

Abstract
Rapamycin and FK506 are macrocyclic natural products with an extraordinary mode of action—they form binary complexes with FKBP through a shared FKBP-binding domain before forming ternary complexes with their respective targets, mTOR and calcineurin, respectively. Inspired by this, we sought to build a rapamycin-like macromolecule library to target new cellular proteins by replacing the effector domain of rapamycin with a combinatorial library of oligopeptides. We developed a robust macrocyclization method using ring-closing metathesis and synthesized a 45,000-compound library of hybrid macrocycles that are named rapafucins using optimized FKBP-binding domains. Screening of the rapafucin library in human cells led to the discovery of rapadocin, an inhibitor of nucleoside uptake. Rapadocin is a potent, isoform-specific and FKBP-dependent inhibitor of the equilibrative nucleoside transporter 1 and is efficacious in an animal model of kidney ischemia reperfusion injury. Together, these results demonstrate that rapafucins are a new class of chemical probes and drug leads that can expand the repertoire of protein targets well beyond mTOR and calcineurin.
Graphical abstract
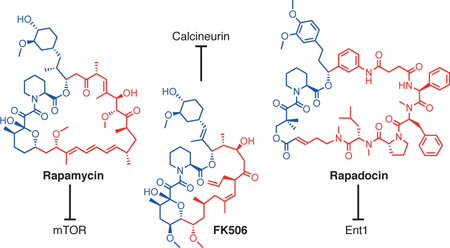
Nature is a bountiful source of bioactive small molecules that display a dizzying array of cellular activities thanks to the evolution process over billions of years. Rapamycin and FK506 comprise a unique structural family of macrocyclic natural products with an extraordinary mode of action1,2. Upon entering cells, both compounds form binary complexes with FKBP12 as well as other members of the FKBP family3,4. The FKBP12-rapamycin complex can then bind to mTOR and block its kinase activity towards downstream substrates such as p70S6K and 4E-BP, while the FKBP12-FK506 complex interacts with calcineurin, a protein phosphatase whose inhibition prevents calcium-dependent signaling and T cell activation5,6. The ability of rapamycin and FK506 to bind FKBPs confers a number of advantages for their use as small molecule probes in biology as well as drugs in medicine. First, the binding of both rapamycin and FK506 to FKBP dramatically increases their effective sizes, allowing for allosteric blockade of substrates to the active sites of mTOR or calcineurin through indirect disruption of protein-protein interactions7–9. Second, the abundance and ubiquitous expression of intracellular FKBPs serves to enrich rapamycin and FK506 in the intracellular compartment and maintain their stability. Third, as macrocycles, FK506 and rapamycin are capable of more extensive interactions with proteins than smaller molecules independent of their ability to bind FKBP. Last but not least, the high-level expression of FKBPs in blood cells renders them reservoirs and carriers of the drugs for efficient delivery in vivo10. It is thus not surprising that both rapamycin and FK506 became widely used drugs in their natural forms without further chemical modifications.
Both rapamycin and FK506 can be divided into two structural and functional domains: an FKBP-binding domain (FKBD) and an effector domain that mediates interaction with mTOR or calcineurin, respectively (Fig. 1a). The structures of the FKBDs of rapamycin and FK506 are quite similar, but their effector domains are different, accounting for their exclusive target specificity. The presence of the separable and modular structural domains of FK506 and rapamycin have been extensively exploited to generate new analogs of both FK506 and rapamycin, including chemical inducers of dimerization and a large number of rapamycin analogs, known as rapalogs, to alter the specificity of rapamycin for mutated FKBP-rapamycin-binding domain of mTOR and to improve the toxicity and solubility profiles of rapamycin11–13. The existence of two distinct FKBD-containing macrocycles with distinct target specificity also raised the intriguing question of whether replacing the effector domains of rapamycin or FK506 could further expand the target repertoire of the resultant macrocycles. In their pioneering work, Chakraborty et al. synthesized several rapamycin-peptide hybrid molecules, which retained high affinity for FKBP but showed no biological activity14. More recently, we and others independently attempted to explore this possibility by making larger libraries of the FKBD-containing macrocycles. In one study, a much larger library of FKBD-containing macrocycles was made with a synthetic mimic of FKBD, but the resultant macrocycles suffered from a significant loss in binding affinity for FKBP12, likely accounting for the lack of bioactive compounds from that library15. Using a natural FKBD extracted from rapamycin, we also observed a significant loss in FKBP binding affinity upon formation of macrocycles (vide infra)16.
Figure 1 |. Design of rapafucin based on the scaffolds of rapamycin and FK506 and general synthetic route to rapafucin.
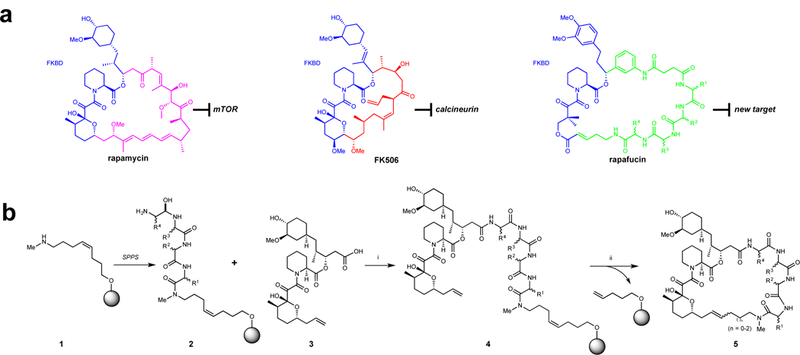
a, Rapafucin design inspired by rapamycin and FK506. Chemical structures of rapamycin and FK506. The effector domains of FK506 and rapamycin are colored red and magenta, respectively. The proposed new oligopeptide effector domain of rapafucin is colored green and the FKBD is colored blue in all of the structures. b, General synthetic route to rapafucins. Reaction conditions: SPPS (solid-phase peptide synthesis) see Supplementary Methods; (i) HATU, DIPEA, DMF, RT, 2h; (ii) Hoveyda-Grubbs catalyst II (30 mol%), 1,2-dichloroethane, 140°C, microwave, 0.5 h.
In a continuing effort to explore the possibility to using FKBD-containing macrocycles to target new proteins, we attempted to optimize and succeeded in identifying FKBDs that allowed for significant retention of binding affinity for FKBP12 upon incorporation into macrocycles. We also established a facile synthetic route for parallel synthesis of a large number of FKBD-containing macrocycles. Using the optimized FKBDs, we synthesized a 45,000-compound library of hybrid macrocycles named rapafucins. From this library, we identified rapadocin, a highly potent and isoform-specific inhibitor of human equilibrative nucleoside transporter (ENT) 1. We demonstrate that inhibition of ENT1 by rapadocin strongly depends on FKBP and resembles the general mode of action of rapamycin and FK506. Moreover, rapadocin treatment protects mice against ischemic reperfusion injury through inhibition of ENT1-mediated adenosine uptake and resulting enhancement of adenosine signaling, suggesting a potential new therapeutic strategy for modulating adenosine signaling.
Results
Synthetic route to rapafucins
We applied solid-phase peptide synthesis with a split-pool strategy17–19 to assemble the tetrapeptide effector domains (2, Fig. 1b). The pre-assembled FKBD (3, Fig. 1b)16 capped with a carboxylic acid at one end and an olefin at the other was subsequently coupled to the tetrapeptide that remained tethered on beads (4, Fig. 1b). To facilitate purification of the newly formed macrocycles, we adopted a coupled macrocyclization and cyclative release strategy whereby the macrocyclization is accompanied by the concurrent release of the macrocyclic products from the solid beads (Fig. 1b). After exploring different macrocyclization methods, we found ring-closing metathesis/cyclative release (RCM) to be a viable strategy for efficient parallel synthesis of different rapafucins.
Although the use of RCM coupled with cyclative release of resultant cyclic peptides has been previously reported20, it remained uncertain whether this strategy is compatible with the olefin-containing functional groups in the context of the rapafucin scaffold. Various combinations of alkene linkers (Fig. 2a) and the terminal alkene on the FKBD scaffold (Fig. 2b) were systematically evaluated. The alkene-containing linkers featured a terminal amino group for coupling with the first amino acid building block. We assessed a choice of different linker lengths and cis versus trans alkene configurations. As initial attempts with the trans-C5 alkene linker gave low to moderate yields for the RCM reaction, we turned to a series of cis alkene-containing linkers, including cis-C4, cis-C6 and cis-C8 (Fig. 2a). Among the different linkers, cis-C6 and cis-C8 linkers gave higher yields than the cis-C4 linker (Supplementary Table 1). Unlike the cis-C6 linker, however, the cis-C8 linkers often gave rise to a mixture of products that appeared to be caused by a shift of the alkene bond during the RCM reaction (Supplementary Fig.1). Thus, we chose the cis-C6 linker for future construction of rapafucin libraries.
Figure 2 |. Optimization of the linker on solid-support and FKBDs to be used for the synthesis of rapafucins.
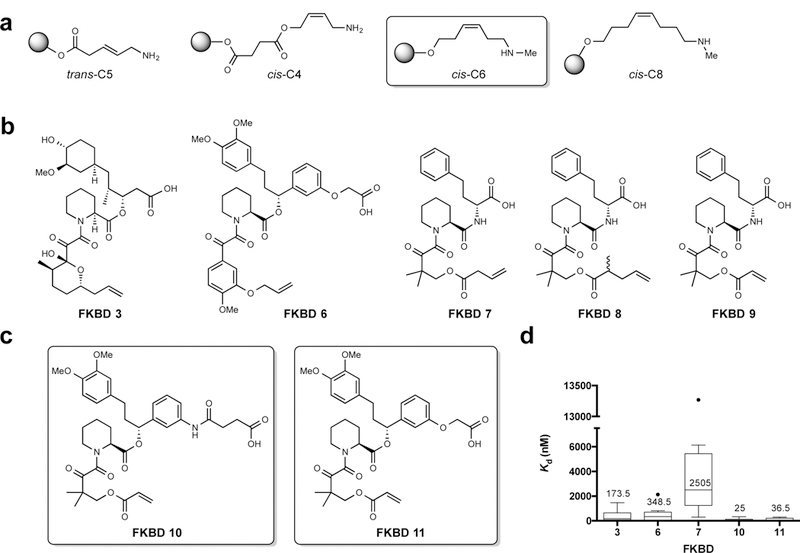
a, Alkene linkers with E or Z configuration and various lengths. Structure of the optimized cis-C6 linker is highlighted. b, Natural and synthetic FKBDs; synthetic FKBD cores 7–9 are coupled next to D-homoPhe on the solid-phase to mimic natural FKBD3. c, Structures of optimized FKBD10 and FKBD11. d Average Kd values for FKBP12 binding with rapafucins made from different FKBDs. n = 14 for FKBD 3, n = 7 for FKBD6, n = 8 for FKBD7, n = 18 for FKBD10, n = 7 for FKBD11, n indicates individual rapafucins. The data points of the second and the third quartiles (25–75%) are enclosed in the box with a band indicating the median. The upper and lower whiskers indicate the highest and lowest values that are not outliers. The outliers are data points with a value higher than 75 % plus 1.5x interquartile range (IQR) or lower than 25% minus 1.5x IQR in each data set. For individual Kd values, see Supplementary Table 4.
Use of the RCM reaction necessitated incorporation of a terminal olefinic group in the FKBD moiety. However, it was unclear at the outset which type of terminal olefins would be optimal. We thus tested several different types of FKBDs. In addition to the rapamycin-derived FKBD3, we also synthesized and tested four additional FKBD mimics (FKBD6–9) (Fig 2b). In all FKBDs, a pipecolic ketoamide fragment was kept, and the nonpolar tetrahydropyran moiety in the FKBD portion of rapamycin was mimicked by a substituted benzene ring (FKBD6) or a geminal dimethyl group (FKBD7-9). In FKBD7–9, the cyclohexyl moiety of natural FKBD is mimicked by D-homoPhe, which was pre-coupled onto the effector domain oligopeptide during solid-phase synthesis, and would lead to a replacement of the ester bond in natural FKBD3 with an amide linkage (Fig. 2b). The various terminal alkene moieties tested included allyl ether, vinylacetic ester, and acrylic ester (Fig. 2b). We found that the structures of the terminal alkene did not significantly affect the overall yields of the RCM reactions.
In addition to the alkene linkers, we also optimized temperature, catalyst loading, and reaction time for the RCM reaction (Supplementary Table 2). A relatively high microwave temperature (120–160 °C) is required to overcome the activation barrier of the RCM reaction (Supplementary Table 2, Entries a-d). However, excessive reaction temperatures (above 160 °C, entry d) or the presence of an excess amount of catalyst loading (50 mol%, entry g) resulted in the formation of undesired byproducts, as judged by HPLC analysis, which would hinder parallel purification of large numbers of compounds. We thus chose a combination of medium temperature and catalyst loading (140 °C, 30 mol% Hoveyda-Grubbs II catalyst, entry c) for the ensuing large-scale synthesis of rapafucin libraries.
Optimization of FKBDs
A key element for the design of novel rapafucins that mimic the mode of action of rapamycin and FK506 is that their shared FKBD should retain high affinity for FKBP independent of the effector domain structure. It has been shown that the ternary FKBP-FK506-calcineurin complex requires the prior formation of the FKBP-FK506 binary complex, and thus high binding affinity between FKBP and FK506 is essential for productive ternary complex formation and inhibition of calcineurin21. In a previous study, the use of a mimic of natural FKBD gave rise to macrocycles that suffered from a significant loss in FKBP binding affinity15. Independently, we began by employing the FKBD derived from rapamycin for the construction of the rapafucin libraries16. However, we subsequently found that the linearized FKBD3 derived from rapamycin had >300-fold lower affinity for FKBP12 than rapamycin itself (Kd values: 327 nM for FKBD3 vs 1 nM for rapamycin) (Supplementary Table 3). Incorporation of FKBD3 into macrocycles resulted in only a modest improvement in affinity for FKBP12 with an average Kd value of 174 nM (Fig. 2d, Supplementary Table 4), suggesting that FKBD3 is not suitable for new library construction. The incorporation of homo-Phe into FKBD7 gave rise to macrocycles with the lowest binding affinity for FKBP12 (Fig. 2d, Supplementary Table 4), ruling out FKBD7-9 as suitable building blocks for making rapafucin library. This observation is in agreement with a previous report and can be attributed to the replacement of the ester bond in FKBD with a more rigid amide moiety15.
A large number of FKBP ligands have been synthesized and characterized, among which a FKBD mimic with a high binding affinity for FKBP stood out and contains a unique meta-substituted phenyl ring linked to the FKBD pipecolate core22,23. We thus designed and synthesized FKBD6 (Fig. 2b) that, in comparison with linearized FKBD3, had significantly enhanced affinity for FKBP12 with a Kd of 62 nM (Supplementary Table 3). However, this improvement was largely lost upon incorporation into macrocycles (Fig. 2d). Next, we modified the lower portion of FKBD6, making two new linear FKBDs, FKBD10 and FKBD11 (Fig. 2c). To our delight, both FKBD10 and FKBD11 possessed high affinity for FKBP12, with Kd values of 4 and 11 nM, respectively (Supplementary Table 3). Importantly, this enhanced affinity was largely retained upon incorporation into macrocycles with average Kd values of 25 and 37 nM, respectively (Fig. 2d). Moreover, there was relatively low variation in binding affinity for FKBP12 among different macrocycles bearing FKBD10 and FKBD11 in comparison with those containing FKBD3 (Supplementary Table 4). These results suggested that both FKBD10 and FKBD11 are tolerant to different effector domain sequences, thus rendering them suitable FKBD building blocks for rapafucin libraries. We also established effective and scalable synthetic routes to both FKBD10 and FKBD11, which permit synthesis of large quantities of both FKBDs (Supplementary Fig. 2).
Synthesis of the rapafucin library
Using the cis-C6 linker and FKBD10/FKBD11 with the optimized microwave-assisted RCM reaction conditions, we next set out to synthesize rapafucin libraries (Fig. 1b). We decided to build rapafucin tetrapeptide effector domains using both natural and unnatural amino acids with hydrophobic sidechains, as the effector domains of both rapamycin and FK506 are mostly hydrophobic (Fig.1a; Fig. 3a, b). Taking a cue from naturally occurring cyclic peptides including cyclosporine A24, we chose to incorporate N-methyl amino acid building blocks at alternating tetrapeptide positions (Fig. 3b) to increase the conformational flexibility and cell permeability while decreasing protease susceptibility. The library synthesis was carried out using a modified partial split-pool solid phase synthesis protocol to construct the combinatorial tetrapeptide library prior to coupling with FKBDs18,19. The numbers of distinct individual amino acid building blocks at each of the four positions were 15, 10, 15 and 10, respectively, giving rise to a total of 22,500 unique variable effector domains (Fig. 3c) and a total of 45,000 individual rapafucins when coupled to the two FKBDs (Fig. 3d). To simplify the purification and facilitate ensuing screening of the rapafucin libraries, we pooled the first 15 individual amino acids upon their coupling to the beads and kept each of the subsequent coupling reaction products separated. This resulted in 3000 distinct pools of rapafucins with each pool containing 15 individual macrocycles differing at the first amino acid building block. Each pool of the rapafucins was individually purified manually using flash chromatography. Separate use of FKBD10 and FKBD11 yielded two structurally distinct rapafucin classes, with 36- (FKBD10) and 34-membered (FKBD11) rings (Fig. 3d).
Figure 3 |. Selection of amino acid building blocks and synthetic strategy used for the construction of the rapafucin library.
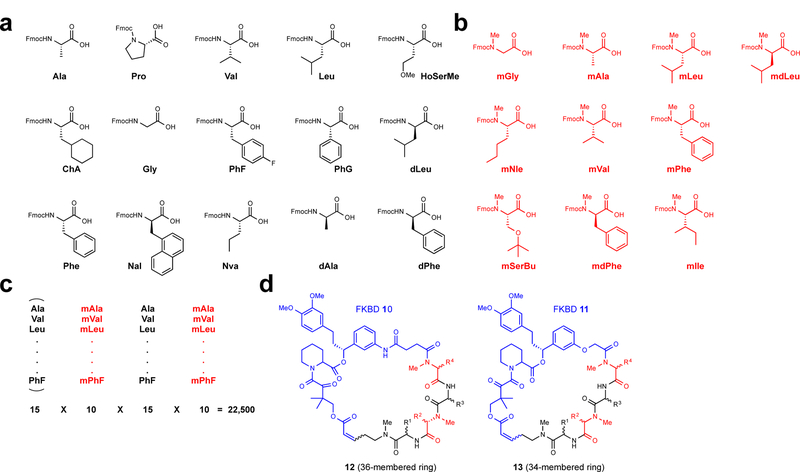
a, Fifteen N-H amino acids (black). b, Ten N-Me amino acids (red). The inclusion of N-Me amino acids was meant to increase the conformational flexibility of the effector domain and decrease susceptibility to protease degradation. c, Partial split-pool strategy for the synthesis of the rapafucin library. The first N-H amino acid building blocks are pooled whereas the rest remained split upon coupling of the individual amino acid building blocks. The pooling of the first amino acid building blocks allowed for the reduction of the total number of final products to be purified. d, Generic structure of rapafucins containing FKBD10 and FKBD11. The two FKBDs differ by two atoms, giving rise to rapafucins with 36- and 34-membered rings, respectively.
To ensure the quality of the rapafucin libraries, we randomly picked 280 pools of rapafucins and assessed the presence of intended macrocycles in each pool using LC-MS analysis. A representative pool of the rapafucins contained all 15 desired rapafucins with the expected molecular mass (Supplementary Fig. 3). The intensity of the peaks varied due to different (M+H)+/(M+Na)+ distribution and overlaps of two pairs of rapafucin isomers. The LC-MS profile further confirmed the identity of each of the desirable rapafucins in that particular pool (Supplementary Fig. 3). Of the 280 pools assessed, only one pool showed incorrect mass spectra. A total of 20 pools (with an additional 19 selected due to low yields) were resynthesized to ensure the quality of the libraries.
Screening of the rapafucin library in a cell-based assay
With 45,000 rapafucins in hand, we set out to perform a cell-based screen of the rapafucin library using the incorporation of [3H]-thymidine as a readout, which has been widely used as an indirect measurement of DNA synthesis during cell proliferation as well as transport of nucleosides. From the initial screen using human umbilical vein endothelial cells (HUVEC), a large number of hits were obtained (Supplementary Fig. 4). Secondary screens of the most potent hits using cell viability and cell cycle analyses revealed two distinct classes of compounds: those that inhibited cell proliferation and those that selectively inhibited thymidine uptake without affecting cell proliferation. In this study, we chose to focus exclusively on the hits that selectively inhibited nucleoside transport. Among those hits, one family made from FKBD10 stood out as having the highest relative potency (Supplementary Fig. 5).
We decoded the hit pool by synthesizing and purifying each of the 15 individual rapafucins contained in the pool and determining their effects on thymidine uptake. This revealed that only three of the rapafucins contained in the pool exhibited submicromolar potency with the most potent hit compound being A15–34-8 with an IC50 of 426 nM (Supplementary Fig. 5). As part of our routine structure-activity relationship (SAR) studies of the initial hits, we reversed the orientation of the effector domain tetrapeptide to determine whether it affected the activity of the hits. To our delight, reversal of the orientation of the same tetrapeptide effector domain, -CO-mLeu-dPro-mPhe-Phe-NH- with respect to FKBD10 led to slight enhancement in potency (SH-13; IC50 = 366 nM). We subsequently performed three rounds of SAR by synthesizing new analogs using different amino acid building blocks using SH-13, alternatively named 95–15, as the new starting point. We synthesized a group of 15 new analogs by varying the sidechains of the fourth amino acid building blocks (some with combined changes in the first, second or third positions) in the effector domain. The most potent analog (95–15-13) that emerged after the second round of SAR inhibited cellular nucleoside uptake with an IC50 of 5.0 nM (Supplementary Fig. 5). Further SAR diversification rounds of the effector domain sequence did not improve potency. We tested 95–15-13 as well as its structurally related analogs for effects on cell viability in the Alamar Blue-based cell viability assay25. At the final concentration of 10 µM, 95–15-13 and most of the analogs had negligible effects on cell viability of HEK293T cells (Supplementary Fig. 6). In addition, no effect on cell cycle progression was observed in HUVEC cells using FACS analysis (Supplementary Fig. 6), consistent with a specific inhibition of nucleoside transport by 95–15-13 and related analogs without overt cytotoxicity.
ENT1 as the target of rapadocin
Nucleoside transporters constitute two families of integral membrane proteins: the concentrative (SLC28) and equilibrative nucleoside transporters (SLC29)26. SLC28 family transporters are active Na+-coupled transporters, whereas SLC29 transporters facilitate passive transport of nucleosides across cellular membranes. These proteins play central roles in cellular physiology as well as in uptake, distribution and elimination of nucleoside-based drugs for treating cancer and several viral diseases including HIV27,28. The SLC29 family consists of four equilibrative nucleoside transporter isoforms (hENT1–4). However, only hENT1 and hENT2 are localized to the plasma membrane and facilitate uptake of exogenous nucleosides29,30. Among the physiological hENT substrates is adenosine, an important signaling molecule that regulates a variety of physiological and pathological processes including vasodilation, platelet aggregation and neuromodulation via its cell-surface adenosine receptors31,32.
The rationale for therapeutic hENT1 inhibition stems from the need to maintain extracellular adenosine reservoirs that mediate intracellular signaling through several adenosine receptors to manifest protective tissue responses during ischemia-reperfusion injury33. Several known nucleoside analog inhibitors of hENT1 have been identified and include S-(4-nitrobenzyl)-6-thioinosine (NBMPR), and clinically relevant draflazine, dilazep and dipyridamole34,35. However, many of these compounds suffer from excess toxicity or poor bioavailability and lack of specificity for different hENT subtypes, highlighting the need for discovery of novel subtype-specific inhibitors of nucleoside transporters.
In our studies, 95-15-13 (hereafter named rapadocin) potently inhibited uptake of [3H]-thymidine in multiple hENT-expressing cell lines (Fig. 4a, b). To further assess the isoform specificity, we used porcine kidney (PK15) cell lines exclusively expressing either hENT1 or hENT236 and determined the [3H]-adenosine uptake in the presence of rapadocin. Interestingly, rapadocin specifically inhibited the hENT1-mediated uptake of [3H]-adenosine in the PK15-ENT1 cell line with an IC50 of 3.2 nM (Fig. 4c), whereas even µM concentrations had little effect on [3H]-adenosine uptake in the PK15-ENT2 cell line, suggesting high specificity of rapadocin for hENT1 (Fig. 4c).
Figure 4 |. Rapadocin is a potent and subtype-selective inhibitor of hENT1.
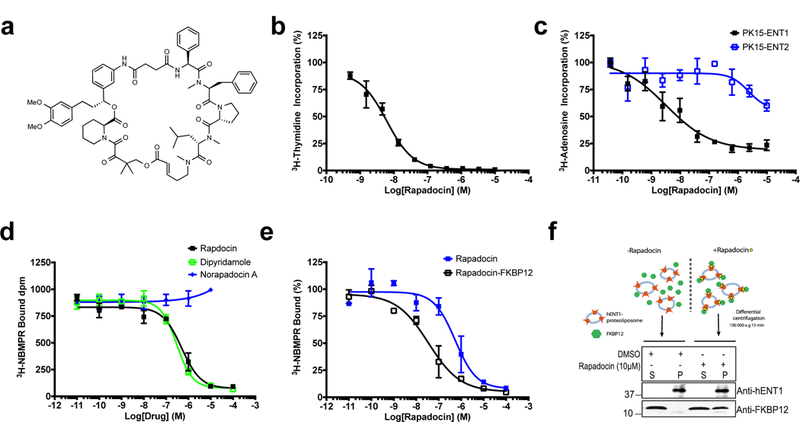
a, Structure of rapadocin. b, Dose-dependent inhibition of [3H]-thymidine uptake in HEK293T cells by rapadocin. Error bars represent s.d.; data are mean ± s.d; n = 3 independent experiments. c, Specific inhibition of [3H]-adenosine uptake in PK15 cell lines exclusively expressing either ENT1 or ENT2. Rapadocin inhibits nucleoside uptake only in ENT1- but not ENT2-expressing cells. Error bars represent s.d.; data are mean ± s.d; n = 3 independent experiments. d, Competitive [3H]NBMPR binding to recombinant hENT1 by rapadocin and dipyridamole on crude membranes from Sf9 insect cells expressing hENT1. hENT1-containing samples were treated with increasing concentrations of either dipyridamole, rapadocin or Norapadocin A before addition of 3.5 nM [3H]NBMPR. Results are shown as dpm [3H]NBMPR bound as a logarithmic function of the concentration of dipyridamole, rapadocin or Norapadocin A. Error bars represent s.e.m.; data are mean ± s.e.m. of three independent experiments (n=3) performed in triplicates yielding Ki values of 95 ± 21 nM and 156 ± 33 nM for dipyridamole and rapadocin, respectively. Norapadocin A, an inactive analog of rapadocin does not compete with [3H]NBMPR binding. e, Competitive [3H]NBMPR binding to purified hENT1 in proteoliposomes by rapadocin and rapadocin-FKBP12 complex. The assay conditions are similar to that described for hENT1 in total Sf9 membranes in (d). The apparent Ki values for rapadocin and rapadocin-FKP12 were 180 ± 27 nM and 5 ± 1.4 nM, respectively. Error bars represent s.e.m.; data are mean ± s.e.m.; n = 3 independent experiments. f, Rapadocin recruits FKBP12 to hENT1-containing proteoliposomes. Rapadocin-FKBP12 complexes were prepared at a final concentrations of 1 µM FKBP12 and 10 µM rapadocin, and mixed with 140 nM recombinant ENT1 reconstituted into proteoliposomes, after which proteoliposomes were pelleted by ultracentrifugation. The amount of co-pelleting FKBP12 and hENT1 was then analyzed by Western blotting. Representative image of n = 2 independent experiments with similar results.
FKBP-dependent interaction between rapadocin and ENT1
To assess the interaction between rapadocin and hENT1, we began with a competitive [3H]-NBMPR binding assay using HEK293 cells. We observed a dose-dependent, saturable increase in binding of [3H]-NBMPR (Supplementary Fig. 7), indicative of specific hENT1 binding. Addition of excess unlabeled NBMPR or rapadocin led to decreased [3H]-NBMPR binding, suggestive of specific and competitive hENT1 binding. To determine whether rapadocin directly interacts with hENT1, we turned to recombinant hENT137. In crude hENT1-expressing Sf9 cell membranes, rapadocin inhibited [3H]-NBMPR binding in a dose-dependent manner similar to dipyridamole (Fig. 4d). In contrast, norapadocin A, an inactive analog of rapadocin (Supplementary Fig. 8), had no effect on [3H]-NBMPR binding.
To evaluate a direct interaction of rapadocin with hENT1 and the effects of FKBP12 for this binding event, we purified heterologously expressed hENT1 from detergent-solubilized Sf9 membranes to homogeneity in a functional and monomeric form (Supplementary Fig. 7). To avoid complications in analyzing ligand interactions with detergent-solubilized hENT138, we reconstituted purified recombinant hENT1 into proteoliposomes in an active form (Supplementary Fig. 9) and carried out [3H]-NBMPR binding assays in the presence of rapadocin or a preformed rapadocin-FKBP12 complex. Consistent with the results obtained from crude hENT1-expressing Sf9 cell membranes (Fig. 4d), rapadocin alone inhibited the binding of [3H]-NBMPR to purified hENT1 dose-dependently with Ki of 150 ± 27 nM. In comparison, the rapadocin-FKBP12 complex competed for [3H]-NBMPR binding to hENT1 with approximately 30-fold increased potency compared to rapadocin alone (Ki of 5 ± 1.4 nM) (Fig. 4e). To determine whether a ternary complex including hENT1, rapadocin and FKBP12 is involved in hENT1 inhibition, we took a complementary approach by determining if rapadocin can recruit FKBP12 to hENT1-containing membranes. Thus, we performed liposome co-pelleting assays using FKBP12 with and without rapadocin. This experiment clearly demonstrated that soluble FKBP12 becomes efficiently recruited to hENT1 liposomes in a manner dependent on rapadocin (Fig 4f). Together, these results suggest that FKBP12 plays a key role in mediating the inhibition of hENT1 by rapadocin, reminiscent of the mode of action of FK506 and rapamycin.
To further characterize the interaction between rapadocin and hENT1, we sought to develop an active affinity probe of rapadocin. Through SAR studies, we found that N-methyl phenylalanine (mPhe), the third amino acid building block in the effector domain (Fig. 4a), tolerated multiple alterations while retaining significant activity (Supplementary Fig. 8). We thus synthesized a biotin-rapadocin conjugate by tethering the biotin moiety via a tyrosine residue in the same position (Supplementary Fig. 8). Because it has been reported that hENT1 is highly expressed in red blood cells, we tested rapadocin alongside its biotin conjugate for adenosine uptake inhibition in red blood cells39. As expected, both rapadocin and biotin-rapadocin dose-dependently inhibited adenosine uptake in red blood cells, with IC50 values of 45 nM and 111 nM, respectively (Supplementary Fig. 10). Using the biotin-rapadocin conjugate and detergent-solubilized plasma membrane fractions prepared from red blood cells, we performed a pulldown experiment followed by Western blot with hENT1-specific antibodies, and found that the biotin-rapadocin conjugate is capable of pulling down hENT1 (Fig. 5a). Importantly, hENT1 binding of the biotin-rapadocin probe is competed by rapadocin, but not appreciably by either rapamycin or FK506, suggesting that rapadocin is capable of specifically binding hENT1 in the absence of FKBP (Fig. 5a).
Figure 5 |. Pulldown of hENT1 from red cell membrane fractions using a biotin-rapadocin conjugate or GST-FKBP12-rapadocin complex, and the FKBP-dependence of hENT1 inhibition by rapadocin in cells.
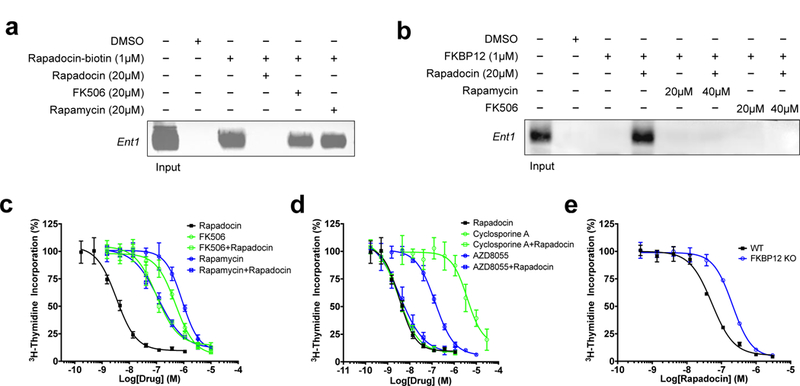
a, Affinity pulldown of detergent-solubilized hENT1 by a biotin-rapadocin conjugate and competition by free rapadocin, FK506 or rapamycin. hENT1 was detected by Western blotting. b, Affinity pulldown of hENT1 by GST-FKBP12 in the presence of rapadocin and competition by free rapadocin, FK506 and rapamycin. c, FK506 and rapamycin antagonize rapadocin inhibition of hENT1-mediated thymidine uptake. The dose-response curves were determined using [3H]-thymidine uptake in PK15-ENT1 cells. d, Cyclosporine A and AZD8055 did not antagonize rapadocin inhibition of hENT1-mediated thymidine uptake. The dose-response curves were determined using [3H]-thymidine uptake in PK15-ENT1 cells. e, Knockout of FKBP12 confers resistance to rapadocin. Dose response curves for inhibition of thymidine uptake by rapadocin in wild type and FKBP12 null Jurkat T cells. Representative image of n = 3 independent experiments with similar results. In all graphs, error bars represent s.d.; data are mean ± s.d; n = 3 independent experiments.
Having shown that FKBP12-rapadocin complex has higher intrinsic affinity for hENT1 than rapadocin itself (Fig. 4e), we determined whether a GST-FKBP12-rapadocin complex is capable of pulling down detergent-solubilized hENT1. While GST-FKBP12 itself does not bind hENT1, GST-FKBP12 pulled down hENT1 in the presence of rapadocin (Fig. 5b). The association between the GST-FKBP12-rapadocin complex and hENT1 is sensitive to competition by rapamycin and FK506. These results corroborate the observed high affinity of the FKBP12-rapafucin complex for hENT1 (Fig. 4e), reminiscent of the interactions between FKBP12-rapamycin and mTOR5, and between FKBP12-FK506 and calcineurin6.
Next, we assessed whether inhibition of hENT1 by rapadocin in a cellular context is also dependent on FKBP. A hallmark of FKBP-dependence is that the cellular effects can be antagonized by other FKBP-binding ligands with no or orthogonal biological activity due to competition for cellular FKBP as has been shown for FK506 and rapamycin40. For unknown reasons, both FK506 and rapamycin showed inhibitory effects on adenosine uptake at high concentrations in HUVEC. Nevertheless, it was still possible to determine mutual antagonism between these drugs and rapadocin using pairwise combinations to determine whether the combinations had additive or antagonistic effects. Both FK506 and rapamycin are significantly less potent inhibitors of nucleoside uptake than rapadocin, with IC50 values in the low micromolar range that are orders of magnitude higher than those for their inhibition of calcineurin and mTOR, respectively. Importantly, combining FK506 with rapadocin significantly decreased the potency of rapadocin (Fig. 5c). The same rapadocin antagonism was also observed with rapamycin. We attribute this observed antagonism to sequestration of FKBP by high concentrations of FK506 and rapamycin, which prevents formation of the much more potent FKBP-rapadocin complex. As negative controls, we tested both cyclosporin A (CsA), a calcineurin inhibitor that is independent of FKBP6, and AZD8055, an FKBP-independent mTOR inhibitor that targets the kinase active site of mTOR41 in combination with rapadocin. Similar to FK506 and rapamycin, CsA inhibited thymidine uptake at higher concentrations (Fig. 5d), but in contrast to FK506 and rapamycin, combining CsA with rapadocin did not affect rapadocin activity. A similar observation was made with the FKBP-independent mTOR inhibitor AZD8055, providing further support for the notion that the mutual antagonism between rapadocin and FK506 or rapamycin resulted from competitive sequestration of intracellular FKBP.
We determined the inhibition constants of rapadocin for different isoforms of FKBP in vitro using the peptidyl prolyl cis-trans isomerase assay42 and found that rapadocin has the highest potency against FKBP12 among five FKBP family members, with a Ki value of 3.7 nM (Supplementary Table 5). In comparison, its potency against FKBP51 and FKBP52 are about 40-fold lower. To further investigate the dependence of rapadocin on endogenous FKBPs, we knocked out FKBP12 and FKBP51 (as a control) using CRISPR-Cas9 in Jurkat T cells (Supplementary Fig. 11). As expected, deletion of FKBP12 led to significant resistance to inhibition of PMA/ionomycin-stimulated activation of an NFAT-luciferase reporter gene by FK506 (Supplementary Fig. 12). Similarly, the FKBP12 null Jurkat T cells also showed resistance to nucleoside uptake inhibition by rapadocin (Fig. 5e), albeit moderate in comparison to the effect on FK506 in the NFAT-luciferase reporter, which is likely attributable to redundancy of other FKBP isoforms. In contrast to rapadocin, dipyridamole inhibited nucleoside uptake equally well in both wild-type and FKBP12 null cells (Supplementary Fig. 13), whereas knockout of FKBP51 had little effect on the sensitivity of cells to either rapadocin or dipyridamole (Supplementary Fig. 14). Together, these results suggest that the cellular activity of rapadocin is dependent, at least in part, on endogenous FKBP12.
Both rapamycin and FK506 are cell-permeable, as their targets calcineurin and mTOR are cytosolic proteins. For rapadocin, however, its target hENT1 is a multi-pass plasma membrane protein, which raises the intriguing question of whether rapadocin acts extracellularly or intracellularly. A more general question concerns whether rapafucins as a class of FKBD-containing hybrid macrocycles are cell permeable. To address these questions, we first employed an Id1-Luc luciferase reporter, which is responsive to BMP receptor activation43. It has been shown that FKBP12 associates with and inhibits BMPR2, and FK506 is capable of activating BMP receptor signaling by binding to FKBP12 and dissociating it from BMPR2, thereby activating downstream Id1 promoter44,45. As the interaction between FKBP12 and BMPR occurs intracellularly, the activation of BMPR signaling necessitates cell penetration by FK506. We thus determined the effects of rapadocin and a number of analogs on the activation of the Id1-Luc reporter. As expected, both FK506 and rapamycin activated the Id1-Luc reporter gene while HP7E3, a carboxylic acid-bearing cell impermeable rapafucin analog did not (Supplementary Fig. 15). Similar to FK506 and rapamycin, rapadocin also activated the reporter gene, albeit to a lesser extent. Interestingly, most of the rapadocin analogs were also capable of activating the Id1-Luc reporter with some showing comparable activity to that of FK506. These results suggest that most rapafucins can penetrate the plasma membrane of mammalian cells to reach intracellular targets, much like rapamycin and FK506. In addition to the reporter gene assay, we also applied LC-MS to detect the uptake of rapadocin into Jurkat T cells. We found that rapadocin, but not HP7E3, can be taken up similar to FK506 (Supplementary Fig. 16). Together, these results suggest that rapadocin and most rapafucin analogs are capable of penetrating into mammalian cells. The observed partial resistance to rapadocin upon knockout of FKBP12 (Fig. 5e) suggests that the hENT1-rapadocin interaction may reside at the cytosolic side of the plasma membrane accessible to FKBP12 and possibly other FKBP isoforms.
Rapadocin protects against ischemic reperfusion injury in vivo
Among the many physiological and pathological activities of adenosine is its protective role in ischemic reperfusion injury46,47. Extracellular adenosine, by activating its receptor A2B, protects renal and hepatic tissues from ischemia-reperfusion injury48. However, extracellular adenosine is short-lived due to uptake through ENT transporters, and thus inhibition of ENT transport activity is expected to sustain extracellular adenosine levels, thereby reducing reperfusion injury. Hence, we determined the effect of rapadocin in an animal model of kidney ischemia-reperfusion injury49. Mice were administered with rapadocin or vehicle control via tail vein injection and preconditioned for 15 min. Reperfusion injury was then induced by removing one kidney and clamping the other for 45 min. The clamp was then released for 24 h before animals were sacrificed, and blood and urine were harvested for analyses. As expected, the levels of creatinine (Fig. 6a) and urea nitrogen in animal blood (Fig. 6b) were significantly increased in animals treated with vehicle alone. Upon treatment with rapadocin at 4 mg/kg, there was a significant inhibition of the release of creatinine and urea nitrogen in the blood (Fig. 6a, b). In contrast, norapadocin B, another inactive analog of rapadocin (Supplementary Fig. 8), had little effect on the levels of either creatinine or blood urea nitrogen. To further confirm that this effect is due to an increase in adenosine, we also coadminstered rapadocin with the specific A2B antagonist PSB111550 and found that PSB1115 reversed the inhibition of the release of creatinine and urea nitrogen by rapadocin, suggesting that the effect on kidney reperfusion injury by rapadocin is due to blockade of adenosine uptake. Together, these results suggest that rapadocin is a promising lead compound for the treatment of ischemia-reperfusion injury.
Figure 6 |. Inhibition of kidney ischemia reperfusion injury by rapadocin in vivo.
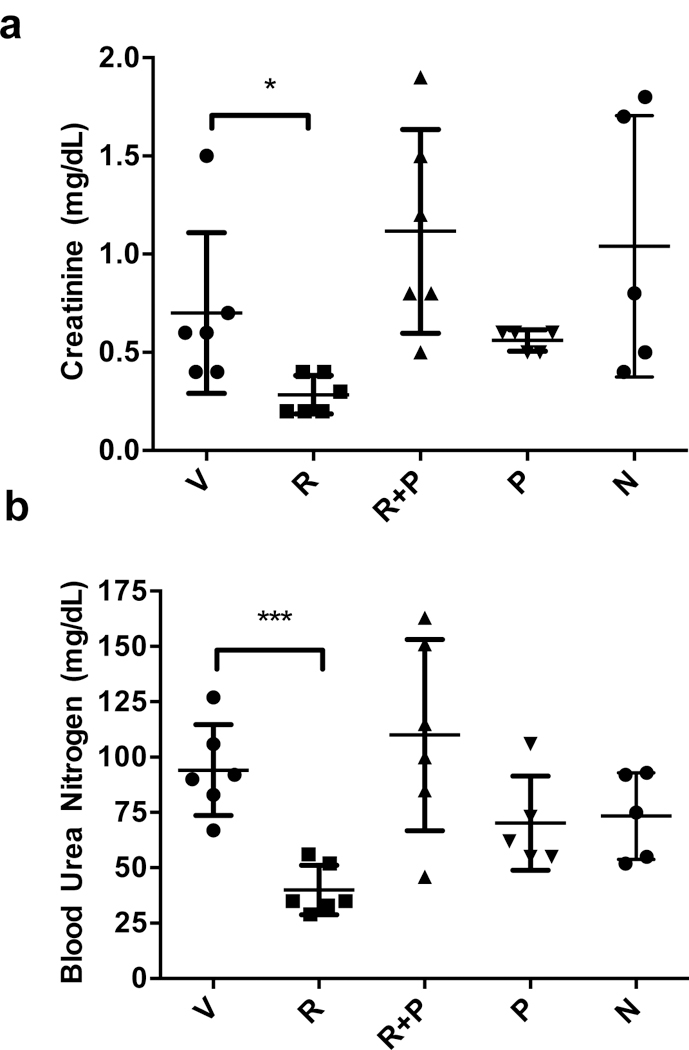
Effects of vehicle (V), rapadocin (R), norapadocin B (N) and the adenosine receptor A2B antagonist PSB1115 (P) on the blood levels of creatinine (a) and urea nitrogen (b) in a mouse model of kidney reperfusion injury. Rapadocin, but not its inactive analog or PSB1115, caused a statistically significant inhibition of the increase in creatinine and urea nitrogen levels. n = 6 for V, R, and R+P; n=5 for P and N; Error bars represent s.d.; data are mean ±s.d.; *p = 0.036 vs vehicle control; *** p = 0.0002 vs vehicle control; P values are from two-sided student t-test.
Discussion
The existence of two distinct FKBD-containing macrocyclic natural products, FK506 and rapamycin, raised the intriguing possibility of discovering analogous compounds with altered target specificity. Although naturally occurring analogs of FK506 such as ascomycin (FR-900520) and FR-900523 have been identified51,52, they bind the same target as FK506. In this work, we attempted to imitate nature by replacing the effector domain of rapamycin with a large collection of oligopeptides in hope of gaining the ability to target other pharmacologically relevant proteins in the human proteome16. We designed and synthesized a 45,000-compound rapafucin library using optimized FKBDs and identified a family of rapadocins that specifically bind and potently inhibit the activity of hENT1, underscoring the wide applicability of the rapafucin library in giving rise to novel ligands against medically relevant and difficult-to-target proteins.
The attainment of the large rapafucin library was made possible by the efficient macrocyclization/cyclative release strategy using an optimized microwave-assisted RCM reaction. Although the yields varied, it was possible to synthesize most rapafucins with tetrapeptide effector domains regardless of the types of amino acid building blocks, including D-amino acids and N-methyl amino acids. The concurrent macrocyclization and release of the cyclized rapafucins from solid phase also greatly simplified the ensuing purification of final rapafucin products. Despite its generality and efficiency, the RCM reaction does have restrictions on substrates, especially those with a Lewis base capable of coordinating with and inactivating the metal-containing catalysts. Alternative macrocyclization methods may help overcome the limitation of the RCM reaction and offer complementary routes to rapafucins.
Aside from the RCM macrocyclization/cyclative release strategy, several other steps played key roles in the successful construction of the rapafucin library. First, optimal FKBDs were identified that were not only compatible with the RCM reaction, but also maintained high affinity for FKBP upon incorporation into macrocycles independent of the effector domain structures. Second, the use of N-methyl amino acids as building blocks of the effector domain conferred the library with stability as well as conformational flexibility. As evident from the structure-activity relationship of rapadocin, replacement of mPhe with Phe at the third position of rapadocin led to over 10-fold reduction in potency (Supplementary Fig. 5, 95–15-13–3).
One of the steps involved in our SAR studies is the reversal of the orientation of the tetrapeptide effect domain with respect of FKBD (Supplementary Fig. 5). This maneuver took advantage of the unique modular structure of rapafucins that contain separable FKBD and effector domains. That the same tetrapeptide effector domain, when fused with FKBD in opposite orientations, can result in enhancement, albeit moderate, in potency is not completely surprising. That rapadocin is capable of binding hENT1 either alone or in complex with FKBP12 in which FKBD moiety is deeply buried within and shielded by FKBP12 suggests that it is the tetrapeptide effector domain that directly interacts with hENT1. As such, it is understandable that analogs with opposite orientation can both bind to hENT1 with slightly different affinity.
The identification of rapadocin from the rapafucin library as an isoform-specific and highly potent inhibitor of hENT1 validates the premise that it is possible to reprogram rapamycin to target other proteins. It is worth pointing out that there are both similarities and differences between rapadocin and rapamycin/FK506 in their mode of action. Like rapamycin and FK506, rapadocin binds to FKBP12 with high affinity and the resultant FKBP12-rapadocin complex is capable of forming a ternary complex with its target hENT1. However, unlike rapamycin and FK506, rapadocin exhibits appreciable affinity for hENT1 in the absence of FKBP (Fig. 4e). These results raised the question of whether FKBP12 makes direct contacts with hENT1 in the FKBP-rapadocin-hENT1 complex similar to the FKBP-FK506-calcineurin and FKBP-rapamycin-mTOR complexes. It is worth noting that a major difference exists in the extent to which FKBP12 interacts with the ultimate targets between FKBP12-FK506-calcineurin and the FKBP12-rapamycin-mTOR complexes, with many fewer contacts between FKBP12 and mTOR than those between FKBP12 and calcineurin, likely due to the larger size of rapamycin over FK506, which may hold mTOR further away from FKBP12. These observations suggest that most of the rapafucins in our current library with ring sizes even larger than rapamycin are not likely to allow extensive direct FKBP interactions with targets. It is possible that the 30-fold enhancement in binding affinity of rapadocin to hENT1 by FKBP12 is mediated through conformational alteration of the effector domain of rapadocin to facilitate ENT1 binding rather than direct FKBP12-hENT1 interaction in the ternary FKBP12-rapadocin-hENT1 complex.
Adenosine plays important roles in a wide variety of physiological and pathological processes53. Upregulaton of extracelluar adenosine and the activation of adenosine receptors have been shown to have therapeutic benefit for a myriad of diseases from asthma to arthritis and from sepsis to ischemic reperfusion injury. There are two complementary strategies to enhance adenosine signaling, activation of adenosine receptors by agonists or inhibition of adenosine reuptake via ENT and other transporters. Of the two, inhibition of ENT may be advantageous in that it will only affect a subset of tissues that have active inflammation and injury accompanied by high extracellular levels of adenosine as opposed to global activation of adenosine receptors by their agonists, which is reminiscent of the inhibition of serotonin reuptake for treating psychiatric disorders54. As such, inhibition of ENT is expected to have fewer side effects in comparison to adenosine receptor agonists, whose development has been limited by toxicity likely associated with global activation of adenosine receptors55. In this manuscript, we have demonstrated the efficacy of rapadocin in an animal of kidney reperfusion injury. Though a number of drugs including dipyridamole have been shown to inhibit ENT1, most cross-inhibit other pharmacological targets26. Rapadocin represents one of the first ENT1 inhibitors with high target specificity. It will be interesting to assess its effects on other disease models and determine whether it indeed is less toxic than other non-specific ENT1 inhibitors and adenosine receptor agonists in the future.
Both rapamycin and FK506 are important drugs in the clinic, but also serve as powerful molecular probes for providing fundamental insights into cellular signaling networks centered around mTOR and calcineurin, respectively56–60. The generation of the rapafucin libraries and identification of rapadocin as an inhibitor for hENT1 with isoform specificity and in vivo efficacy in treating kidney ischemia reperfusion injury demonstrate that it is possible to reprogram rapamycin to target distinct pharmacologically relevant proteins. It is noteworthy that our screen also identified a large number of uncharacterized rapafucin hits that inhibit cell proliferation without affecting nucleoside uptake (Supplementary Fig. 4). Those compounds most likely target proteins other than hENT1, suggesting that ligands for a multitude of other proteins can be discovered from the existing and expanded versions of the rapafucin libraries, which will serve as novel leads for drug development and molecular probes for investigating various cellular processes.
Methods
See Supplementary Information for a detailed Methods section.
Supplementary Material
Acknowledgements
This work was made possible by the NIH Director’s Pioneer Award, the Flight Attendant Medical Research Institute and a generous gift from Mr. Shengjun Yan and Ms. Hongju Mao (J.O.L.), a Damon Runyon Postdoctoral Fellowship (H.P.) and NIH Postdoctoral Training Award (M.D.). V.O.P. is supported by the Academy of Finland (Grant 289737) and Sigrid Juselius Foundation. We wish to thank Dr. Sarah A. Head for critical comments on the manuscript. Correspondence and requests for materials should be addressed to J.O.L.
Footnotes
Competing financial interests.
The authors declare conflict of interest. Patent applications covering the rapafucin library and rapadocin have been filed by Johns Hopkins University and licensed to Rapafusyn Pharmaceuticals, Inc. J.O.L. is a co-founder of, as well as a Scientific Advisory Board Member for, Rapafusyn Pharmaceuticals, Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
Data availability
The data that support the findings of this study are available from the authors on reasonable request; see author contributions for specific data sets.
References
- 1.Sehgal SN, Baker H & Vezina C Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 28, 727–732 (1975). [DOI] [PubMed] [Google Scholar]
- 2.Tanaka H et al. Structure of FK506, a novel immunosuppressant isolated from Streptomyces. J. Am. Chem. Soc 109, 5031–5033 (1987). [Google Scholar]
- 3.Harding MW, Galat A, Uehling DE & Schreiber SL A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341, 758–760 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Siekierka JJ, Hung SH, Poe M, Lin CS & Sigal NH A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341, 755–757 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Heitman J, Movva NR & Hall MN Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253, 905–909 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Liu J et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815 (1991). [DOI] [PubMed] [Google Scholar]
- 7.Yang H et al. mTOR kinase structure, mechanism and regulation. Nature 497, 217–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffith JP et al. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82, 507–522 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Kissinger CR et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature 378, 641–644 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Marinec PS et al. FK506-binding protein (FKBP) partitions a modified HIV protease inhibitor into blood cells and prolongs its lifetime in vivo. Proc. Natl. Acad. Sci. U S A 106, 1336–1341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klemm JD, Schreiber SL & Crabtree GR Dimerization as a regulatory mechanism in signal transduction. Annu. Rev. Immunol 16, 569–592 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Bayle JH et al. Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem. Biol 13, 99–107 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Guduru SKR & Arya P Synthesis and biological evaluation of rapamycin-derived, next generation small molecules. Med. Chem. Commun 9, 27–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty TK, Weber HP & Nicolaou KC Design and synthesis of a rapamycin-based high affinity binding FKBP12 ligand. Chem. Biol 2, 157–161 (1995). [DOI] [PubMed] [Google Scholar]
- 15.Wu X et al. Creating diverse target-binding surfaces on FKBP12: synthesis and evaluation of a rapamycin analogue library. ACS Comb. Sci 13, 486–495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Bhat S & Liu JO A simple and efficient route to the FKBP-binding domain from rapamycin. Tetrahedron Lett 52, 5070–5072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furka A, Sebestyen F, Asgedom M & Dibo G General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res 37, 487–493 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Houghten RA et al. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 354, 84–86 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Lam KS et al. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 354, 82–84 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Reichwein JF, Wels B, Kruijtzer JA, Versluis C & Liskamp RM Rolling Loop Scan: An Approach Featuring Ring-Closing Metathesis for Generating Libraries of Peptides with Molecular Shapes Mimicking Bioactive Conformations or Local Folding of Peptides and Proteins. Angew. Chem. Int. Ed 38, 3684–3687 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Liu J et al. Inhibition of T cell signaling by immunophilin-ligand complexes correlates with loss of calcineurin phosphatase activity. Biochemistry 31, 3896–3901 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Halt DA et al. Design, Synthesis and Kinetic Evaluation of High-Affinity FKBP Ligands and the X-ray Crystal Structures of Their Complexes with FKBP12. J. Am. Chem. Soc 115, 9925–9938 (1993). [Google Scholar]
- 23.Clackson T et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. U S A 95, 10437–10442 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sagan S, Karoyan P, Lequin O, Chassaing G & Lavielle S N- and Calpha-methylation in biologically active peptides: synthesis, structural and functional aspects. Curr. Med. Chem 11, 2799–2822 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Ahmed SA, Gogal RM Jr. & Walsh JE A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J. Immunol. Methods 170, 211–224 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Young JD, Yao SY, Baldwin JM, Cass CE & Baldwin SA The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med 34, 529–547 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Owen RP et al. Functional characterization and haplotype analysis of polymorphisms in the human equilibrative nucleoside transporter, ENT2. Drug Metab. Dispos 34, 12–15 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Boswell-Casteel RC & Hays FA Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids 36, 7–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao JC, Zhang TP & Zhao YP Human equilibrative nucleoside transporter 1 (hENT1) predicts the Asian patient response to gemcitabine-based chemotherapy in pancreatic cancer. Hepatogastroenterology 60, 258–262 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Meijer LL, Puik JR, Peters GJ, Kazemier G & Giovannetti E hENT-1 Expression and Localization Predict Outcome After Adjuvant Gemcitabine in Resected Cholangiocarcinoma Patients. Oncologist 21, e4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson KA & Gao ZG Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov 5, 247–264 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loffler M, Morote-Garcia JC, Eltzschig SA, Coe IR & Eltzschig HK Physiological roles of vascular nucleoside transporters. Arterioscler. Thromb. Vasc. Biol 27, 1004–1013 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Headrick JP & Lasley RD Adenosine receptors and reperfusion injury of the heart. Handb. Exp. Pharmacol 189–214 (2009). [DOI] [PMC free article] [PubMed]
- 34.Cass CE & Paterson AR Inhibition by nitrobenzylthioinosine of uptake of adenosine, 2’-deoxyadenosine and 9-beta-D-arabinofuranosyladenine by human and mouse erythrocytes. Biochem. Pharmacol 24, 1989–1993 (1975). [DOI] [PubMed] [Google Scholar]
- 35.Scholtissek C Studies on the uptake of nucleic acid precursors into cells in tissue culture. Biochim. Biophys. Acta 158, 435–447 (1968). [DOI] [PubMed] [Google Scholar]
- 36.Ward JL, Sherali A, Mo ZP & Tse CM Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J. Biol. Chem 275, 8375–8381 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Rehan S & Jaakola VP Expression, purification and functional characterization of human equilibrative nucleoside transporter subtype-1 (hENT1) protein from Sf9 insect cells. Protein Expr. .Purif 114, 99–107 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Rehan S, Ashok Y, Nanekar R & Jaakola VP Thermodynamics and kinetics of inhibitor binding to human equilibrative nucleoside transporter subtype-1. Biochem. Pharmacol 98, 681–689 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Hammond JR Interaction of a series of draflazine analogues with equilibrative nucleoside transporters: species differences and transporter subtype selectivity. Naunyn Schmiedebergs Arch. Pharmacol 361, 373–382 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Bierer BE et al. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. U S A 87, 9231–9235 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chresta CM et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70, 288–298 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T & Schmid FX Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337, 476–478 (1989). [DOI] [PubMed] [Google Scholar]
- 43.Korchynskyi O & ten Dijke P Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem 277, 4883–4891 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Kugimiya F et al. Mechanism of osteogenic induction by FK506 via BMP/Smad pathways. Biochem. Biophys. Res. Commun 338, 872–879 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Spiekerkoetter E et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest 123, 3600–3613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day YJ, Huang L, Ye H, Linden J & Okusa MD Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am. J. Physiol. Renal. Physiol 288, F722–731 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Lappas CM, Day YJ, Marshall MA, Engelhard VH & Linden J Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J. Exp. Med 203, 2639–2648 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grenz A et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med 5, e137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu M et al. Acute kidney injury leads to inflammation and functional changes in the brain. J. Am. Soc. Nephrol 19, 1360–1370 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan L & Muller CE Preparation, properties, reactions, and adenosine receptor affinities of sulfophenylxanthine nitrophenyl esters: toward the development of sulfonic acid prodrugs with peroral bioavailability. J. Med. Chem 47, 1031–1043 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Arai T, Kouama Y, Suenaga T & Honda H Ascomycin, an antifungal antibiotic. J. Antibiot. (Tokyo) 15, 231–232 (1962). [PubMed] [Google Scholar]
- 52.Hatanaka H et al. FR-900520 and FR-900523, novel immunosuppressants isolated from a Streptomyces. II. Fermentation, isolation and physico-chemical and biological characteristics. J. Antibiot. (Tokyo) 41, 1592–1601 (1988). [DOI] [PubMed] [Google Scholar]
- 53.Hasko G, Linden J, Cronstein B & Pacher P Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov 7, 759–770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaswani M, Linda FK & Ramesh S Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Progress in neuro-psychopharmacology & biological psychiatry 27, 85–102 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Chen JF, Eltzschig HK & Fredholm BB Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov 12, 265–286 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laplante M & Sabatini DM mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dazert E & Hall MN mTOR signaling in disease. Curr. Opin. Cell Biol 23, 744–755 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Liu JO Calmodulin-dependent phosphatase, kinases, and transcriptional corepressors involved in T-cell activation. Immunol. Rev 228, 184–198 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schreiber SL & Crabtree GR The mechanism of action of cyclosporin A and FK506. Immunol. Today 13, 136–142 (1992). [DOI] [PubMed] [Google Scholar]
- 60.Rao A, Luo C & Hogan PG Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol 15, 707–747 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.