

Abstract
Protective immune responses to viral infection are initiated by innate immune sensors that survey extracellular and intracellular space for foreign nucleic acids. The existence of these sensors raises fundamental questions about self/non-self discrimination because of the abundance of self-DNA and self-RNA that occupy these same compartments. Recent advances have revealed that enzymes that metabolize or modify endogenous nucleic acids are essential for preventing inappropriate activation of the innate antiviral response. In this review, we discuss rare human diseases caused by dysregulated nucleic acid sensing, focusing primarily on intracellular sensors of nucleic acids. We summarize lessons learned from these disorders, we rationalize the existence of these diseases in the context of evolution, and we propose that this framework may also apply to a number of more common autoimmune diseases for which the underlying genetics and mechanisms are not yet fully understood.
Keywords: Type I interferons (IFNs), Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), cGAS-STING, Aicardi-Goutières Syndrome (AGS), Systemic lupus erythematosus (SLE)
Introduction
A fundamental property of the immune system is to distinguish self from non-self and only mount a specific and robust immune response against the latter (1). In 1989, Charles Janeway, Jr proposed the theory of pattern recognition that remains the predominant conceptual framework for understanding how the innate immune system accomplishes this self/non-self discrimination (2). Pattern recognition theory postulated the existence of pattern recognition receptors (PRRs) that evolved to detect features of microbes that are distinct from our own physiology. Importantly, these features must be essential to the microbe and evolutionarily constrained so that they cannot readily mutate away from PRR detection. For bacteria, such features are abundant: cell wall lipopolysaccharides and peptidoglycans offer structures that are starkly different from nearly anything found in a human cell. Similarly, the unique sugars of fungal cell walls offer an unambiguous target for PRRs. Viruses, however, present a unique challenge because every component of a virion is made from the infected host cell. What feature of viruses is sufficiently distinct from the host to provide an attractive, faithful target for innate immune receptors?
Remarkable advances over the past ~17 years have revealed that the answer to this question is a compromise that challenges the fidelity of self/non-self discrimination. Most, if not all, antiviral responses are initiated by innate immune receptors that detect viral nucleic acids (3, 4). On the one hand, this makes sense because all viruses have a genome composed of either DNA or RNA, and viruses cannot replicate without a genome. On the other hand, DNA and RNA are among the most abundant macromolecules in all of our cells, which means that nucleic acid sensors have perhaps the highest potential among PRRs for encountering endogenous ligands.
In parallel with the definition of the sensors and signaling pathways that mediate nucleic acid detection, it has become increasingly apparent that these same sensors that protect us from viral infection can also drive specific human autoimmune diseases when they are inappropriately activated by endogenous nucleic acids. This dichotomy between protective immunity and autoimmune disease driven by the same innate immune response is the subject of this review. We will first summarize our current understanding of the nucleic acid sensing pathways, as well as the emerging framework of accessory enzymes that aid in establishing thresholds of self/non-self discrimination. We then evaluate rare human disorders in which endogenous nucleic acids trigger a self-directed antiviral response with severe pathological consequences. Finally, we summarize important lessons learned from the parallel studies of innate antiviral immunity and rare human diseases and propose that this framework may offer a useful paradigm for considering the confluence of genetics and environmental factors that underlie common autoimmune diseases like systemic lupus erythematosus (SLE).
Innate immune sensors of nucleic acids
The identification of the sensors of foreign nucleic acids that control the inducible inflammatory and antiviral response dates back to the year 2000, when Akira and colleagues reported that Toll-like receptor 9 (TLR9) is activated by DNA (5). The identification of TLR3 as a sensor of double-stranded RNA followed shortly thereafter (6). In 2004, three groups demonstrated that TLR7, originally characterized as the receptor responsible for the antiviral properties of small imidazoquinoline compounds (7), is a sensor of single-stranded RNA (8–10). TLR8, which is present in primates but not mice, similarly detects ssRNA (8).
The nucleic acid-sensing TLRs have been studied extensively over the past 15 years, and a number of excellent reviews summarize the signaling pathways, cell biology, and functions of these receptors (3, 11, 12). Together, these TLRs detect viruses and other microbes by sampling endocytosed material. In other words, the source of the nucleic acids that activate these TLRs is distinct from the cells that detect them. Here, we highlight three key points about these TLRs. First, the nucleic acid-sensing TLRs are not expressed on the surface of cells; rather, they are localized to endosomes by a process that requires the trafficking chaperone protein UNC93B1 (Figure 1; 13–15). This endosomal localization is essential for preventing autoreactivity against self nucleic acids (16, 17). Second, the endosomal localization of TLRs 3, 7, 8, and 9 is coupled to proteolytic processing that eliminates a significant portion of their leucine-rich repeat-containing ectodomains (15, 18). This processing is important for receptor function, revealing an additional mechanism that restricts signaling by the nucleic acid-sensing TLRs to endosomal compartments. Third, the inducible gene expression activated by these TLRs is “wired” differently depending on cell type. In conventional dendritic cells (DCs) and macrophages, activation of TLR7 or TLR9 drives a strong pro-inflammatory response, but in plasmacytoid DCs these same receptors trigger a potent type I interferon (IFN) response (19, 20). Interestingly, expression of TLR7 and TLR9 by B cells is important for the production of autoantibodies against DNA, chromatin, and ribonucleoproteins, which are hallmarks of SLE (21–23).
Figure 1: Pathway-specific negative regulators of nucleic acid sensing.
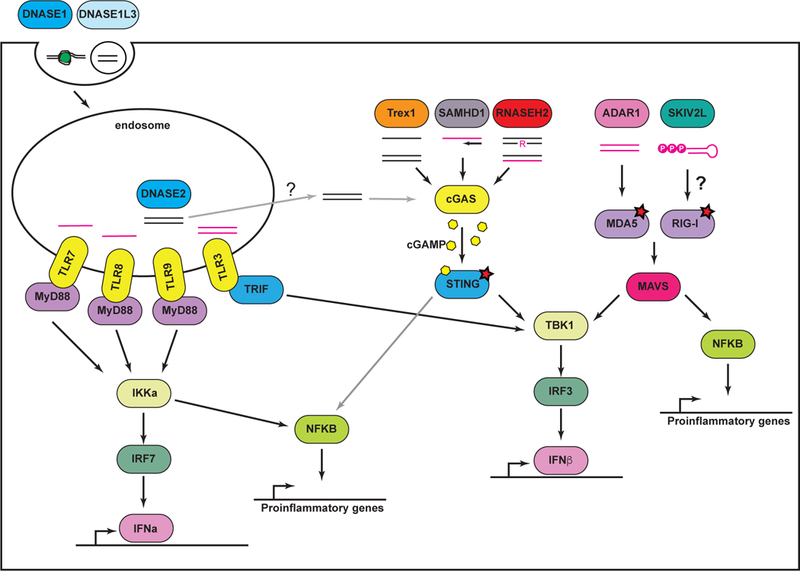
Endosomal TLRs and sensors of cytosolic nucleic acids are depicted, together with key negative regulators. See text for details on each.
In addition to the TLRs, a second system of receptors is positioned in the cytosol to detect foreign nucleic acids within infected cells. The RNA sensors are the RIG-I and MDA5 helicases, known as RIG-I-like receptors (RLRs). RLRs detect structural features of viral RNA that are scarce within host RNAs and activate a potent antiviral response that includes the inducible production of type I IFNs (4, 24). In particular, RIG-I binds to double-stranded RNAs that contain 5’ triphosphates or diphosphates and mediates the antiviral response to several classes of RNA viruses (25–27). MDA5 is activated by long, double-stranded RNA, the precise nature of which is unknown, and it is the non-redundant sensor that activates the IFN response to picornavirus infection (28). These RLRs signal through the adaptor protein MAVS that is found on the surface of mitochondria and links RNA detection to the activation of the TBK1 kinase and the IRF3 transcription factor that are important for IFN production (Figure 1; 29–32).
Shortly after the RLRs were discovered, an analogous pathway for type I IFN induction triggered by cytosolic DNA was described (33, 34). The principal intracellular DNA sensor is cGAS (35), which binds to DNA and catalyzes the synthesis of cyclic GMP-AMP (cGAMP; 36), a 2’, 3’-linked dinucleotide (37–39). cGAMP then binds to and activates STING, a transmembrane adapter protein on the endoplasmic reticulum (ER) that links DNA detection to TBK1/IRF3 (40–42). In general, the cGAS-STING pathway is important for the antiviral response triggered by DNA viruses and retroviruses (43, 44).
Together, the TLR, RLR, and the cGAS-STING pathways are essential for host defense against viral infection. However, the potent antiviral responses triggered by these nucleic acid sensors come with the high potential cost of autoreactivity against the abundant self nucleic acids that reside within every host cell. Indeed, recent advances in our understanding of rare human autoimmune diseases have illuminated several key enzymes that regulate the activation of these sensors by preventing the formation or accumulation of self-derived immunostimulatory nucleic acids. Below we describe these enzymes and the diseases that arise when they are mutated. We focus primarily on the intracellular nucleic acid sensing pathways because the potential contributions of TLRs to autoimmunity are well established and excellent comprehensive reviews covering this topic already exist (45–48). Building from these recent studies of rare monogenic disorders, we extract lessons that can be applied to the study of more common autoimmune diseases, with implications for the development of promising new therapies.
Rare disorders of nucleic acid sensing in humans
As described above, much progress has been made in identifying and characterizing the nucleic acid sensors that trigger the inducible antiviral response. In parallel with the definition of these innate immune pathways, the genetic diagnoses of several rare monogenic disorders in humans has illuminated the fundamental importance of negative regulation of nucleic acid sensors. These disorders, collectively referred to as type I interferonopathies (49), are characterized by the constitutive production of type I IFNs and an inflammatory response directed at self tissues. Broadly, the type I interferonopathies can be categorized into four classes. First, and perhaps best studied, Aicardi-Goutières Syndrome (AGS) is caused by mutations in the genes encoding one of five enzymes, four of which regulate the accumulation of intracellular nucleic acids that activate either the cGAS-STING pathway or the RLR-MAVS pathway. Additional examples of enzymes that regulate intracellular nucleic acid sensing add to our understanding of these interferonopathies. Second, mutations that result in hypersensitive or constitutively active RLR-MAVS or cGAS-STING pathways can cause diseases that may not require ligand-dependent activation of the pathways. Third, rare mutations in extracellular nucleases cause severe, SLE-like disease phenotypes that are likely driven primarily by TLRs, not intracellular sensors. Finally, failure to control signaling by the type I IFN receptor can result in disease that may not be driven by any single innate immune pathway. These diseases and their underlying mechanisms have been the subject of excellent recent reviews (50–52). Below, we highlight some key points about them and the conceptual framework that has emerged from studying them.
Aicardi-Goutières Syndrome and metabolism of intracellular nucleic acids
Aicardi-Goutières Syndrome (AGS) was first described in the 1980s as an inherited encephalopathy accompanied by elevated levels of type I IFNs that resembled the sequelae of congenital virus infection (53, 54). AGS symptoms include calcifications in the white matter of the brain, leukocyte infiltration into the central nervous system, profound psychomotor retardation owing to loss of motor neurons, and skin rashes known as chilblains (51). AGS is a rare disorder, with approximately 500 affected families known worldwide. Starting in 2006, Yanick Crow and his colleagues have identified mutations in seven genes that cause AGS, accounting for the majority of clinically diagnosed cases of this disease. While there is significant phenotypic overlap in patients with mutations in each of these genes, inflammation in certain tissues appears to be associated with mutations in some AGS genes but not others. These shared and unique clinical features of AGS have been extensively characterized (51, 55, 56), and for the purpose of this review we focus on a few broad insights illuminated by each of the five known AGS enzymes.
1. Trex1.
Trex1 is a DNA exonuclease, the activity of which was discovered by Tomas Lindahl almost 50 years ago in extracts prepared from rabbit bone marrow (57). Cloning of the TREX1 gene revealed its homology to proofreading DNA exonucleases in bacteria (58, 59). However, unlike the proofreading exonucleases, Trex1 is anchored by its c-terminus to the cytosolic face of the ER membrane (60, 61). Trex1 is active against single stranded DNA and the nicked strand of double-stranded DNA (62, 63). The identification of TREX1 mutations in AGS and familial chilblain lupus (64, 65) revealed a role for Trex1 as a key regulator of the antiviral response. Importantly, all of the AGS mutations in TREX1 can be classified as classic loss of function alleles that severely compromise Trex1 exonuclease activity (63), leading to the simple proposition that Trex1 metabolizes immunostimulatory DNA.
The mouse model of Trex1 deficiency has been particularly instructive in defining its role as a key negative regulator of the innate antiviral response. Similar to AGS, Trex1−/− mice have an elevated type I interferon signature and develop profound inflammation in multiple tissues, leading to significant mortality. All aspects of this disease require cGAS, STING, the IRF3 transcription factor, the type I IFN receptor, and lymphocytes, revealing a specific genetic pathway that links Trex1 deficiency to lethal autoimmunity (61, 66–68). Interestingly, Trex1−/− mice uniformly develop inflammatory myocarditis and lack detectable inflammation in the brain, whereas AGS in humans affects the brain but only rarely the heart (55). The reasons that underlie the difference in target tissues between Trex1 deficiency in humans and mice remain unclear.
The studies of Trex1−/− mice strongly implicate endogenous DNA molecules as the cause of this form of AGS. What is the source of this DNA? There are at least three possibilities that are not mutually exclusive. First, Trex1 is a potent anti-retroviral enzyme that targets cDNA formed by reverse transcription of retroelements (61). Supporting this notion is the observation that Trex1 can also metabolize the cDNA of HIV (69). Moreover, SAMHD1, another AGS enzyme, is a potent HIV restriction factor (see below). Finally, treatment of Trex1−/− mice with a cocktail of reverse transcriptase inhibitors ameliorates disease (70). A second possibility is that Trex1 is important for eliminating DNA byproducts of DNA replication or DNA repair (71, 72). Until quantitative analysis of the DNA that accumulates in Trex1-deficient cells is performed, the precise origins of these DNAs will continue to be debated. A third possibility is that Trex1 regulates inflammation independent of its DNA exonuclease activity (73). However, the fact that the disease in Trex1−/− mice is entirely cGAS- and STING-dependent implicates DNA metabolism as the primary function of Trex1 (66–68).
2. RNase H2.
Mutations in the genes encoding any of the three subunits of the heterotrimeric RNase H2 enzyme cause AGS (74). Interestingly, and unlike the TREX1 mutations, AGS-mutant RNase H2 enzymes retain all or most of their measurable catalytic activity in vitro (75). RNase H2 is capable of two distinct activities: incising the RNA strand of an RNA-DNA hybrid, and cleaving the phophodiester backbone adjacent to a single ribonucleotide in double-stranded DNA (76). Interestingly, RNase H2-deficient mice, generated by deletion of the Rnaseh2b gene, are embryonic lethal early in development (76, 77). This lethality is caused by defective removal of single ribonucleotides from replicating genomic DNA, which results in massive genome instability and p53-dependent cell cycle arrest. Remarkably, misincorporation of ribonucleotides into replicating DNA occurs at a frequency of approximately 1 in 7,000 base pairs in the absence of RNase H2 activity, making them the most common base lesions in genomic DNA (76). Interestingly, and likely because of the early lethality, RNase H2-deficient mice do not show evidence for a systemic type I IFN signature (76). However, two recently generated knock-in mice that express AGS alleles of Rnaseh2a and Rnaseh2b have an IFN signature that is cGAS- and STING-dependent (Figure 1; 78, 79). The nature of the immunostimulatory nucleic acids that accumulate in RNase H2-deficient cells and trigger cGAS remains incompletely defined, although evidence suggests that they are related to the damage caused by ribonucleotide incorporation into DNA (78, 80).
3. SAMHD1.
When mutations in the SAMHD1 gene were originally identified in AGS, its function remained unknown (81). Two years later, two groups identified SAMHD1 as the essential myeloid restriction factor for HIV-1 that is targeted for degradation by the Vpx accessory protein of HIV-2 (82, 83). SAMHD1 is a dNTP triphosphoydrolase that starves the HIV reverse transcriptase of the dNTPs required for generation of cDNA (84, 85). A number of studies have suggested that SAMHD1 is also an RNA exonuclease and that this activity is important for HIV restriction (86–89). However, this RNase activity may be a contaminant that copurifies with SAMHD1 (90, 91). Moreover, a recent study demonstrated that the IFN signature in Samhd1−/− mice, which do not develop overt autoimmune disease, is dependent on the cGAS-STING DNA sensing pathway (Figure 1; 92), which is consistent with the targeting of dNTPs by SAMHD1. One possible explanation for why Samhd1−/− mice do not develop AGS-like disease is the observation that resting dNTP levels in mouse myeloid cells are higher than in human cells, rendering these mouse cells more refractory to SAMHD1-mediated dNTP depletion (93).
As with all of the AGS enzymes, the identity of the cGAS ligands that accumulate in SAMHD1-mutant cells remains incompletely defined. The well-characterized function of SAMHD1 as an HIV restriction factor that blocks cDNA synthesis (82–85), together with its similar ability to block retroelements (94), suggests that retroelement cDNA is a relevant source of cGAS ligands. However, others have shown that SAMHD1-mutant cells accumulate DNA damage, suggesting a distinct origin of the immunostimulatory DNAs regulated by SAMHD1 (95). Intriguingly, one AGS patient with SAMHD1 mutations developed chronic lymphocytic leukemia (96), and somatic SAMHD1 mutations have been discovered in other CLL cases that are not accompanied by AGS (97). Although these remain the only known associations of any AGS genes with cancer, the connections between nucleic acid metabolism, DNA damage, and innate immunity require further study.
4. ADAR1.
The three AGS enzymes described above are all regulators of the cGAS-STING pathway. The identification in 2012 of AGS mutations in the ADAR gene that encodes the ADAR1 RNA editing enzyme suggested a disease mechanism independent of DNA sensing (98). ADAR1 is a deaminase that converts adenosines to inosines within RNA (99). Inosine is decoded as a guanosine, leading to amino acid changes, RNA splice site disruption, and read-through of stop codons (99). There are two isoforms of ADAR1: the smaller p110 isoform that is constitutively and ubiquitously expressed, and the larger p150 isoform that is strongly IFN-inducible (99, 100). Adar−/− mice are embryonic lethal and develop a massive type I IFN signature that precedes this lethality (101). Remarkably, this embryonic lethality is driven by the MDA5-MAVS pathway (102–104), and not the RIG-I-MAVS pathway (104), revealing a monogamous relationship between ADAR1 and MDA5. Moreover, as suggested by the existence of AGS mutations that are found only in the p150 isoform of ADAR1 (98), ADAR1 p150 is uniquely responsible for regulation of MDA5, whereas the p110 isoform contributes to development (104).
What are the RNA ligands of MDA5 that are edited by ADAR1? Studies have implicated the RNAs of the highly structured Alu retroelements as the primary targets of ADAR1 in cells, which suggests the possibility that failure to edit these sequences creates RNA ligands that are specifically detected by MDA5 (105). Alus are the most prolific of the retroelements, with millions of copies that together account for approximately 11% of the DNA content of the human genome (106). Other potential sources of endogenous MDA5 ligands include structured regions of mRNA 3’ untranslated regions (UTRs) that are known to be edited by ADAR1 (103). A clearer understanding of the intersection of RNA editing and the MDA5-dependent antiviral response promises to provide important insight into the origins of AGS, as well as a number of other human autoimmune diseases in which polymorphisms in the IFIH1 (MDA5) gene have been found (107–110).
5. IFIH1/MDA5.
The dominant (heterozygous) AGS-causing mutations in the IFIH1/MDA5 gene are the only known gain of function mutations in this disease, and they directly affect the activation thresholds of MDA5 by endogenous RNAs (111). As such, the mechanism of disease associated with these IFIH1 mutations is distinct from the enzymes described above, which prevent the formation or accumulation of immunostimulatory nucleic acids. Thus, these IFIH1 mutations can be considered together with additional known mutations in RNA and DNA sensing pathways that cause their constitutive activation, as described in more detail below.
Two other non-AGS enzymes illustrate the importance of metabolism of intracellular nucleic acids as a key mechanism to prevent autoreactivity. The SKIV2L component of the cytosolic RNA exosome is important for the elimination of damaged RNAs and the turnover of mRNAs (112). Interestingly, SKIV2L also is a potent negative regulator of the RLR-MAVS pathway (Figure 1; 113). Biallelic mutations in the human SKIV2L gene cause a rare disease called tricohepatoenteric syndrome (THES), which is characterized by growth retardation, craniofacial abnormalities, and intestinal dysfunction (114). Most of these symptoms likely reflect the role of SKIV2L in RNA turnover, but it is interesting to note that SKIV2L-THES patients have a peripheral blood IFN signature that is as robust as that observed in AGS (113).
Lastly, mice deficient in the lysosomal DNase 2 develop a potent, cGAS- and STING-dependent type I IFN signature during development that results in embryonic lethality (Figure 1; (68, 115–118). This implies that in the absence of DNase 2, failure to degrade lysosomal DNA results in “leakage” of this DNA to the cytosol and activation of cGAS. To date, there are no known mutations in the human DNASE2 gene that are associated with disease.
Mutations in sensors that drive constitutive activation
A second class of human interferonopathies is caused by activating mutations in the nucleic acid sensors or key adaptor proteins themselves. Such mutations render the pathways constitutively active in the absence of ligand, more sensitive to lower amounts of ligand, or both. As described above, the AGS mutations in IFIH1/MDA5 fall into this class (111). Other heterozygous, gain of function mutations in either DDX58/RIG-I or IFIH1/MDA5 are also found in another rare disorder called Singleton-Merten Syndrome (SMS), which is characterized by premature tooth loss, calcifications in the aorta, and overproduction of type I IFNs (119–121). Although the disease symptoms of AGS and SMS are largely non-overlapping, one particular mutation in IFIH1/MDA5 results in a clinical presentation that has both AGS-like and SMS-like features (122), revealing the potential for phenotypic overlap in these diseases.
STING-associated vasculopathy with onset in infancy (SAVI) is a rare inflammatory disease caused by dominant activating mutations in TMEM173, which encodes the STING adaptor of the intracellular DNA sensing pathway (123). SAVI is characterized by severe vasculitis in the extremities, and some cases involves severe lung inflammation. These symptoms are distinct from those of AGS, but the recent discovery of additional heterozygous STING mutations associated with SLE-like features and chilblain lupus highlight a spectrum of outcomes that can be driven by STING activation (124–126). These mutations are thought to bypass the need for cGAMP binding to STING, leading to its dimerization and constitutive activation in the absence of cGAS activation by DNA (123–125).
Metabolism of extracellular nucleic acids
All of the mutations described in the first two classes of interferonopathies described above influence the activation of the cGAS-STING or the RLR-MAVS pathways. In contrast, mutations in two extracellular DNases cause diseases resembling SLE that likely involve dysregulated activation of TLRs, rather than intracellular sensors. First, mice deficient for DNase 1, the most abundant secreted deoxyribonuclease, develop high titer anti-nuclear autoantibodies (ANA) and glomerulonephritis (127). Moreover, heterozygous mutations in DNASE1 have been identified in rare cases of SLE that present with high-titer ANAs (128). Although the precise nucleic acid sensors that drive these disease features have not yet been tested, the extracellular location of DNase 1, together with the well established role for TLRs in the detection of extracellular complexes of proteins and nucleic acids, strongly implicates DNase 1 as a key regulator of TLR signaling (46).
Recently, human mutations in the DNASE1L3 gene were discovered in families with inherited cases of severe SLE (129), as well as in an extremely rare disorder called hypocomplementemic urticarial vasculitis syndrome (HUVS), which can be accompanied by severe SLE (130). A recent study of DNase1L3-deficient mice demonstrated that this enzyme is uniquely capable of digesting DNA contained within membrane-associated “microparticles” that circulate in blood, and found that the potent anti-chromatin autoantibody response is entirely MyD88-dependent, strongly implicating DNase1L3 as a key regulator of TLR signaling (131).
Finally, human mutations in the ACP5 gene that encodes the tartrate-resistant acid phosphatase (TRAP) cause spondyloenchondrodysplasia (SPENCD) (132), a disease characterized by disturbances in bone homeostasis that also presents with a type I IFN signature and variable systemic autoimmune phenotypes. Although TRAP is not itself a nuclease, it appears to regulate TLR signaling (133), revealing another potential mechanism for limiting the activation of TLRs.
“Generic” regulators of type I IFN signaling
In addition to the pathway-specific regulators of nucleic acid sensors described above, newly characterized human interferonopathies reveal a fourth mechanism of disease in which signaling by the type I IFN receptor becomes dysregulated, leading to amplification of IFN signaling and overproduction of IFNs. Loss of function mutations in the ISG15 gene that encodes an IFN-inducible, ubiquitin-like molecule result in an interferonopathy that is associated with failure to accumulate the USP18 protease that removes ISG15 conjugates from proteins and is a potent negative regulator of type I IFN receptor signaling (134). Similarly, mutations in the USP18 gene cause pseudo-TORCH syndrome, which has some overlapping clinical features with AGS, including intracranial calcifications (135). Because these mutations affect IFN receptor signaling, not a specific pathway, it is probable that no single IFN-inducing pathway is responsible for the dysregulated IFN response.
Sidebar: Autoimmune versus autoinflammatory disease?
The words autoimmune and autoinflammatory are sometimes used interchangeably to describe a particular disease state, often because the precise underlying mechanisms that drive disease remain unknown or controversial. It is useful to understand the distinctions between these two terms and what they describe (136). An autoinflammatory disease is one in which the clinical presentation is driven by the innate immune system. In other words, neither T cells nor B cells are essential for the disease process. Perhaps the best examples of autoinflammatory diseases are the cryopyrin-associated periodic syndromes (CAPS), which are caused by gain of function mutations in the human NLRP3 gene (50, 137). Mouse models of these CAPS-associated NLRP3 alleles reveal that the disease is independent of both T and B cells (138). In contrast, autoimmune diseases require T cells and/or B cells as essential mediators of the pathophysiology. Importantly, such autoimmune diseases can originate specifically within the innate immune system, yet their clinical manifestation requires lymphocytes. In AGS, arguments can be made for both autoinflammatory and autoimmune components (51), highlighting another important point: some disease symptoms can be solely due to dysregulated innate immunity, whereas others can be driven by T and B cells. AGS patients develop abundant autoantibodies that only partially overlap with those found in SLE (51, 139). In the Trex1-deficient mouse model of AGS, T and B cells are absolutely required for disease, and tissue-specific autoantibodies are readily apparent (61, 66). While the human disease could be more autoinflammatory in nature than its mouse model, it will be important to understand the relative contributions and potential crosstalk between autoinflammatory and autoimmune mechanisms in AGS.
Lessons learned from rare human autoimmune disorders
The diseases described above, together with the discovery of the gene mutations that cause them, offer a number of broad lessons that can inform the way we think about not only these rare disorders, but also other, more common autoimmune diseases that share clinical symptoms. We summarize some of these key lessons below.
Nucleic acid metabolism is essential for preventing autoimmune disease
The use of innate immune sensors of nucleic acids to detect viral infection fits with the Janeway PAMP-PRR paradigm because one feature shared by all viruses is a genome composed of nucleic acids. While some nucleic acid sensors detect fairly precise ligand structures, like the 5’ phosphate moieties and double-stranded character of RIG-I RNA ligands, others have more generic specificities, like that of cGAS for any double-stranded DNA of sufficient size. Because foreign and self nucleic acids are made of the same four bases joined by the same phosphodiester bonds, the selectivity of these sensors for foreign nucleic acids over host nucleic acids is constrained by the structural features and locations of foreign nucleic acids that can be used to distinguish them from self (52). This imperfect selectivity, in turn, necessitates additional mechanisms that set thresholds for the activation of these sensors. The enzymes that metabolize nucleic acids described above are clear examples of trans-acting factors that establish thresholds by eliminating or preventing the formation of immunostimulatory nucleic acids.
A corollary to this lesson is that there are many potential sources of endogenous immunostimulatory nucleic acids, each with its own dedicated mechanism(s) of regulation. For example, DNase 1 and DNase1L3, despite both being extracellular nucleases, have distinct roles in the elimination of extracellular DNA (131). Similarly, the identification of three enzymes that each non-redundantly regulate the cGAS-STING pathway strongly implies multiple sources (and possibly distinct structures) of intracellular DNA ligands of cGAS. We predict that there are additional enzymes and pathways, not yet discovered, that serve a similar purpose by targeting other sources of nucleic acids. Understanding the full breadth of these pathways will solidify the framework established in the context of the monogenic interferonopathies and will offer new places to look for genetic causes of orphan rare diseases.
Interferon-driven diseases can be classified into specific pathways
As shown in Figure 1 and described above, nearly all of the currently known type I interferonopathy genes can each be paired with one of four specific pathways: TLRs, cGAS-STING, RLR-MAVS, or IFN receptor signaling. It is interesting to note that some of these gene mutations were identified before the sensors and key signaling adapters of the relevant pathway were discovered, for example the mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, and SAMHD1 that are now known to be upstream of the cGAS-STING pathway (64, 74). Thus, the rare diseases have informed our understanding of the innate immune pathways, and vice versa. A simple but very important lesson is that the genetics of each specific case of disease implicate one – and only one – innate immune pathway as its underlying cause. This has fundamental implications for treating each case of disease based on the specific pathway affected. Moreover, as discussed below, we propose that this lesson can be extrapolated to other, more common autoimmune diseases that are accompanied by a type I IFN signature.
Multiple individual pathways can converge on a similar clinical presentation
One surprise that has emerged from the study of mouse models of AGS is that it can be caused by mutations that affect either the cGAS-STING pathway or the MDA5-MAVS pathway. This is particularly interesting in light of the fact that cGAS and MDA5 bear no resemblance to each other, they are activated by completely different ligands, and each evolved to detect completely distinct classes of pathogens. How, then, could unrestrained signaling by either of these separate pathways result in a clinical presentation that, upon initial diagnosis, is highly similar? Importantly, careful, long-term study of hundreds of AGS patients has revealed certain clinical features that are uniquely associated with specific AGS genotypes (55, 56), but the shared features are both striking and surprising. Just as defining the breadth of regulatory mechanisms for each pathway will increase our understanding of these rare diseases, the convergence of multiple pathways on a similar disease presentation also increases the genetic “space” into which a particular disease-causing mutation can fall.
There is an evolutionary advantage to an exuberant, sensitive antiviral response
Humans and their primate ancestors have been under intense selective pressure due to viral infections for tens of millions of years (140). Viruses like influenza, polio and smallpox have strongly shaped human evolution in the recent past of recorded history, which only spans a few thousand years. Thankfully, vaccines have either dramatically reduced (polio) or completely eliminated (smallpox) the devastating consequences of some of these viruses. However, these viruses and others have left indelible imprints on the current human population. Antiviral proteins that are at the front lines of defense are the principal targets of virus-encoded antagonists. In turn, these host proteins are under strong selective pressure to escape antagonism by altering amino acids at key sites of interaction with the virus-encoded proteins. Such escape mutants, which appear randomly in each generation, restore the antiviral response and provide an advantage to the host in combating a specific viral infection. This ongoing conflict of virus-mediated antagonism and host escape by mutation is referred to as an evolutionary “arms race,” the signatures of which can be identified by comparative analysis of primate genes and inference of the rate of host protein evolution (140).
It is not surprising that nearly all of the antiviral pathways implicated in the rare human autoimmune diseases discussed above are under strong positive selection in primates: cGAS, STING, the RLRs, and MAVS are clear examples (141–144). Although the precise viruses and antagonists that have driven this positive selection are often difficult to identify, key examples illustrate the selective advantages that are conferred by “new” alleles that escape specific viral antagonists (145, 146).
A simple model has emerged from the study of host-virus arms races: viral antagonists reduce the function of specific antiviral pathways (often incompletely), and escape mutations in host sensors restore the function of the pathway by evading antagonism (Figure 2A). However, there is another way for the host to escape viral antagonism without physical evasion of the antagonist: by increasing the sensitivity and amplitude of the pathway itself. This could be accomplished in at least two ways. First, mutations in sensors that make them more active at lower thresholds of ligand confer a hypersensitive pathway that could function even in the presence of a viral antagonist; the IFIH1 mutations in AGS are clear examples of this (111). Second, hypomorphic mutations in pathway-specific negative regulators can similarly enhance the strength of the antiviral response (Figure 2B). For example, consider an antiviral pathway (MDA5-MAVS) that is important for host defense against a specific picornavirus that places strong selective pressure on humans (e.g., poliovirus). A virus-encoded antagonist could reduce the MDA5-MAVS pathway to 20% of normal within infected cells, which would strongly favor viral replication and dissemination. Hypomorphic mutation in a key, MDA5-specific negative regulator (ADAR1) could triple the strength of the MDA5-MAVS pathway, thus restoring the antiviral response to 60% of normal, which would confer increased resistance even in the presence of continued antagonism (Figure 2B).
Figure 2: Nucleic acid sensing and the host-virus conflict.
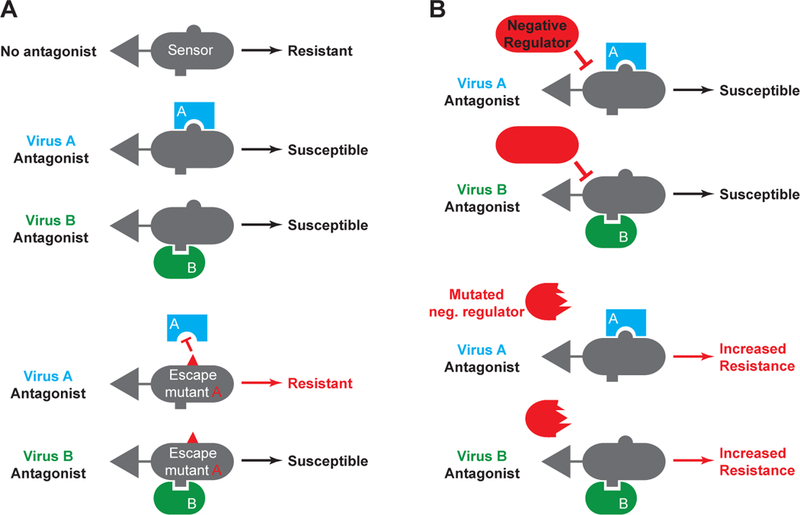
A: Two viruses are shown, each of which encodes a distinct antagonist of the same sensing pathway. An escape mutation in the sensor that overcomes antagonism by virus A does not impact continued antagonism by virus B. B: A mutation in a pathway-specific negative regulator increases the overall strength of the sensing pathway, conferring increased resistance to both viruses even in the presence of their antagonists.
There are three important features of mutations in negative regulators that distinguish them from escape mutations in the direct host targets of viral antagonists. First, there are many ways to create a hypomorphic negative regulator by mutation, the simplest of which is to acquire a catastrophic frameshift mutation that destroys the encoded protein (if that protein does not have another essential function). In contrast, useful escape mutations in sensors are not only largely confined to the specific physical interface between the host protein and the antagonist, they must also preserve functionality of the host protein itself. In other words, mutations that escape direct antagonism are more constrained than mutations in pathway specific negative regulators, yet both can have a similar effect in the context of host defense. Second, hypomorphic mutations in negative regulators would, in principle, yield enhanced protection against ALL viruses for which that particular pathway is important. In contrast, specific escape mutations in a sensor can solve antagonism by one particular virus-encoded protein, but perhaps create vulnerability to a second, unrelated virus. For example, the human allele of the TRIM5α lentiviral restriction factor efficiently prevents infection by an ancient, extinct retrovirus, but may have rendered human cells uniquely susceptible to HIV, which entered the human population millions of years later (147). Finally, negative regulators, unlike activating sensors, are usually not direct targets of virus-encoded antagonists, which means that they can exert their effects without specific interference by the virus.
We propose that the unique solutions to the host-virus conflict offered by hypomorphic mutations in negative regulators of antiviral pathways, in the context of antagonism of these same pathways by viruses, may explain the prevalence and potential maintenance of mutations in some AGS genes. For example, the A177T AGS allele of RNASEH2B and the P193A allele of ADAR are present in the human population at frequencies of ~1/800 and ~1/460, respectively (Y. Crow, personal communication). One key prediction raised by this proposal is that carriers of AGS alleles will have a “heterozygous advantage” in host defense against specific viruses. Although this has not yet been tested, the emergence of new mouse models and the definition of the pathways regulated by each AGS enzyme will allow for the design of such experiments. Another important point, the implications of which are discussed in more detail below, is that rare alleles that emerged recently in evolutionary time can confer both an advantage in host defense and a strong predisposition to autoimmune disease.
A framework for understanding genetics and mechanisms of common autoimmune diseases
One interesting aspect of the rare diseases described above is that the study of their underlying mechanisms paralleled the characterization of the innate immune sensors of nucleic acids. Although the specific pathways implicated in these monogenic disorders are, in most cases, clearly established, a key outstanding question is whether these examples are informative for a number of more common diseases that share some underlying features. The best example of this is systemic lupus erythematosus (SLE), which remains particularly vexing in both diagnosis and treatment. SLE, like AGS, is a disorder of nucleic acid detection, characterized by autoantibodies specific for DNA, chromatin, and ribonucleoproteins. These autoantibodies are thought to be responsible for the variable symptoms of the disease, which include skin and joint inflammation, kidney pathology, and in some cases heart and brain involvement. Diagnostic criteria include 11 factors, any four of which are sufficient for SLE diagnosis in a particular individual (148). Thus, SLE can be considered a constellation of related disorders, and heterogeneity in clinical phenotype remains a key obstacle to understanding this disease. Moreover, despite much progress in defining innate immune mechanisms, the principal treatments that manage inflammation in SLE have remained the same for the past few decades and solely treat the symptoms of the disease, not its underlying mechanism(s).
Can AGS provide insight into SLE? One possibility is that the rare, isolated diseases described above are just that: isolated examples that are not informative for explaining either the genetics or the mechanisms of a common disease like SLE. This would mean that the mechanisms that regulate the antiviral response uncovered by these rare diseases similarly do not apply to SLE. However, such lack of overlap, genetically or mechanistically, seems unlikely, particularly in light of the identification and strong genetic association of TREX1 and RNASEH2 mutations with SLE (80, 149, 150), as well as a specific TREX1 mutation found in both AGS and SLE cases (150). Instead, we propose that the conceptual framework illuminated by rare interferonopathies not only applies to SLE, but also offers new insights that can inform clinical diagnosis, patient phenotyping, ongoing genetic studies, and the development and patient-specific utilization of new therapeutics.
SLE Genetics: Common versus rare alleles
SLE, while much more prevalent than the monogenic disorders described above, is not a very common disease: current estimates place the incidence of SLE at approximately 25–150 per 100,000 people, with large variation among geographic and ethnic populations and a 9:1 ratio of affected females to males (151). In contrast, type I diabetes is approximately ten times more prevalent than SLE in the United States (152). SLE has a major genetic component, with monozygotic twin concordance rates of up to 40% (153). However, most efforts to understand the genetic contributions to SLE have focused on the “common disease, common allele” framework (154), which holds that common diseases reflect the cumulative contributions of many common alleles, each of which has a small effect on the disease. In other words, each case of SLE is thought to be the summation of many “small” problems. For example: a slightly enhanced innate immune response plus a minor deficiency in T cell negative selection plus a marginal increase in sensitivity of B cell receptor signaling plus a small defect in the clearance of apoptotic cells. Such a model is not only difficult to prove genetically or experimentally, it also offers little insight into the design and implementation of novel therapies. As stated in a recent review about autoimmunity, “when the proposed solution to a problem is more complicated than the problem itself, there is often something wrong (155).”
Genome wide association studies (GWAS) of SLE have densely scanned patient and control populations for common alleles associated with disease. The single nucleotide polymorphisms (SNPs) selected as “tags” for GWAS are considered “common” because they are generally present in the human population at frequencies of ~5% or greater. Because of their prevalence, most of these alleles are ancient, originating in humans greater than 50,000 years ago (156, 157). While over 40 genomic regions relating to expected innate and adaptive immune pathways have been implicated through GWAS (158), these loci account for less than a quarter of the proposed heritability of SLE, and the functional consequences of most identified SNPs remain uncharacterized. The question remains: where is the missing genetic contribution to SLE? Will more GWAS in more patients slowly chip away at the remaining 75% of unexplained genetics? Or is there another solution to the complexity of SLE genetics that cannot be found among common allelic variation?
What if each case of SLE, instead of being a conglomeration of small problems with many disparate pathways, is more like the monogenic interferonopathies in that it is driven by one big problem with one pathway? What if the relatively “common” occurrence of SLE reflects the number of individual pathways that can lead to SLE when compromised, particularly since we now know from the monogenic interferonopathies that multiple pathways can converge on a similar clinical presentation? What if the underlying genetics of SLE reflect the contributions of rare alleles with a strong effect on disease? Lastly, what if these alleles are recent and not ancient? Importantly, such alleles would be invisible to GWAS because of their rarity in the human population and lack of consistent linkage to more common “tag” SNPs (157). A similar view has recently been proposed and clearly articulated by others (159). In light of the evolutionary model described above that rationalizes the existence of alleles that strongly predispose to autoimmunity, it is worth considering this possibility and its implications in more detail. Specifically, we will discuss the IFN signature of SLE in the context of the “one big problem with one pathway” framework, the potential intersection of genetics with specific environmental triggers of SLE, the need for careful, patient-specific clinical phenotyping to enrich emerging genetic data, and finally the implications for development and implementation of new therapies. Progress in each of these areas will inform the others and will provide a clearer framework for treating SLE.
The sources of the IFN signature in SLE: finite choices
One key feature of SLE that overlaps with the monogenic interferonopathies is the fact that most SLE patients have a detectable IFN signature that can be sampled in their peripheral blood (160, 161). The definition of IFN-inducing innate immune sensors over the past 16 years has revealed that there are three – and likely only three – specific sensing pathways that activate IFN production: cGAS-STING, RLR-MAVS, or TLRs (TLRs 3, 4, 7/8, 9). A fourth mechanism, illuminated by rare human mutations in ISG15 and USP18 (134, 135), is that a failure to control IFN receptor signaling leads to overamplification of IFN production that is not initiated by a specific innate immune pathway. We propose that the IFN signature in each SLE patient will likely be driven by one – and only one – of these pathways. Knowing which pathway is responsible for disease in each patient will transform our understanding of how to treat SLE, as discussed below.
The intersection of genetics and environmental triggers
Despite the high monozygotic twin concordance and familial segregation of SLE, environmental factors strongly influence the development of disease in susceptible individuals. Since SLE is a disorder of nucleic acid detection, virus infections are attractive candidates for environmental triggers of disease. A number of viruses that are common in the human population, including Epstein-Barr Virus (EBV), have been proposed to contribute to development of SLE (162, 163). One difficulty in assigning causality for a common virus like EBV is the fact that EBV is nearly ubiquitous in the human population (164), but only a small fraction of humans develop SLE. New insights into the finite sources of the IFN signature may offer a new way to consider environmental triggers of SLE as pathway-specific instigators of disease. Put simply, individuals with a hyperactive cGAS-STING pathway might progress to disease in response to infections that are distinct from those that trigger disease in individuals with an enhanced MDA5-MAVS pathway (Figure 3A). In the case of cGAS-STING, such triggers may include EBV, human cytomegalovirus (HCMV), or other DNA viruses. For MDA5-MAVS, triggers may be restricted to those viruses for which this specific pathway is important, including common picornaviruses like the enteroviruses (28). In each case, the dysregulated, pathway-specific antiviral response could result in heightened IFN production, increased presentation of self antigens in a prolonged inflammatory setting, and enhanced activation of lymphocytes.
Figure 3: Pathway-specific and generic triggers of autoimmune disease.
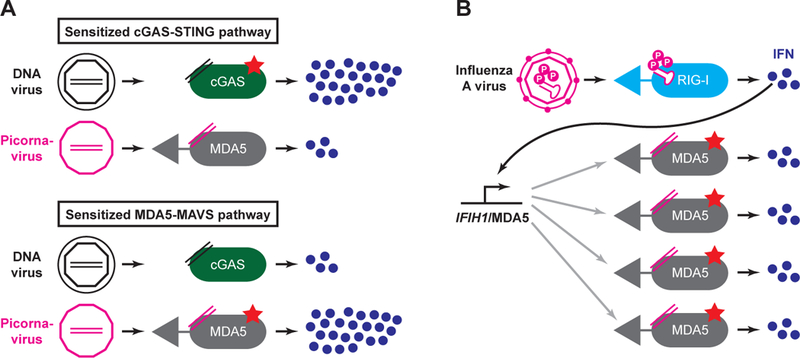
A: In individuals with a sensitized cGAS-STING pathway, the enhanced response to DNA viruses might predispose to autoimmunity, but the normal response to infection with picornavirus, an RNA virus, might not. Conversely, individuals with a sensitized MDA5 pathway may be triggered by picornavirus infection, not DNA virus infection. B: In individuals with a sensitized MDA5 pathway, an infection that causes systemic type I IFN production may increase the abundance of MDA5 within cells, leading to autoreactivity against endogenous ligands.
An alternative possibility is that a “generic” infection that causes a systemic type I IFN response could serve as a trigger for pathway-specific autoimmune disease (Figure 3B). For example, the IFIH1 gene that encodes MDA5 is a highly IFN-inducible gene that is maintained at low levels in uninfected cells (28). Mutations that sensitize the MDA5 pathway could become particularly hyperactive in the context of IFN-mediated upregulation, such that the increased expression of MDA5 would drive more sensitive detection of endogenous RNA ligands and instigate autoreactivity. In this scenario, the trigger virus does not necessarily need to be matched to the sensitized pathway in each patient, and a virus like Influenza A virus (which is primarily sensed by RIG-I and not MDA5; 28), could still unleash MDA5-mediated autoreactivity through a systemic IFN response (Figure 3B).
SLE patient clinical phenotyping
As discussed above, SLE is characterized by marked clinical heterogeneity, exemplified by the 11 criteria for SLE diagnosis that show considerable variability among patients (148). Moreover, SLE is a chronic, relapsing disease in which certain clinical symptoms appear and then resolve, only to flare again at a later date (165). If each case of SLE represents a strong predisposition caused by one of a finite number of possibilities, it is possible that the clinical phenotypes caused by mutations in each pathway might share certain features that may allow more accurate connection of genotype to phenotype, with important predictive power in both directions.
The phenotypic parameters that are simplest in principle are unique and easily accessible biomarkers that would identify pathway-specific dysregulation. The most promising of such biomarkers is the cGAMP product that is formed by cGAS after detection of DNA (36). Quantitation of cGAMP abundance by mass spectrometry is relatively straightforward and sensitive, and mouse models of AGS have demonstrated markedly enhanced cGAMP abundance in affected tissues (67, 68). Adaptation of this protocol to a clinical test on peripheral blood samples holds considerable promise for identifying patients with elevated cGAMP levels that would strongly implicate the cGAS-STING pathway as an important determinant of the disease process in this subset of individuals.
Apart from the clear diagnostic utility of cGAMP measurements, there are no currently known additional biomarkers that would unambiguously identify any of the remaining nucleic acid pathways. The gene expression profiles induced by the RLR-MAVS pathways and the cGAS-STING pathways are remarkably similar (33), and the IFN and inflammatory signatures identified in peripheral blood can be shared to some extent by all of the pathways described above. However, using differential gene expression as a tool to classify patients into groups can enable stratification of disease states, as evidenced by a recent longitudinal study of 158 SLE cases (166). Combining these sophisticated analyses with deeper genetic profiling of patients will pave the way for new breakthroughs in molecular diagnosis of SLE.
Another potential source of pathway-specific biomarkers would be revealed if the endogenous nucleic acids that trigger each pathway can be unambiguously identified and reliably measured. For example, in the case of ADAR1, the extent of editing of specific (and currently incompletely characterized) RNAs could provide a means to identify insufficient RNA editing as a biomarker that would directly implicate the MDA5-MAVS pathway as a key contributor to specific cases of disease. For Trex1, RNase H2, and SAMHD1, accumulated cytosolic DNAs, perhaps of specific and restricted origins, could also be measured. For extracellular enzymes like DNase1L3, a clinical test for degradation of microparticle DNA using serum samples from SLE patients, like the prototype described in a recent study (131), could implicate dysregulation of extracellular DNA metabolism in a subset of patients. Although assays to measure the instigating nucleic acids as a specific proxy of disease remain hypothetical for now, the potential utility of such diagnostic tools highlights the need for further study of the sources of endogenous ligands for antiviral nucleic acid sensors.
Implications for therapies
In the past 50 years, only a single new drug has been approved by the FDA for the treatment of SLE: belimumab, which is a humanized monoclonal antibody that blocks Blys/BAFF (167). With the rapid advancements in our understanding of the innate immune pathways that can contribute to SLE, more new drugs are now in trials for SLE than ever before. These include blockade of type I IFNs and inflammatory cytokines, and depletion of B cells (167). However, many obstacles remain. Perhaps the most daunting challenge is confronting the clinical heterogeneity of SLE and developing predictive models to determine which patients would benefit from which therapy. This is especially important for new clinical trials because if a small number of patients benefit from a specific therapy but the majority do not (or if some get worse), the trial will fail.
The framework developed by the study of AGS and related diseases offers a starting point for considering targets of new therapies and strategies for implementing them. Restating our key prediction, we propose that each case of SLE is likely caused by dysregulation of one of a small number of finite innate immune pathways. Advances in understanding the genetics of SLE, together with patient phenotyping and identification of new biomarkers that implicate specific pathways, will offer new avenues for creating therapies. Of course, the question of whether such new therapies would ameliorate disease, particularly in the context of established inflammation, is another key obstacle. Perhaps more importantly, the confluence of these areas will enable selective treatment of those patients who are most likely to benefit from a specific therapy.
One illustrative example, hinted at by several mouse studies, underscores the importance of knowing which pathway is dysregulated in a particular disease state. The cGAS-STING pathway drives the type I IFN signature in mouse models of three AGS enzymes: Trex1, RNase H2, and SAMHD1 (61, 66–68, 78, 79, 92, 168). In the case of Trex1-deficient mice, both cGAS and STING are essential for all aspects of the autoimmune disease (66–68). These findings make cGAS a tantalizing therapeutic target that could benefit patients with mutations in five of the seven known AGS genes. However, in three separate mouse models of autoimmunity, genetic ablation of the cGAS-STING pathway not only has no positive effect, it actually makes the disease worse. First, deleting Tmem173 (STING) in the MRL.Faslpr mouse model of SLE, which is driven primarily by TLRs (22), worsens clinical symptoms, increases autoantibody titers, and accelerates mortality (169). Second, crossing Adar-deficient mice, in which the IFN response is driven exclusively by MDA5-MAVS (103, 104), to Tmem173−/− mice results in an exacerbated IFN signature compared to plain Adar−/− embryos (104). Finally, autoantibodies in DNase1L3-deficient mice, which are MyD88-dependent, are elevated in Dnase1l3−/−;Tmem173−/− mice (131). These studies illuminate an important point regarding the potential for cGAS-targeted therapies: they should only be used when disease is cGAS-dependent. Treating the wrong patients with an otherwise promising drug might actually worsen disease, which further emphasizes the need to link genetics to pathways and clinical phenotypes in SLE.
Concluding Remarks
We have attempted to highlight the remarkable recent progress that has been made in the identification of nucleic acid sensing pathways, together with rare human diseases caused by their inappropriate activation. As we learn more about the genetics and mechanisms of rare human interferonopathies, we will be able to apply these lessons to the study of more common autoimmune diseases in which type I IFNs play a central role.
Acknowledgements
We are grateful to Yanick Crow and to members of the Stetson lab for helpful discussions. DBS is a scholar of the Rita Allen Foundation, a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease, a Howard Hughes Medical Institute Faculty Scholar, and a past recipient of a Lupus Research Institute Novel Research Grant. EEG was supported by a Cancer Research Institute Irvington postdoctoral fellowship. HEV was supported by a Jane Coffin Childs postdoctoral fellowship.
Footnotes
Disclosure Statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1.Burnet FMS. 1969. Self and not-self. Melbourne: Melbourne University Press; VIII, 318 S pp. [Google Scholar]
- 2.Janeway CA Jr., 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54 Pt 1: 1–13 [DOI] [PubMed] [Google Scholar]
- 3.Barbalat R, Ewald SE, Mouchess ML, Barton GM. 2011. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29: 185–214 [DOI] [PubMed] [Google Scholar]
- 4.Goubau D, Deddouche S, Reis ESC. 2013. Cytosolic sensing of viruses. Immunity 38: 855–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408: 740–5 [DOI] [PubMed] [Google Scholar]
- 6.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413: 732–8 [DOI] [PubMed] [Google Scholar]
- 7.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. 2002. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 3: 196–200 [DOI] [PubMed] [Google Scholar]
- 8.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303: 1526–9 [DOI] [PubMed] [Google Scholar]
- 9.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303: 1529–31 [DOI] [PubMed] [Google Scholar]
- 10.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101: 5598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda K, Kaisho T, Akira S. 2003. Toll-like receptors. Annu Rev Immunol 21: 335–76 [DOI] [PubMed] [Google Scholar]
- 12.Iwasaki A, Medzhitov R. 2004. Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987–95 [DOI] [PubMed] [Google Scholar]
- 13.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7: 156–64 [DOI] [PubMed] [Google Scholar]
- 14.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. 2013. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2: e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyake K, Shibata T, Ohto U, Shimizu T. 2016. Emerging roles of the processing of nucleic acids and Toll-like receptors in innate immune responses to nucleic acids. J Leukoc Biol [DOI] [PubMed] [Google Scholar]
- 16.Barton GM, Kagan JC, Medzhitov R. 2006. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 7: 49–56 [DOI] [PubMed] [Google Scholar]
- 17.Mouchess ML, Arpaia N, Souza G, Barbalat R, Ewald SE, Lau L, Barton GM. 2011. Transmembrane mutations in Toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity 35: 721–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. 2008. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456: 658–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434: 1035–40 [DOI] [PubMed] [Google Scholar]
- 20.Pichlmair A, Reis e Sousa C. 2007. Innate recognition of viruses. Immunity 27: 370–83 [DOI] [PubMed] [Google Scholar]
- 21.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416: 603–7 [DOI] [PubMed] [Google Scholar]
- 22.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. 2006. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25: 417–28 [DOI] [PubMed] [Google Scholar]
- 23.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. 2013. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity 38: 528–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–7 [DOI] [PubMed] [Google Scholar]
- 25.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5’-Triphosphate RNA is the ligand for RIG-I. Science 314: 994–7 [DOI] [PubMed] [Google Scholar]
- 26.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314: 997–1001 [DOI] [PubMed] [Google Scholar]
- 27.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis e Sousa C. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 514: 372–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441: 101–5 [DOI] [PubMed] [Google Scholar]
- 29.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669–82 [DOI] [PubMed] [Google Scholar]
- 30.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature [DOI] [PubMed] [Google Scholar]
- 31.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA Is an Adapter Protein Required for Virus-Triggered IFN-beta Signaling. Mol Cell 19: 727–40 [DOI] [PubMed] [Google Scholar]
- 32.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6: 981–8 [DOI] [PubMed] [Google Scholar]
- 33.Stetson DB, Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24: 93–103 [DOI] [PubMed] [Google Scholar]
- 34.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. 2006. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7: 40–8 [DOI] [PubMed] [Google Scholar]
- 35.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. 2013. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153: 1094–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498: 380–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3: 1355–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29: 538–50 [DOI] [PubMed] [Google Scholar]
- 42.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461: 788–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341: 903–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Q, Sun L, Chen ZJ. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17: 1142–9 [DOI] [PubMed] [Google Scholar]
- 45.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. 2005. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev 204: 27–42 [DOI] [PubMed] [Google Scholar]
- 46.Marshak-Rothstein A, Rifkin IR. 2007. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol 25: 419–41 [DOI] [PubMed] [Google Scholar]
- 47.Christensen SR, Shlomchik MJ. 2007. Regulation of lupus-related autoantibody production and clinical disease by Toll-like receptors. Semin Immunol 19: 11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosen A, Casciola-Rosen L. 2016. Autoantigens as Partners in Initiation and Propagation of Autoimmune Rheumatic Diseases. Annu Rev Immunol 34: 395–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crow YJ. 2011. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci 1238: 91–8 [DOI] [PubMed] [Google Scholar]
- 50.de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. 2015. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 33: 823–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crow YJ, Manel N. 2015. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–40 [DOI] [PubMed] [Google Scholar]
- 52.Roers A, Hiller B, Hornung V. 2016. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 44: 739–54 [DOI] [PubMed] [Google Scholar]
- 53.Aicardi J, Goutieres F. 1984. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol 15: 49–54 [DOI] [PubMed] [Google Scholar]
- 54.Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. 1988. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci 84: 201–8 [DOI] [PubMed] [Google Scholar]
- 55.Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, et al. 2015. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A: 296–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livingston JH, Crow YJ. 2016. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutieres Syndrome and Beyond. Neuropediatrics [DOI] [PubMed] [Google Scholar]
- 57.Lindahl T, Gally JA, Edelman GM. 1969. Properties of deoxyribonuclease 3 from mammalian tissues. J Biol Chem 244: 5014–9 [PubMed] [Google Scholar]
- 58.Hoss M, Robins P, Naven TJ, Pappin DJ, Sgouros J, Lindahl T. 1999. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. Embo J 18: 3868–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazur DJ, Perrino FW. 1999. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3’-->5’ exonucleases. J Biol Chem 274: 19655–60 [DOI] [PubMed] [Google Scholar]
- 60.Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J. 2006. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23: 133–42 [DOI] [PubMed] [Google Scholar]
- 61.Stetson DB, Ko JS, Heidmann T, Medzhitov R. 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134: 587–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mazur DJ, Perrino FW. 2001. Excision of 3’ termini by the Trex1 and TREX2 3’-->5’ exonucleases. Characterization of the recombinant proteins. J Biol Chem 276: 17022–9 [DOI] [PubMed] [Google Scholar]
- 63.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. 2008. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem 283: 31649–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. 2006. Mutations in the gene encoding the 3’−5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38: 917–20 [DOI] [PubMed] [Google Scholar]
- 65.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. 2007. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet 80: 811–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr.., Barber GN, Stetson DB 2012. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity 36: 120–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gray EE, Treuting PM, Woodward JJ, Stetson DB. 2015. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol 195: 1939–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, Chen ZJ. 2015. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112: E5699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 11: 1005–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beck-Engeser GB, Eilat D, Wabl M. 2011. An autoimmune disease prevented by anti-retroviral drugs. Retrovirology 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang YG, Lindahl T, Barnes DE. 2007. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 131: 873–86 [DOI] [PubMed] [Google Scholar]
- 72.Ahn J, Ruiz P, Barber GN. 2014. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J Immunol 193: 4634–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, Li QZ, Atkinson JP, Morse HC 3rd, Lehrman MA, Yan N. 2015. Cytosolic Nuclease TREX1 Regulates Oligosaccharyltransferase Activity Independent of Nuclease Activity to Suppress Immune Activation. Immunity 43: 463–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. 2006. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–6 [DOI] [PubMed] [Google Scholar]
- 75.Perrino FW, Harvey S, Shaban NM, Hollis T. 2009. RNaseH2 mutants that cause Aicardi-Goutieres syndrome are active nucleases. J Mol Med 87: 25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. 2012. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, Kilanowski F, Grimes G, Fluteau A, Devenney PS, Hill RE, Reijns MA, Jackson AP. 2016. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35: 831–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ. 2016. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med 213: 329–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, Tungler V, Chara O, Lee YA, Hubner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL, Cunninghame Graham DS, Bubeck D, Leitch A, Ralston SH, Blackburn EA, Gahr M, Witte T, Vyse TJ, Melchers I, Mangold E, Nothen MM, Aringer M, Kuhn A, Luthke K, Unger L, Bley A, Lorenzi A, Isaacs JD, Alexopoulou D, Conrad K, Dahl A, Roers A, Alarcon-Riquelme ME, Jackson AP, Lee-Kirsch MA. 2015. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. 2009. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474: 654–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474: 658–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480: 379–82 [DOI] [PubMed] [Google Scholar]
- 85.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, Margottin-Goguet F. 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 13: 223–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, Yakunin AF. 2013. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J Biol Chem 288: 8101–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim SY, Seo D, Kim J, White TE, Brandariz-Nunez A, Diaz-Griffero F, Yun CH, Hollenbaugh JA, Kim B, Baek D, Ahn K. 2014. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med 20: 936–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi J, Ryoo J, Oh C, Hwang S, Ahn K. 2015. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 12: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ryoo J, Hwang SY, Choi J, Oh C, Ahn K. 2016. SAMHD1, the Aicardi-Goutieres syndrome gene and retroviral restriction factor, is a phosphorolytic ribonuclease rather than a hydrolytic ribonuclease. Biochem Biophys Res Commun 477: 977–81 [DOI] [PubMed] [Google Scholar]
- 90.Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, Stivers JT. 2015. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res 43: 6486–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antonucci JM, St Gelais C, de Silva S, Yount JS, Tang C, Ji X, Shepard C, Xiong Y, Kim B, Wu L. 2016. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat Med 22: 1072–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel J. 2016. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep 16: 1492–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, Bieniasz PD, Towers GJ, Moita LF, Crow YJ, Bonthron DT, Reis e Sousa C. 2013. SAMHD1-dependent retroviral control and escape in mice. Embo J 32: 2454–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, Wei W, Evans SL, Li L, Zhang W, Cheung LE, Wang G, Kazazian HH, Jr., Yu XF. 2013. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep 4: 1108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M, Luksch H, Nguyen LA, Kim B, Alexopoulou D, Dahl A, Rapp A, Cardoso MC, Shevchenko A, Lee-Kirsch MA. 2014. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, Wright Colopy G, Dreau H, Sigaux F, Judde JG, Rotger M, Telenti A, Lin YL, Pasero P, Maelfait J, Titsias M, Cohen DR, Henderson SJ, Ross MT, Bentley D, Hillmen P, Pettitt A, Rehwinkel J, Knight SJ, Taylor JC, Crow YJ, Benkirane M, Schuh A. 2014. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 123: 1021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, Vartanov A, Fernandes SM, Saksena G, Cibulskis K, Tesar B, Gabriel S, Hacohen N, Meyerson M, Lander ES, Neuberg D, Brown JR, Getz G, Wu CJ. 2013. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W Jr., Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bass BL. 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Patterson JB, Samuel CE. 1995. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15: 5376–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O’Connell MA. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43: 933–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. 2004. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res 14: 1719–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goodier JL, Kazazian HH Jr., 2008. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135: 23–35 [DOI] [PubMed] [Google Scholar]
- 107.Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, Savage DA, Walker NM, Clayton DG, Todd JA. 2006. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38: 617–9 [DOI] [PubMed] [Google Scholar]
- 108.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jonsen A, Bengtsson AA, Rantapaa-Dahlqvist S, Baechler EC, Brown EE, Alarcon GS, Edberg JC, Ramsey-Goldman R, McGwin G Jr., , Reveille JD, Vila LM, Kimberly RP, Manzi S, Petri MA, Lee A, Gregersen PK, Seldin MF, Ronnblom L, Criswell LA, Syvanen AC, Behrens TW, Graham RR. 2009. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41: 1228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. 2009. Rare Variants of IFIH1, a Gene Implicated in Antiviral Responses, Protect Against Type 1 Diabetes. Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sutherland A, Davies J, Owen CJ, Vaikkakara S, Walker C, Cheetham TD, James RA, Perros P, Donaldson PT, Cordell HJ, Quinton R, Pearce SH. 2007. Genomic polymorphism at the interferon-induced helicase (IFIH1) locus contributes to Graves’ disease susceptibility. J Clin Endocrinol Metab 92: 3338–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR, Lebon P, Li C, Livingston JH, Lourenco CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O’Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ. 2014. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46: 503–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–26 [DOI] [PubMed] [Google Scholar]
- 113.Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, Hartley JL, Crow YJ, Stetson DB. 2014. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 15: 839–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fabre A, Charroux B, Martinez-Vinson C, Roquelaure B, Odul E, Sayar E, Smith H, Colomb V, Andre N, Hugot JP, Goulet O, Lacoste C, Sarles J, Royet J, Levy N, Badens C. 2012. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet 90: 689–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, Nagata S. 2001. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292: 1546–9 [DOI] [PubMed] [Google Scholar]
- 116.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. 2005. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 6: 49–56 [DOI] [PubMed] [Google Scholar]
- 117.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. 2005. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 202: 1333–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ahn J, Gutman D, Saijo S, Barber GN. 2012. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A 109: 19386–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Singleton EB, Merten DF. 1973. An unusual syndrome of widened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr Radiol 1: 2–7 [DOI] [PubMed] [Google Scholar]
- 120.Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, Lee J, Jeong YM, Kim CH, Kim OH, Sohn S, Nam SH, Hong Y, Lee YS, Chang SA, Jang SY, Kim JW, Lee MS, Lim SY, Sung KS, Park KT, Kim BJ, Lee JH, Kim DK, Kee C, Ki CS. 2015. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am J Hum Genet 96: 266–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, Nurnberg P, Hohne W, Crow YJ, Feigenbaum A, Hennekam RC. 2015. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet 96: 275–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O’Sullivan J, Williams SG, Bodemer C, Fraitag S, Gebhard F, Leheup B, Lemelle I, Oojageer A, Raffo E, Schmitt E, Rice GI, Hur S, Crow YJ. 2015. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutieres and Singleton-Merten syndromes. Br J Dermatol 173: 1505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. 2014. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371: 507–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, Blanche S, Picard C, Rice GI, Crow YJ, Manel N, Fischer A, Bader-Meunier B, Rieux-Laucat F. 2014. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 124: 5516–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Konig N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tungler V, Alvarez HA, Chara O, Engel K, Goldbach-Mansky R, Gunther C, Lee-Kirsch MA. 2016. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis [DOI] [PubMed] [Google Scholar]
- 126.Kim H, Sanchez GA, Goldbach-Mansky R. 2016. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J Mol Med (Berl) 94: 1111–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. 2000. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25: 177–81 [DOI] [PubMed] [Google Scholar]
- 128.Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y. 2001. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 28: 313–4 [DOI] [PubMed] [Google Scholar]
- 129.Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, Al Sonbul A, Sewairi W, Qari A, Abdallah E, Al-Owain M, Al Motywee S, Al-Rayes H, Hashem M, Khalak H, Al-Jebali L, Alkuraya FS. 2011. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 43: 1186–8 [DOI] [PubMed] [Google Scholar]
- 130.Ozcakar ZB, Foster J 2nd, , Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, Tekin M. 2013. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum 65: 2183–9 [DOI] [PubMed] [Google Scholar]
- 131.Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, Rashidfarrokhi A, Yeste A, Panea C, Chida AS, Bogunovic M, Ivanov II, Quintana FJ, Sanz I, Elkon KB, Tekin M, Yalcinkaya F, Cardozo TJ, Clancy RM, Buyon JP, Reizis B. 2016. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 166: 88–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, Baskar K, Baskar S, Baudouin V, Beresford MW, Black GC, Dearman RJ, de Zegher F, Foster ES, Frances C, Hayman AR, Hilton E, Job-Deslandre C, Kulkarni ML, Le Merrer M, Linglart A, Lovell SC, Maurer K, Musset L, Navarro V, Picard C, Puel A, Rieux-Laucat F, Roifman CM, Scholl-Burgi S, Smith N, Szynkiewicz M, Wiedeman A, Wouters C, Zeef LA, Casanova JL, Elkon KB, Janckila A, Lebon P, Crow YJ. 2011. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 43: 127–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.An J, Briggs TA, Dumax-Vorzet A, Alarcon-Riquelme ME, Belot A, Beresford M, Bruce I, Carvalho C, Chaperot L, Frostegard J, Plumas J, Rice GI, Vyse TJ, Wiedeman A, Crow YJ, Elkon KB. 2016. Tartrate-Resistant Acid Phosphatase Deficiency in the Predisposition to Systemic Lupus Erythematosus. Arthritis Rheumatol [DOI] [PubMed] [Google Scholar]
- 134.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uze G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, Garcia-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S. 2015. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 517: 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, Lequin MH, Kros JM, Stam W, Hermann M, Willemsen R, Brouwer RW, Van IWF, Martin-Fernandez M, de Coo I, Dudink J, de Vries FA, Bertoli Avella A, Prinz M, Crow YJ, Verheijen FW, Pellegrini S, Bogunovic D, Mancini GM. 2016. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 213: 1163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McGonagle D, McDermott MF. 2006. A proposed classification of the immunological diseases. PLoS Med 3: e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Masters SL, Simon A, Aksentijevich I, Kastner DL. 2009. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 27: 621–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, Soroosh P, Watford WT, O’Shea JJ, Kastner DL, Hoffman HM. 2009. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 30: 875–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cuadrado E, Vanderver A, Brown KJ, Sandza A, Takanohashi A, Jansen MH, Anink J, Herron B, Orcesi S, Olivieri I, Rice GI, Aronica E, Lebon P, Crow YJ, Hol EM, Kuijpers TW. 2015. Aicardi-Goutieres syndrome harbours abundant systemic and brain-reactive autoantibodies. Ann Rheum Dis 74: 1931–9 [DOI] [PubMed] [Google Scholar]
- 140.Daugherty MD, Malik HS. 2012. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46: 677–700 [DOI] [PubMed] [Google Scholar]
- 141.Hancks DC, Hartley MK, Hagan C, Clark NL, Elde NC. 2015. Overlapping Patterns of Rapid Evolution in the Nucleic Acid Sensors cGAS and OAS1 Suggest a Common Mechanism of Pathogen Antagonism and Escape. PLoS Genet 11: e1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mozzi A, Pontremoli C, Forni D, Clerici M, Pozzoli U, Bresolin N, Cagliani R, Sironi M. 2015. OASes and STING: adaptive evolution in concert. Genome Biol Evol 7: 1016–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lemos de Matos A, McFadden G, Esteves PJ. 2013. Positive evolutionary selection on the RIG-I-like receptor genes in mammals. PLoS One 8: e81864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Patel MR, Loo YM, Horner SM, Gale M Jr., Malik HS. 2012. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol 10: e1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Elde NC, Child SJ, Geballe AP, Malik HS. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457: 485–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Compton AA, Malik HS, Emerman M. 2013. Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos Trans R Soc Lond B Biol Sci 368: 20120496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kaiser SM, Malik HS, Emerman M. 2007. Restriction of an extinct retrovirus by the human TRIM5alpha antiviral protein. Science 316: 1756–8 [DOI] [PubMed] [Google Scholar]
- 148.Hochberg MC. 1997. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40: 1725. [DOI] [PubMed] [Google Scholar]
- 149.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. 2007. Mutations in the gene encoding the 3’−5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 39: 1065–7 [DOI] [PubMed] [Google Scholar]
- 150.Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP. 2011. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun 12: 270–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Danchenko N, Satia JA, Anthony MS. 2006. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus 15: 308–18 [DOI] [PubMed] [Google Scholar]
- 152.Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. 2010. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 39: 481–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM. 1992. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum 35: 311–8 [DOI] [PubMed] [Google Scholar]
- 154.Risch N, Merikangas K. 1996. The future of genetic studies of complex human diseases. Science 273: 1516–7 [DOI] [PubMed] [Google Scholar]
- 155.Anderson MS, Casanova JL. 2015. More than Meets the Eye: Monogenic Autoimmunity Strikes Again. Immunity 42: 986–8 [DOI] [PubMed] [Google Scholar]
- 156.Cavalli-Sforza LL, Feldman MW. 2003. The application of molecular genetic approaches to the study of human evolution. Nat Genet 33 Suppl: 266–75 [DOI] [PubMed] [Google Scholar]
- 157.McClellan J, King MC. 2010. Genetic heterogeneity in human disease. Cell 141: 210–7 [DOI] [PubMed] [Google Scholar]
- 158.Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, Ma J, Qi YY, Kim-Howard X, Motghare P, Bhattarai K, Adler A, Bang SY, Lee HS, Kim TH, Kang YM, Suh CH, Chung WT, Park YB, Choe JY, Shim SC, Kochi Y, Suzuki A, Kubo M, Sumida T, Yamamoto K, Lee SS, Kim YJ, Han BG, Dozmorov M, Kaufman KM, Wren JD, Harley JB, Shen N, Chua KH, Zhang H, Bae SC, Nath SK. 2016. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet 48: 323–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crow YJ. 2011. Lupus: how much “complexity” is really (just) genetic heterogeneity? Arthritis Rheum 63: 3661–4 [DOI] [PubMed] [Google Scholar]
- 160.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100: 2610–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197: 711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. 2005. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 11: 85–9 [DOI] [PubMed] [Google Scholar]
- 163.James JA, Harley JB, Scofield RH. 2006. Epstein-Barr virus and systemic lupus erythematosus. Curr Opin Rheumatol 18: 462–7 [DOI] [PubMed] [Google Scholar]
- 164.Gratama JW, Ernberg I. 1995. Molecular epidemiology of Epstein-Barr virus infection. Adv Cancer Res 67: 197–255 [PubMed] [Google Scholar]
- 165.Merrill JT, Buyon JP, Utset T. 2014. A 2014 update on the management of patients with systemic lupus erythematosus. Semin Arthritis Rheum 44: e1–2 [DOI] [PubMed] [Google Scholar]
- 166.Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, Cepika AM, Acs P, Turner J, Anguiano E, Vinod P, Khan S, Obermoser G, Blankenship D, Wakeland E, Nassi L, Gotte A, Punaro M, Liu YJ, Banchereau J, Rossello-Urgell J, Wright T, Pascual V. 2016. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165: 1548–50 [DOI] [PubMed] [Google Scholar]
- 167.Borba HH, Funke A, Wiens A, Utiyama SR, Perlin CM, Pontarolo R. 2016. Update on Biologic Therapies for Systemic Lupus Erythematosus. Curr Rheumatol Rep 18: 44. [DOI] [PubMed] [Google Scholar]
- 168.Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V. 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192: 5993–7 [DOI] [PubMed] [Google Scholar]
- 169.Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, Huyler AH, Nundel K, Mohan C, Berg LJ, Shlomchik MJ, Marshak-Rothstein A, Fitzgerald KA. 2015. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci U S A 112: E710–7 [DOI] [PMC free article] [PubMed] [Google Scholar]