Ikpot IZ; Stonik J; Drake SK; Amar M; Osei-Hwedieh DO; Piszczek G; Turner S; Remaley AT, Helix Stabilization of Amphipathic Peptides by Hydrocarbon Stapling Increases Cholesterol Efflux by the Abca1 Transporter. Biochem Biophys Res Commun 2011, 410 (3), 446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Q; Chen J; Zhang Z; Zheng G, Lipid-Based Nanoparticles in the Systemic Delivery of siRNA. Nanomedicine (Lond) 2014, 9 (1), 105–20. [DOI] [PubMed] [Google Scholar]
- 40.Ng KK; Lovell JF; Zheng G, Lipoprotein-Inspired Nanoparticles for Cancer Theranostics. Acc Chem Res 2011, 44 (10), 1105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gessner A; Lieske A; Paulke BR; Muller RH, Functional Groups on Polystyrene Model Nanoparticles: Influence on Protein Adsorption. Journal of Biomedical Materials Research Part A 2003, 65A (3), 319–326. [DOI] [PubMed] [Google Scholar]
- 42.Lundqvist M; Stigler J; Elia G; Lynch I; Cedervall T; Dawson KA, Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc Natl Acad Sci U S A 2008, 105 (38), 14265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tenzer S; Docter D; Kuharev J; Musyanovych A; Fetz V; Hecht R; Schlenk F; Fischer D; Kiouptsi K; Reinhardt C; Landfester K; Schild H; Maskos M; Knauer SK; Stauber RH, Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nature Nanotechnology 2013, 8 (10), 772–U1000. [DOI] [PubMed] [Google Scholar]
- 44.Cedervall T; Lynch I; Foy M; Berggard T; Donnelly SC; Cagney G; Linse S; Dawson KA, Detailed Identification of Plasma Proteins Adsorbed on Copolymer Nanoparticles. Angew Chem Int Ed Engl 2007, 46 (30), 5754–6. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal P; Hall JB; McLeland CB; Dobrovolskaia MA; McNeil SE, Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Advanced Drug Delivery Reviews 2009, 61 (6), 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corbo C; Molinaro R; Parodi A; Toledano Furman NE; Salvatore F; Tasciotti E, The Impact of Nanoparticle Protein Corona on Cytotoxicity, Immunotoxicity and Target Drug Delivery. Nanomedicine (Lond) 2016, 11 (1), 81–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.