

Abstract
Dysbiosis, an imbalance in microbial communities, is linked with disease when this imbalance disturbs microbiota functions essential for maintaining health or introduces processes that promote disease. Dysbiosis in disease is predicted when microbiota differ compositionally from a healthy control population, but only truly defined when these differences are mechanistically related to adverse phenotypes. For the human gut microbiota, dysbiosis varies across diseases. One common manifestation is replacement of the complex community of anaerobes typical of the healthy adult gut microbiome with a community of lower overall microbial diversity and increased facultative anaerobes. Here we review diseases in which low-diversity dysbiosis has been observed and mechanistically linked with disease, with a particular focus on liver disease, inflammatory bowel disease, and Clostridium difficile infection.
Introduction:
The bacterial community of the human distal intestine is one of the richest microbial environments on earth, consisting of >1012 cells and hundreds of species [1]. The healthy adult gut microbiota is typically dominated by anaerobic members of the Firmicutes and Bacteroidetes phyla and is known to provide key functions for maintaining health, including production of metabolites that promote immune homeostasis and competitive exclusion of pathogens [2]. This complex community establishes during the first two to three years of life through a process similar to ecological succession [2], in which the community follows a path from sterility through a systematic series of turnover of early successional “pioneer” species before reaching a relatively stable complex community that is dominated by anaerobes (Fig. 1). Remarkably, this path is very conserved in diverse geographic and socioeconomic settings [3]. In the perinatal period, facultative anaerobes such as Proteobacteria, Lactobacilli and Enterococcus species colonize as well as particular anaerobes such as Bifidobacteria. With the introduction of solid foods, dramatic shifts occur with expansion of strict anaerobes such as the Clostridiales and Bacteroidales (Table 1) [4].
Figure 1. Recovery from disturbance through secondary succession:
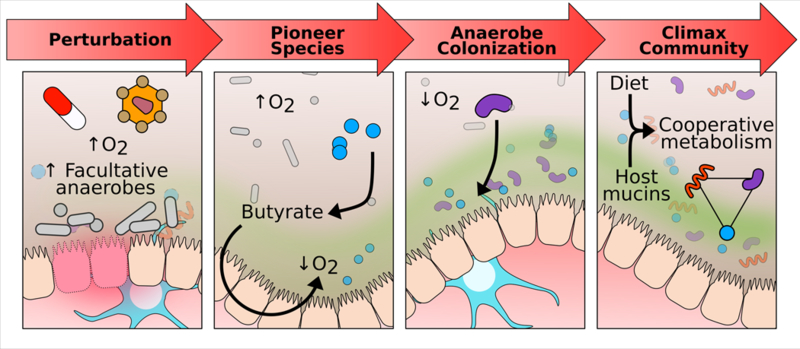
An insult (e.g. antibiotics) raises intraluminal oxygen concentrations leading to a bloom of facultative anaerobes. One factor in recovery may be oxygen-tolerant anaerobes that are able to colonize and reestablish butyrate production. Colonocyte metabolism of butyrate depletes luminal oxygen allowing for further colonization by anaerobes. Interdependent metabolic networks of the anaerobes are restarted and the mature, complex climax community of the healthy adult gut is reached.
Table 1:
Bacterial families that dominate the adult human gut in health and during low-diversity dysbiosis.
| Healthy | Low-diversity dysbiosis |
|---|---|
| Firmicutes Clostridia Clostridiales Lachnospiraceae (Clostridia XIVa) Ruminococcaceae (Clostridia IV) |
Firmicutes Bacilli Lactobacillales Lactobacillaceae Streptococcaceae Enterococcaceae |
| Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Prevotellaceae |
Proteobacteria Gammaproteobacteria Enterobacteriales Enterobacteraceae |
Dysbiosis, or imbalance of the microbiome, may have diverse compositional and functional attributes in different disease contexts [5]. In many different diseases, however, the dysbiotic gut microbiome has been described to have a reduction in the proportion of anaerobes that are typically abundant in health and an increased proportion of facultative anaerobes, including Proteobacteria and Bacilli. Such low-diversity, disease-associated microbiomes can resemble the gut microbiome of infants (younger than ~2 years of age) compositionally [2,6,7]. While it may be surprising that the gut microbiome of a very sick adult would resemble that of a perfectly healthy infant, this observation may be explained by the concept of “secondary succession” where a dramatic disturbance that wipes out a complex community (such as a forest fire) results in the observation of similar early succession, or “pioneer” species [2,6]. A low diversity, facultative anaerobe-dominated community observed in the adult gut thus may be considered a bioindicator of disturbance with age-dependent implications for health. Low-diversity microbiota, with increases in proportions of facultative anaerobes, have been observed with acute diarrheal disease [8], Inflammatory Bowel Disease (IBD) [7], C. difficile infection (CDI) [9], liver disease [10,11], and in cancer patients [12]. In cancer patients who undergo allogeneic stem cell transplantation, this “infant-like” microbiome was associated with all-cause mortality following stem cell transplant [12]. Since this particular type of dysbiosis may be common in many disease settings, research into drivers, functional consequences, and therapeutic strategies of recovery may have particularly far reaching implications. Dysbiosis can potentially take many different forms, therefore we use the term “low-diversity dysbiosis” to specifically refer to a microbiome characterized by low diversity and increased proportions of facultative anaerobes (Table 1).
Drivers of Low-diversity Dysbiosis:
Low-diversity dysbiosis may be driven by many factors that differ by disease context. One known contributor to low-diversity dysbiosis is broad-spectrum antibiotics. Individuals with recurrent CDI (rCDI) often have microbiome states consistent with low-diversity dysbiosis [9] and CDI is strongly associated with antibiotic exposure [13]. Epidemiologic studies have identified clindamycin, cephalosporins, and fluoroquinolones as significant risk factors for CDI [14]. However, CDI is also more common in certain populations, including the elderly, individuals with IBD [15], with liver disease [16] and with blood cancers [17]. Thus, the factors that predispose an individual to low-diversity dysbiosis and associated opportunistic infections are varied.
One factor that may also lead to dysbiosis in disease contexts is host genetics, such as in the case of IBD. IBD is characterized by chronic, relapsing inflammation with an unknown etiology and is currently separated into ulcerative colitis (UC) and Crohn’s disease (CD) [18]. Individuals with IBD can have microbial compositions characteristic of low-diversity dysbiosis [18,19]. However, the dysbiosis observed with IBD can be more complex: for instance, one study showed low-diversity dysbiosis to be commonly observed in ileal CD but not colonic CD or UC [7,20]. Drivers of dysbiosis in the context of IBD are not known, but studies have illustrated a genetic component as one factor. 20–50% of monozygotic twins are concordant for CD versus 10% concordance in dizygotic twins [21] and more than 200 susceptibility loci have been identified as associated with either UC or CD. Many of these loci are implicated in other immune-related diseases and are linked with innate immunity, epithelial barrier function, and microbial recognition [22,23]. For example, NOD2 is involved in peptidoglycan recognition and variations in the gene are linked to increased risk of CD [24].
Cross-talk between the gut and other organs may also drive the development of low-diversity dysbiosis (Fig. 2). Liver cirrhosis is characterized by compositional features typical of low-diversity dysbiosis; namely a reduction in anaerobic bacterial families that typically dominate the healthy gut with corresponding increases in facultative anaerobes (Table 1) [10,11,25]. One factor that may drive gut microbiota shifts in low-diversity dysbiosis is alteration of the quantity of bile salts excreted into the gut [26]. Animal models of both non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD) have demonstrated the central role of dysbiosis as a driver of hepatic inflammation and the ability to transfer disease susceptibility via fecal microbial transplantation (FMT) from diseased mice into non-diseased mice [27–29]. These findings parallel extensive human data demonstrating dysbiosis as not only a biomarker for progression of liver disease, but also as an independent predictor of disease severity and clinical outcomes in patients with cirrhosis [11,25,30].
Figure 2. Gut-liver axis.
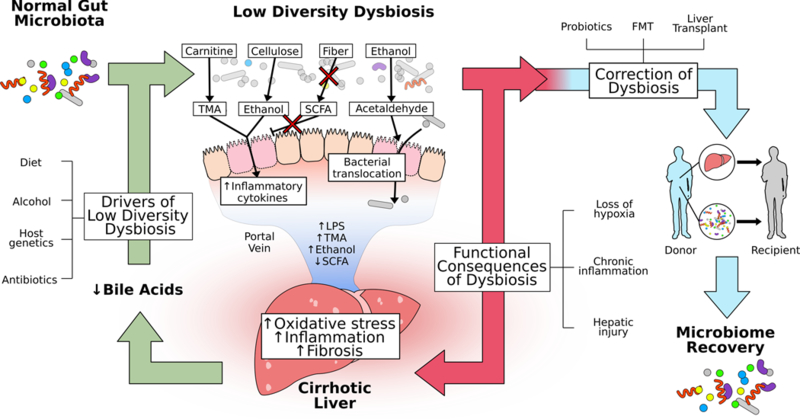
Low-diversity dysbiosis is a significant contributor to the development and progression of chronic liver disease. Drivers of low-diversity dysbiosis include both environmental (diet, alcohol, antibiotics) and genetic determinants. Low-diversity dysbiosis is characterized by relative increases in facultative anaerobes, possibly due to loss of intraluminal hypoxia leading to changes not only in microbiota composition but also function. Carnitine is metabolized by facultative anaerobic bacteria and as this population expands, production of hepatotoxic trimethylamine (TMA) increases. Ethanol, both as a fermentation product of cellulosic substrates and as a fermentation substrate leading to acetaldehyde, disrupts intestinal tight junctions and is a mediator of both mucosal and hepatic inflammation. Reciprocal loss of butyrate-producing bacteria leads to reduction in butyrate formation, further compromising colonocytes and promoting bacterial translocation. Collectively, these functional changes lead to increases in bacterial load in the portal vein and delivery of inflammatory mediators (TMA, LPS, ethanol) to the liver with reduction in anti-inflammatory metabolites (SCFA). Within the liver, this leads to increased oxidative stress, marked inflammation, and fibrogenesis. As liver disease progresses, there is impaired hepatic synthesis of both primary and secondary bile acids, further driving low-diversity dysbiosis. Therapeutic interventions aimed to facilitate microbiome recovery include probiotics and FMT. Liver transplantation also leads to microbiome recovery and is a novel clinical model to assess the kinetics of recovery of low-diversity dysbiosis and its functional consequences. (TMA = trimethylamine; LPS = lipopolysaccharide; FMT = fecal microbial transplant)
Functional Consequences of Dysbiosis:
With the establishment of low-diversity dysbiosis there are changes to the bacterial metabolic flux and direct interactions with the host immune system. Many anaerobes of the healthy gut ferment complex sugars to short chain fatty acids (SCFAs), including many species in Lachnospiraceae and Ruminococcaceae such as Faecalibacterium prausnitzii. These are among the groups whose relative abundance markedly decrease in low-diversity dysbiosis (Table 1). The SCFA butyrate has been shown to have both local and systemic anti-inflammatory effects and thus its loss may mediate immune phenotypes in disease [31]. For instance, liver cirrhosis results in the disproportionate loss of butyrate producing species, representing a possible mechanism by which low-diversity dysbiosis leads to hepatic inflammation [10,32]. Consistent with this hypothesis, butyrate administration has been shown to ameliorate innate immune activation in liver injury models [33].
Butyrate is also a primary calorie source for the colonic epithelium [34] and its consumption has been shown to be crucial for maintaining hypoxia in the lumen of the colon [35], thereby limiting colonization of pathogenic facultative anaerobes such as Salmonella enterica [36]. Taken together, this suggests that loss of butyrate producers plays a role in a compositional shift towards facultative anaerobes that occurs in low-diversity dysbiosis. Although butyrate-producers tend to be late colonizers of the gut, we have previously shown that certain butyrate-producing Clostridia, namely Clostridium symbiosum and Anaerostipes caccae, show ‘early successional’ distribution and also select for genes in their genomes for tolerance of oxidative stress [6]. These early colonizers of the gut that produce butyrate have the potential to be “ecosystem drivers”, because by depleting oxygen they may make the environment more favorable to the strict anaerobes that dominate the healthy adult gut (Fig. 1).
With an increase in luminal oxygen and the associated proportional increase in facultative anaerobes, the production of certain metabolites negatively associated with health may increase. For instance, the cntA gene, which is in the pathway that converts dietary carnitine to the atherogenic compound trimethylamine, has been linked with microbiomes rich in Gamma Proteobacteria, in particular Escherichia coli [37,38]. Endogenous alcohol production by intestinal facultative anaerobes has also been suggested as a contributor to non-alcoholic steatohepatitis [39]. Conversely, with a selection against anaerobes, other important metabolites for maintaining health that are produced through anaerobic metabolism may also be lost. One such example is secondary bile acids and CDI. In vitro work has shown that the primary bile salt taurocholate is a germination agent for C. difficile spores while secondary bile acids arrest the growth of vegetative C. difficile [40]. The secondary bile acids are believed to be mainly produced by a small number of Gram-positive anaerobes, notably Clostridium scindens [41], which are depleted during antibiotic treatment. A role for secondary bile acid metabolism in CDI treatment is suggested as C. scindens was shown to be protective in a mouse model of CDI [42] and clinical data from FMT trials support a central role for normalization of bacterial bile acid metabolism in resolution of rCDI [43]. Additionally, patients with first-time CDI showed an intermediate bile acid composition compared to patients with rCDI and healthy controls, suggesting fecal bile acid analysis may have a role in future diagnostics [44].
Correction of Dysbiosis:
Correction of low-diversity dysbiosis can be achieved by interventions as simple as supportive care, in the case of acute diarrheal disease, or as complex as liver transplantation. FMT from healthy donors into patients with rCDI, was first performed in the 1950’s to treat severe pseudomembranous colitis [44]. Over the past two decades FMT has been increasingly viewed as an effective treatment with clinical trials showing efficacy of >90% [45,46] and durability of treatment lasting years [47].
Microbiome analysis of patients undergoing FMT for rCDI has revealed low-diversity dysbiosis prior to FMT and marked restoration after FMT. As described above, the microbiome prior to FMT shows low-diversity dysbiosis as described in Table 1 [9,45]. These studies uniformly show marked shifts in composition from the pre-FMT state towards a healthy composition of anaerobes (Table 1).
With the incidence of IBD increasing worldwide much attention is directed toward FMT [48–50]. A meta-analysis of randomized control trials showed 49% of UC patients had a clinical response and 28% of patients went into clinical remission after FMT [50]. Response rates may be influenced by factors such as time since UC diagnosis, with one study showing higher response rates in recently diagnosed patients. Also, one study indicated high UC remission rates with stool from one particular donor, indicating that specific microbiome configurations in donor stools may be most efficacious [51]. This result is in contrast to the results of FMT in rCDI, where success rates are high regardless of donor stool. Identifying the microbes specific to donor stools with high remission rates would inform more targeted therapeutic strategies for UC. These promising results support the 30+ clinical trials currently underway, but other studies show possible adverse effects including disease flares and severe infections [52] underscoring the need for continued study of both efficacy and safety [48,49].
Liver transplantation, currently the only definitive therapy for patients with decompensated cirrhosis, provides a unique clinical model of microbial recovery following low-diversity dysbiosis. Because early innate immune activation impacts subsequent adaptive immunity in the allograft, the gut-liver axis represents an attractive target for intervention [53]. Observational data has demonstrated partial recovery of dysbiosis following liver transplant [54–56]. Furthermore, persistence of dysbiosis has been associated with post-transplant infectious complications and acute cellular rejection [54]. These observations are further supported by animal models demonstrating microbial signatures associated with acute cellular rejection (↓ Faecalibacterium) and modulation of microbial metabolites (i.e. SCFAs) to ameliorate liver injury post-transplant [33,57]. Taken together, these data support the emerging role for targeted, microbial-based therapeutics to facilitate recovery of dysbiosis in liver transplant recipients.
Conclusion:
We describe how dysbiosis in different disease systems can have a common manifestation of low diversity coupled with a proportional increase in facultative anaerobes and decrease in the typical anaerobes that dominate the adult healthy gut. We would like to emphasize that diversity or richness of the adult gut microbiome alone may be a poor marker of dysbiosis as it also may be driven by other factors such as transit time [58]. Also, as we have noted, dysbiosis can come in many forms, some of which have no change in or even increased diversity [58].
Loss of health-associated strict anaerobes may occur for different reasons in different diseases ranging from use of broad spectrum antibiotics, break-down in host-microbe interactions as facilitated by host genetics or other factors, or liver malfunction. New research has shown that butyrate plays a key role in maintaining hypoxia in the gut, suggesting that a loss of butyrate producers for any of the above reasons could result in a shared phenotype in which luminal oxygen increases and facultative anaerobes come to dominate. This results in a greatly altered metabolic environment that may favor increased production of detrimental metabolites such as TMA and alcohol and reduced production of metabolites that maintain health such as SCFAs and bile acid metabolites. Although a shared low-diversity dysbiosis phenotype in diverse diseases may suggest shared strategies for recovery, it is also important to consider that the driving factors of low-diversity dysbiosis vary and may be more persistent for some diseases than for others. As an example, FMT may be more effective for treatment of rCDI than IBD because the main driving factor of rCDI (antibiotics) is no longer present. In contrast, host genetic attributes that pre-dispose an individual to IBD do not change over time. Thus, efforts to correct low-diversity dysbiosis must consider both commonalities and unique attributes of the diseases in which it is observed.
Highlights:
Drivers of low-diversity dysbiosis include antibiotics, host genetics, physiology
Dysbiosis results in shifts in microbial composition and function
Restoration of eubiosis is an emerging treatment in CDI, IBD, and liver disease
Funding
This research was supported by the National Institutes of Health [grant number 5T32DK067009-12]
References:
- 1.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI: Host-bacterial mutualism in the human intestine. Science 2005, 307:1915–1920. [DOI] [PubMed] [Google Scholar]
- 2.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R: Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. : Human gut microbiome viewed across age and geography. Nature 2012, 486:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis MY, Zhang H, Brannan LE, Carman RJ, Boone JH: Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome 2016, 4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ: Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 2017, 8:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J, Gordon JI, Knight R: Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res 2012, 22:1974–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vazquez-Baeza Y, Jansson JK, Gordon JI, Knight R: Meta-analyses of studies of the human microbiota. Genome Res 2013, 23:1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David LA, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, Begum Y, Qadri F, LaRocque RC, Turnbaugh PJ: Gut microbial succession follows acute secretory diarrhea in humans. MBio 2015, 6:e00381–00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ: High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 2013, 4:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. : Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513:59–64. [DOI] [PubMed] [Google Scholar]
- 11.Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, et al. : Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol 2014, 60:940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C, Frank DN, Horch M, Chau S, Ir D, Horch EA, Tretina K, van Besien K, Lozupone CA, Nguyen VH: Associations between acute gastrointestinal GvHD and the baseline gut microbiota of allogeneic hematopoietic stem cell transplant recipients and donors. Bone Marrow Transplant 2017, 52:1643–1650. [DOI] [PubMed] [Google Scholar]
- 13.Loo VG, Bourgault AM, Poirier L, Lamothe F, Michaud S, Turgeon N, Toye B, Beaudoin A, Frost EH, Gilca R, et al. : Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med 2011, 365:1693–1703. [DOI] [PubMed] [Google Scholar]
- 14.Kavanagh K, Pan J, Marwick C, Davey P, Wiuff C, Bryson S, Robertson C, Bennie M: Cumulative and temporal associations between antimicrobial prescribing and community-associated Clostridium difficile infection: population-based case-control study using administrative data. J Antimicrob Chemother 2017, 72:1193–1201. [DOI] [PubMed] [Google Scholar]
- 15.Hourigan SK, Oliva-Hemker M, Hutfless S: The prevalence of Clostridium difficile infection in pediatric and adult patients with inflammatory bowel disease. Dig Dis Sci 2014, 59:2222–2227. [DOI] [PubMed] [Google Scholar]
- 16.Trifan A, Stoica O, Stanciu C, Cojocariu C, Singeap AM, Girleanu I, Miftode E: Clostridium difficile infection in patients with liver disease: a review. Eur J Clin Microbiol Infect Dis 2015, 34:2313–2324. [DOI] [PubMed] [Google Scholar]
- 17.Vehreschild MJ, Weitershagen D, Biehl LM, Tacke D, Waldschmidt D, Tox U, Wisplinghoff H, Von Bergwelt-Baildon M, Cornely OA, Vehreschild JJ: Clostridium difficile infection in patients with acute myelogenous leukemia and in patients undergoing allogeneic stem cell transplantation: epidemiology and risk factor analysis. Biol Blood Marrow Transplant 2014, 20:823–828. [DOI] [PubMed] [Google Scholar]
- 18.Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A: Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol 2017. [DOI] [PubMed] [Google Scholar]
- 19.Knoll RL, Forslund K, Kultima JR, Meyer CU, Kullmer U, Sunagawa S, Bork P, Gehring S: Gut microbiota differs between children with Inflammatory Bowel Disease and healthy siblings in taxonomic and functional composition: a metagenomic analysis. Am J Physiol Gastrointest Liver Physiol 2017, 312:G327–G339. [DOI] [PubMed] [Google Scholar]
- 20.Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK: Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J 2008, 2:716–727. [DOI] [PubMed] [Google Scholar]
- 21.Halfvarson J, Jess T, Magnuson A, Montgomery SM, Orholm M, Tysk C, Binder V, Jarnerot G : Environmental factors in inflammatory bowel disease: a co-twin control study of a Swedish-Danish twin population. Inflamm Bowel Dis 2006, 12:925–933. [DOI] [PubMed] [Google Scholar]
- 22.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. : Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. : Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015, 47:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hampe J, Cuthbert A, Croucher PJP, Mirza MM, Mascheretti S, Fisher S, Frenzel H, King K, Hasselmeyer A, MacPherson AJS, et al. : Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. The Lancet 2001, 357:1925–1928. [DOI] [PubMed] [Google Scholar]
- 25.Bajaj JS, Betrapally NS, Hylemon PB, Thacker LR, Daita K, Kang DJ, White MB, Unser AB, Fagan A, Gavis EA, et al. : Gut Microbiota Alterations can predict Hospitalizations in Cirrhosis Independent of Diabetes Mellitus. Sci Rep 2015, 5:18559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schubert K, Olde Damink SWM, von Bergen M, Schaap FG: Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol Rev 2017, 279:23–35. [DOI] [PubMed] [Google Scholar]
- 27.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. : Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. : Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62:1787–1794. [DOI] [PubMed] [Google Scholar]
- 29.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, Puchois V, Martin JC, Lepage P, Le Roy T, et al. : Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65:830–839. [DOI] [PubMed] [Google Scholar]
- 30.Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, Dulai PS, Caussy C, Bettencourt R, Highlander SK, et al. : Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab 2017, 25:1054–1062 e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA: Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology 2016, 5:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L: Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54:562–572. [DOI] [PubMed] [Google Scholar]
- 33.Qiao YL, Qian JM, Wang FR, Ma ZY, Wang QW: Butyrate protects liver against ischemia reperfusion injury by inhibiting nuclear factor kappa B activation in Kupffer cells. J Surg Res 2014, 187:653–659. [DOI] [PubMed] [Google Scholar]
- 34.Roediger WE: Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. : Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, et al. : Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rath S, Heidrich B, Pieper DH, Vital M: Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith K: Microbiota: Gut microbiota produce alcohol in patients with NASH. Nat Rev Gastroenterol Hepatol 2012, 9:687. [DOI] [PubMed] [Google Scholar]
- 39.Schnabl B, Brenner DA: Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sorg JA, Sonenshein AL: Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 2008, 190:2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wells JE, Berr F, Thomas LA, Dowling RH, Hylemon PB: Isolation and characterization of cholic acid 7alpha-dehydroxylating fecal bacteria from cholesterol gallstone patients. J Hepatol 2000, 32:4–10. [DOI] [PubMed] [Google Scholar]
- 42.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. : Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517:205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weingarden AR, Chen C, Bobr A, Yao D, Lu Y, Nelson VM, Sadowsky MJ, Khoruts A: Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am J Physiol Gastrointest Liver Physiol 2014, 306:G310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, Gerber GK, Bry L, Clish CB, Alm E, Korzenik JR: Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment Pharmacol Ther 2016, 43:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly CR, Khoruts A, Staley C, Sadowsky MJ, Abd M, Alani M, Bakow B, Curran P, McKenney J, Tisch A, et al. : Effect of Fecal Microbiota Transplantation on Recurrence in Multiply Recurrent Clostridium difficile Infection: A Randomized Trial. Ann Intern Med 2016, 165:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. : Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 2013, 368:407–415. [DOI] [PubMed] [Google Scholar]
- 47.Mamo Y, Woodworth MH, Wang T, Dhere T, Kraft CS: Durability and Long-Term Clinical Outcomes of Fecal Microbiota Transplant (FMT) Treatment in Patients with Recurrent Clostridium difficile Infection. Clin Infect Dis 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Browne AS, Kelly CR: Fecal Transplant in Inflammatory Bowel Disease. Gastroenterol Clin North Am 2017, 46:825–837. [DOI] [PubMed] [Google Scholar]
- 49.Khajah MA: The potential role of fecal microbiota transplantation in the treatment of inflammatory Bowel disease. Scand J Gastroenterol 2017, 52:1172–1184. [DOI] [PubMed] [Google Scholar]
- 50.Costello SP, Soo W, Bryant RV, Jairath V, Hart AL, Andrews JM: Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment Pharmacol Ther 2017, 46:213–224. [DOI] [PubMed] [Google Scholar]
- 51.Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, Armstrong D, Marshall JK, Kassam Z, Reinisch W, et al. : Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149:102–109.e106. [DOI] [PubMed] [Google Scholar]
- 52.Bak SH, Choi HH, Lee J, Kim MH, Lee YH, Kim JS, Cho YS: Fecal microbiota transplantation for refractory Crohn’s disease. Intest Res 2017, 15:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bromberg JS, Fricke WF, Brinkman CC, Simon T, Mongodin EF: Microbiota-implications for immunity and transplantation. Nat Rev Nephrol 2015, 11:342–353. [DOI] [PubMed] [Google Scholar]
- 54.Kato K, Nagao M, Miyamoto K, Oka K, Takahashi M, Yamamoto M, Matsumura Y, Kaido T, Uemoto S, Ichiyama S: Longitudinal Analysis of the Intestinal Microbiota in Liver Transplantation. Transplant Direct 2017, 3:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H, He J, Wu Z, Xu W, Zhang H, Ye P, Yang J, Zhen S, Li L: Assessment of microbiome variation during the perioperative period in liver transplant patients: a retrospective analysis. Microb Ecol 2013, 65:781–791. [DOI] [PubMed] [Google Scholar]
- 56.Bajaj JS, Fagan A, Sikaroodi M, White MB, Sterling RK, Gilles H, Heuman D, Stravitz RT, Matherly SC, Siddiqui MS, et al. : Liver transplant modulates gut microbial dysbiosis and cognitive function in cirrhosis. Liver Transpl 2017, 23:907–914. [DOI] [PubMed] [Google Scholar]
- 57.Ren Z, Jiang J, Lu H, Chen X, He Y, Zhang H, Xie H, Wang W, Zheng S, Zhou L: Intestinal microbial variation may predict early acute rejection after liver transplantation in rats. Transplantation 2014, 98:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falony G, Vieira-Silva S, Raes J: Richness and ecosystem development across faecal snapshots of the gut microbiota. Nat Microbiol 2018, 3:526–528. [DOI] [PubMed] [Google Scholar]