

Abstract
High mobility group A1 (HMGA1) chromatin remodeling proteins are enriched in aggressive cancers and stem cells, although their common function in these settings has remained elusive until now. Recent work in murine intestinal stem cells (ISC) revealed a novel role for Hmga1 in enhancing selfrenewal by amplifying Wnt signaling, both by inducing genes expressing Wnt agonist receptors and Wnt effectors. Surprisingly, Hmga1 also “builds” a stem cell niche by upregulating Sox9, a factor required for differentiation to Paneth cells; these cells constitute an epithelial niche by secreting Wnt and other factors to support ISCs. HMGA1 is also highly upregulated in colon cancer compared with nonmalignant epithelium and SOX9 becomes overexpressed during colon carcinogenesis. Intriguingly, HMGA1 is overexpressed in diverse cancers with poor outcomes, where it regulates developmental genes. Similarly, HMGA1 induces genes responsible for pluripotency and self-renewal in embryonic stem cells. These findings demonstrate that HMGA1 maintains Wnt and other developmental transcriptional networks and suggest that HMGA1 overexpression fosters carcinogenesis and tumor progression through dysregulation of these pathways. Studies are nowneeded to determine more precisely how HMGA1 modulates chromatin structure to amplify developmental genes and how to disrupt this process in cancer therapy.
Chromatin and Cell Fate
Emerging evidence underscores the key role for chromatin binding proteins in maintaining nuclear organization critical for stem cell properties, both during development and oncogenesis. Indeed, nuclear structure is the most important feature that distinguishes a cancer cell from a normal cell histologically (1). Both stem cells and poorly differentiated cancer cells harbor enlarged nuclei with open, “poised” chromatin (2), which may endow them with plasticity or potential for multiple cell fate decisions. While the molecular underpinnings of chromatin structure and stem cell transcriptional programs are beginning to emerge, a better understanding of these networks promises to provide insight into cancer and development. Here, we focus on HMGA1 in “stemness” and carcinogenesis.
HMG Proteins
Remarkably, eukaryotic DNA is condensed from >2 meters to <20 mm by association with nuclear proteins. Nucleoproteins and DNA comprise chromatin, and histones are the most abundant chromatin binding proteins. Histones compact DNA by creating positively charged octamer “spools” around which negatively charged DNA fibers wrap. HMG proteins are the next most abundant class of chromatin binding proteins (3–6), which were discovered in the 1970s in calf thymus using salt extraction and solubility in trichloroacetic acid (7–8). Ten years later, HMGA proteins were separated from other HMG proteins by their phosphorylation status in cancer cells (9–10). Intriguingly, histone H1 and HMGA share significant homology in plants and lower organisms, suggesting that they evolved from the same ancestral protein (11).
Today, HMG proteins are classified into 3 families: HMGB, HMGN, HMGA. All are basic, low-molecular-weight proteins that migrate rapidly through polyacrylamide gel, hence the name high mobility group. They all contain an acidic carboxyl terminus, although each family is defined by unique DNA or nucleosome binding motifs. All modify chromatin structure, but each has distinct functions.
HMGB
HMGB (HMGB1, HMGB2, HMGB3, and HMGB4) are the most abundant HMG proteins. They are distinguished by 2 HMG-box motifs that mediate binding to DNA without sequence specificity (5, 12–13). HMG boxes are formed by 3 alpha helices that fold into an L-shape that penetrates the minor groove of DNA, inducing a sharp bend. Unlike other HMG proteins, HMGB proteins function as cytokines mediating paracrine signaling (12–13). “HMG-box proteins” comprise a larger class of proteins that includes HMGB, and less abundant proteins with one or more HMG-boxes (SOX9, SRY, LEF1, TCF). In contrast to HMGB, proteins with only one HMG-box bind DNA with sequence specifically. The acidic carboxyl terminus modulates their affinity for different DNA structures. HMGB proteins participate in cell fate decisions, DNA damage responses, and senescence. Increasing evidence also implicates HMGB as key signaling molecules in cancer (13).
HMGN
HMGN proteins (HMGN1, HMGN2, HMGN3, HMGN4, and HMGN5) are found only in vertebrates. They lack an HMG-box, but contain a positively charged, nucleosome-binding “N” domain that mediates binding to nucleosomes (5). The acidic carboxyl terminus or “chromatin unfolding domain” alters DNA architecture, inducing changes in local organization and higher order structure. HMGN proteins “relax” DNA by competing for nucleosome binding with histone H1, which tends to compact DNA (14). HMGN proteins also recruit active histone marks (histone 3 lysine 14 acetylation; H3K14Ac; ref. 15), deplete repressive marks (H3K17trimethylation; ref. 16), and oppose ATP-dependent remodeling proteins that restrict nucleosome motility (17). Recent work in Down syndrome leukemia found that triplication of chromosome 21 causes HMGN1 overexpression, which contributes to leukemogenesis (16).
HMGA
HMGA proteins—the focus of this review—are distinguished from other HMG families by 3 AT-hook motifs that mediate binding to the minor groove of B-form DNA at AT-rich sequences (3–6, 18–20). This family includes HMGA1a/HMGA1b isoforms, encoded by the HMGA1 gene (human chr 6p21) through alternatively spliced mRNA; HMGA1c, encoded by a rare splice variant found in testes; and (iii) HMGA2, an HMGA1 homolog encoded by HMGA2 (human chr 12q14). Like HMGN, these proteins lack an HMG-box, but in contrast to HMGN or HMGB, they bind to DNA with sequence specifically (18–26). By NMR structural analysis, the AT-hooks are unstructured, although they transition to a highly ordered structure after binding DNA and/or protein partners (23). Like HMG boxes, AT-hooks penetrate the minor groove to induce bending. HMGA proteins also harbor an amino-terminal serine-, threonine-rich domain, although its function is unclear. The acidic carboxyl terminus mediates protein–protein interactions (5). Similar to HMGN, HMGA1 competes with histone H1 for DNA binding in vitro (21). After displacing histone H1, HMGA widens or “opens” the minor groove, facilitating recruitment of transcription factor complexes and chromatin modifiers to modulate gene expression (Fig. 1A; refs. 4–6, 27–35).
Figure 1.
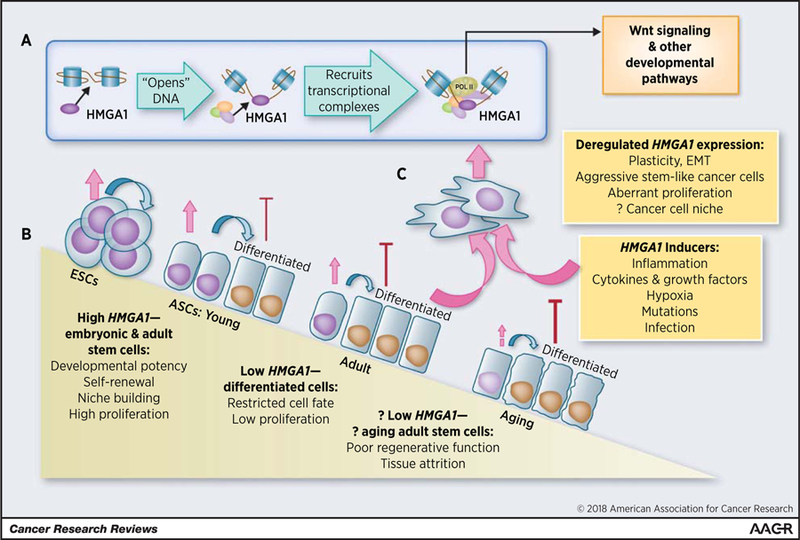
HMGA1 remodels chromatin to drive developmental transcriptional networks in cancer and stem cells. A, HMGA1 binds to DNA, “opens” chromatin, and recruits transcriptional complexes to activate Wnt genes and other developmental transcriptional networks. B, In ESCs and adult stem cells, HMGA1 is highly expressed, where it fosters plasticity, regenerative function, self-renewal, niche building, and proliferation, whereas HMGA1 is low or silenced in differentiated cells. Data in murine and human adult stem cells suggest that HMGA1/Hmga1 levels decline with aging, which could contribute to decreased regenerative function and tissue attrition. C, In contrast, HMGA1 is induced by many factors, which, in the setting of an aged and/or mutated genome, could drive plasticity, EMT, neoplastic transformation, and cancer stem cell properties. It may also help to establish a “cancer cell niche.” This model predicts that tightly regulated HMGA1 is essential for normal regenerative function, and possibly “normal” aging, whereas deregulated overexpression fosters tumor initiation and progression.
HMGA1 Portends Poor Outcomes
The first evidence linking HMGA1/2 to cancer was their discovery in extraordinarily proliferative HeLa cervical cancer cells (10). Subsequent studies showed that HMGA1/2 become overexpressed in diverse tumors arising from all three germ layers (18–20). While the list is expanding, HMGA1 is overexpressed in cancers of the brain, head and neck, esophagus, thyroid, lung, breast, prostate, colon, rectum, pancreas, liver, uterine corpus, cervix, skin, and hematopoietic system (reviewed in ref. 19). HMGA2 is also overexpressed in diverse cancers, although less broadly (18–20, 36–42). With the advent of microarray and RNAsequencing technology, it became clear that HMGA1/2 overexpression correlates with poor differentiation and adverse clinical outcomes in diverse tumors (43–44). Indeed, the first study using mRNA microarrays revealed that HMGA1 is among the genes associated with poor survival in medulloblastoma (43). High HMGA1 also correlates with relapse in childhood leukemia (44). HMGA1/2 are highly expressed during embryogenesis with low or undetectable levels in adult, differentiated tissues (45, 46). Similarly, HMGA1/2 genes are enriched in ESCs (46–49) and many tissue-specific, adult stem cells (49–51). In fact, HMGA1 was identified among a signature of 13 transcription factor genes most enriched in human ESCs (48). Strikingly, this signature predicts poor outcomes in breast, bladder, and brain cancer (48). Immunohistochemical analysis of HMGA proteins in primary tumors further validated gene expression studies, demonstrating high levels with poor differentiation and metastatic progression (52–53). To illustrate, HMGA1 immunoreactivity is present in >90% of pancreatic ductal adenocarcinomas (53–55) and correlates positively with poor differentiation and decreased survival (53). While HMGA1 was undetectable in normal tissue and early precursor lesions, immunoreactivity occurs in late precursor lesions and invasive tumors, suggesting a role in tumor progression (53). In contrast, HMGA2 immunoreactivity is present in 30% of tumors, where it associated with lymph node metastases and poor survival (41). In general, tumors with high HMGA1 lacked HMGA2 and vice versa. In contrast, studies in lung (39–40, 52), breast (42, 56), colon (42, 57–59), and other malignancies showed overexpression of both HMGA1/2 genes and proteins, suggesting that they collaborate in these settings. Together, these studies highlight HMGA1/2 as potential biomarkers and therapeutic targets in diverse tumors.
While HMGA1/2 genes are overexpressed in cancer, the mechanisms that mediate their expression are only beginning to emerge. Both Hmga1/2 are robustly induced by serum or growth factors (FGF, EGF, and PDGF) in murine fibroblasts rendered quiescent by serum deprivation (60). Similarly, HMGA1/Hmga1 is upregulated by EGF or phorbol esters in MCF-7 breast cancer cells (61) or by IL2 in mouse T cells (CTLL; ref. 60). In fibroblasts, Hmga1/2 display delayed-early kinetics with maximal induction within 5 to 10 hours following growth factor stimulation (60). Their transcription requires new protein synthesis, suggesting that immediate-early transcription factors induce HMGA1/2 expression. In fact, HMGA1 is a direct transcriptional target of MYC oncoproteins, and HMGA1 is overexpressed in tumors driven by MYC (Burkitt lymphoma, neuroblastoma; refs. 62–63). AP1 transcription factors also induce HMGA1 (64, 65) and HMGA1 is upregulated in tumors linked to inflammation (colon, refs. 57–59, 66; esophageal, ref. 67; cervical carcinomas, ref. 68) as well as experimental models of viral infection (27–34). Following viral infection, HMGA1 recruits NF-kB to enhancer complexes, where it transactivates IFNb (27–34). HMGA1 also upregulates other proinflammatory genes, including STAT3 (69, 70) and COX-2 (71, 72). STAT3 may induce HMGA1, providing a feed-forward loop to maintain HMGA1 (69–70). Hypoxia also induces HMGA1 in vascular endothelial cells, which promotes angiogenesis through COX-2 (73). In rare cases, chromosomal duplication or translocations cause HMGA1 overexpression, although translocations and fusion genes involve HMGA2 more commonly (36, 74). Loss of tumor suppressor microRNAs, such as Let-7, induces HMGA2 (75–76). APC mutations, common early events in colorectal cancer, repress mir-26, resulting in overexpression of Hmga1 in murine intestinal epithelium (77). Together, these findings suggest a model whereby hyperactive growth factor signaling, mutations, infection, and inflammation converge on HMGA1/2 to upregulate their expression.
Cancer and Embryogenesis
Following their discovery, functional studies of HMGA1/2 revealed potent oncogenic and stem cell properties. Forced expression of Hmga1a, Hmga1b, or Hmga2 induces oncogenic transformation (62, 78–80), recapitulating cMYC phenotypes in immortalized cells, including anchorage-independent cell growth and xenograft tumorigenesis in immunosuppressed mice. Trangenic mice overexpressing murine Hmga1a from the H-2K promoter and m enhancer develop aggressive lymphoid tumors (80). During tumorigenesis, Hmga1 upregulates genes involved in proliferation, inflammation, and hematologic development (81). Female transgenics also develop uterine sarcomas that depend, at least in part, on COX-2 upregulation (71). Transgenics overexpressing human HMGA1b from the CMV promoter develop lymphoid tumors and pituitary adenomas (82), while those overexpressing a truncated HMGA2 develop lipomatosis and gigantism (83–84). Functional studies demonstrated that blocking HMGA1 expression profoundly disrupts cancer phenotypes. For example, silencing HMGA1 in breast cancer cells halts proliferation and reprograms invasive, mesenchymal cells into noninvasive, epithelial-like cells (56). Both orthotopic tumorigenesis and metastatic progression to the lungs were disrupted. HMGA1 silencing also depletes tumor initiator/ cancer stem cells in limiting dilution tumor assays and prevents three-dimensional (3D) sphere formation (56). Similar results were observed in cancer cells from colon (57), pancreas (54), lung (52), and others. In some cancer cells (breast, colon, and pancreatic), tumor progression, epithelial–mesenchymal transition (EMT), and cancer stem cell properties depend upon HMGA2 (42, 58, 75, 85). These studies showed that HMGA1/2 genes promote tumor progression, at least in part, by hijacking EMT and other stem cell pathways (Fig. 1B and C).
Studies in human ESCs revealed that HMGA1 decreases with differentiation and parallels that of the pluripotency factors, while forced Hmga1 expression blocks differentiation (46). When included with the Yamanaka reprogramming cocktail, HMGA1 enhances the derivation of induced pluripotent stem cells (iPSC), resulting in larger, more abundant colonies (46). Mechanistic studies showed that HMGA1 occupies promoters of pluripotency genes and induces their expression. Intriguingly, another group showed that HMGA2 slightly decreases reprogramming efficiency to iPSCs by the Yamanaka factors (47). Mice lacking Hmga2 exhibit a pygmy phenotype with decreased fat tissue (86). Hematopoietic stem cells lacking Hmga2 have defective fetal hematopoiesis with slower proliferation and self-renewal rates, whereas Hmga2 is dispensable for adult hematopoiesis (87). Mice deficient in Hmga1 have been described, with decreased spermatogenesis and infertility in one model (88), and a diabetes-like phenotype (89) with cardiomegaly and aberrant hematopoiesis (90), in another. In preliminary studies, our group found premature aging and partial embryonic lethality in Hmga1-deficient mice (91). Further research is needed to better understand the function of HMGA1/2 during embryogenesis and aging.
Hmga1 Amplifies Wnt and Self-Renewal
Recent work uncovered a unique role for Hmga1 in maintaining both stem cells and the niche compartment in intestinal epithelium (Fig. 2; ref. 51). Hmga1a transgenic mice develop hyperproliferation, aberrant crypt formation, and polyposis involving small and large intestines (51). To determine how this occurs, crypt cultures from Hmga1 mice were compared with those from wild-type controls, revealing a marked increase in organoid formation, organoid and bud size, and bud number from Hmga1 cells (51). Because ISCs localize to bud tips recapitulating in vivo crypt organization, bud number is a surrogate for ISC number and/or function (92). To enumerate ISCs, Hmga1 mice were crossed to EGFP-Lgr5+ mice, which mark crypt basilar Lgr5+ ISCs with green fluorescent protein (GFP; ref. 92). ISC frequency was increased in all regions of small intestine in Hmga1 mice compared with controls. In both controls and transgenic mice, Hmga1 mRNA and protein are enriched in Lgr5+ ISCs, but with higher levels in the transgenic model (51). To determine whether Hmga1 regulates self-renewal, purified Lgr5+ ISC cultures were followed using time-lapsed imaging and demonstrated an increase in self-renewal rates. Colony formation and replating efficiency were also increased, further demonstrating enhanced stem cell function (51). In contrast, silencing Hmga1 in crypt cultures disrupts organization of 3D organoids and bud formation, while overexpressing Hmga1 in wild-type crypt cells phenocopies the organoids from Hmga1 mice. Intriguingly, Hmga1- overexpressing organoids exposed to Wnt exhibit exaggerated responses, forming larger cystic structures with more Lgr5+ ISCs compared with controls (51). Conversely, Hmga1 crypts are relatively impervious to Wnt inhibitors, suggesting that Hmga1 amplifies Wnt signals, possibly by upregulating Wnt signaling genes. To test this, canonical Wnt pathway gene expression was interrogated in Lgr5+ ISCs. Strikingly, Hmga1 upregulated genes encoding both Wnt agonist receptors and downstream Wnt effectors (51). While the mechanisms are not yet known, Hmga1 could directly transactivate Wnt effector gene expression or indirectly upregulate their expression through changes in chromatin architecture.
Figure 2.
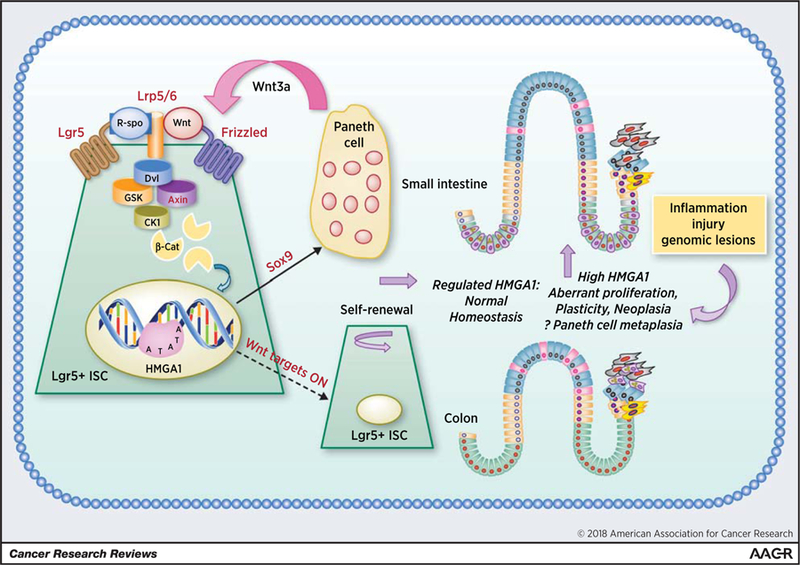
Tightly regulated HMGA1 fosters balanced self-renewal and “builds a niche” in normal intestinal homeostasis (left). In contrast, deregulated, overexpressed HMGA1 drives aberrant plasticity, EMT, cancer stem cell properties, proliferation/polyposis, and may also “build” a “cancer stem cell niche” through Paneth cell metaplasia in the colon (right). Proteins encoded by genes that are upregulated in Hmga1 transgenic Lgr5+ ISCs are indicated by red text. (Figure adapted from reference 51).
HMGA1 “Builds” a Niche
Surprisingly, Hmga1 also helps “build” a Paneth cell niche by upregulating Sox9, a Wnt target gene that is essential for Paneth cell differentiation (51, 93, 94). This was unexpected because Paneth cells are terminally differentiated; they also provide an epithelial-specific niche for ISCs by secreting Wnt. Additionally, Paneth cell granules protect the intestinal epithelium from bacteria and other pathogens by releasing lysozyme and other enzymes. Hmga1 binds directly to the Sox9 promoter to induce its expression (51). Accordingly, both Sox9 mRNA and protein are upregulated in Hmga1 transgenic intestinal epithelium and organoid cultures. Further, Sox9 overexpression in organoids is sufficient for Paneth cell expansion in this model (51). This was the first example of Hmga1 promoting terminal differentiation to establish a stem cell niche.
Hmga1/2 in Colorectal Carcinogenesis
In human colonic epithelium, HMGA1 and SOX9 are positively correlated and both become markedly upregulated during colorectal carcinogenesis [data from The Cancer Genome Atlas (TCGA); ref. 51]. In cancer, however, their correlation is lost, which likely occurs because colonic epithelium acquires multiple mutations during carcinogenesis, some of which enhance SOX9 or HMGA1 independently (95). Prior studies identified HMGA1 among the genes most upregulated in colon cancer compared with nonmalignant epithelium (66). These data support a model whereby tightly regulated HMGA1 and SOX9 collaborate in intestinal homeostasis, whereas both become deregulated and overexpressed in cancer.
Intriguingly, transgenic mice overexpressing Lin28 in the intestinal epithelium develop Hmga2 overexpression and findings similar to Hmga1 mice with intestinal hyperproliferation and polyps (58). Accelerated adenomas and adenocarcinomas also form and these phenotypes depend upon repression in Let-7b/c by knockout of the mirLet7c2/mirLet7b locus in the intestinal epithelium in concert with Lin28 overexpression (59). In contrast to Hmga1 mice, Paneth cells are depleted, both in Lin28 mice and in Let-7b/c-deficient mice, suggesting that a threshold level of Let-7b/c is needed for Paneth cell development (58–59). Wnt signaling and stem cell genes are upregulated in intestinal epithelium in both Lin28-overexpressing and Let-7–deficient models. In addition to Hmga2, Igf2bp1, and to a lesser extent, Hmga1, are induced in intestinal crypts from both models (58–59). Hmga2 overexpression in organoids phenocopies the Lin28 organoids whereas heterozygous loss of Hmga2 decreases tumorigenesis in Lin28 transgenic mice (59). Human colorectal tumors overexpress HMGA1 or HMGA2, and HMGA1/2 overexpression associates with poor survival in a subset of tumors (TCGA data; ref. 59). Together, these findings indicate that Hmga2 or Hmga1 confer proliferative and stem-like programs when overexpressed in mouse intestinal epithelium, while human tumor data further implicate HMGA1/2 as potential therapeutic targets in colorectal cancer.
Implications and Future Directions
The recent work highlighted here revealed for the first time that intestinal overexpression of Hmga1 or Hmga2 amplifies Wnt signaling, causing hyperproliferation and polyposis. Hmga1 drives ISC expansion by enhancing self-renewal, although it is not yet known whether Hmga2 alters the ISC number or function (52). Hmga1 also upregulates Wnt agonist receptors. Recent work also uncovered an unexpected role for Hmga1 in “building a niche” by fostering Paneth cell differentiation through Sox9. In contrast, mice overexpressing Lin28 repress Let-7b/c and deplete Paneth cells, despite an upregulation in Hmga1/2 and other stem cell genes, indicating that Paneth cell development may require Let-7. HMGA1/2 genes are enriched in human ISCs (50–51) and other tissue-specific stem cells, including hematopoietic (49) and mesenchymal (96); they may be critical regulators in diverse adult stem cells. Prior studies showed declining Hmga1/HMGA1 in murine and human hematopoietic stem cells with aging (97, 98), which could contribute to decreasing regenerative function with age. Our knockout mouse model also suggests that Hmga1 deficiency causes aging phenotypes, possibly through attrition in stem cell number or function (91). Because HMG proteins foster “open” chromatin, it is plausible that HMGA1/2 promote epigenetic alterations and a chromatin state that permits multiple cell fate decisions, plasticity, and regenerative function, not only in adult stem cells but also in aggressive, stem-like cancers.
Overexpression of HMGA1/2 and SOX9 in colonic epithelium could also collaborate in tumor initiation and progression. Multiple genetic lesions are acquired during colon carcinogenesis, such as APC mutations and other genes in this pathway, including SOX9 (95). APC mutations generally occur early and may upregulate HMGA1 by repressing miR-26 as demonstrated in murine models (77). Functional studies show that HMGA1 is required for metastatic progression and stem cell properties in colorectal cancer models (57). Thus, mutant APC could induce both HMGA1 and SOX9 during tumor progression. These data, together with results showing that Tcf4 binds to the HMGA1 promoter in colorectal cancer cells (99), suggest that HMGA1 orchestrates a “feed-forward” loop whereby Wnt/ Tcf4/b-catenin induces HMGA1, and HMGA1, in turn, amplifies Wnt and other developmental pathways to drive tumor progression. Wnt signaling also upregulates HMGA1 in gastric cancer (100), and there are likely to be many developmental pathways linked to HMGA1 during tumorigenesis. HMGA2 also promotes tumor progression and stem cell properties (40, 58–59, 75–76).
While Paneth cells are absent in normal colon, Paneth cell “metaplasia” occurs with inflammatory bowel disease, adenomas, and in a subset of colon cancers (101), and HMGA1 could foster their development. Although their function is unknown, colonic Paneth-like cells could provide a “cancer niche” to support and nurture malignant cells. Disrupting this pathway may provide a unique opportunity to target stem-like cancer cells, although further work is needed to test this.
As Siddhartha Mukherjee so eloquently wrote in the Emperor of all Maladies (102), “cancer cells are distortions of our normal selves.” Indeed, cancer cells distort normal development and HMGA proteins could represent fundamental “distorters” where signals converge, but become amplified and warped to foster hyperactive Wnt signaling, stem-like networks, niche development, and tumor progression. During normal development, HMGA1 also serves as a discriminating conductor, precisely orchestrating Wnt and stem cell pathways. Studies are now needed to dissect mechanisms that distinguish normal regeneration from distorted processes manifest in cancer cells and their microenvironment. Through this work, we will hopefully gain the capacity to slay this malevolent emperor and harness the regenerative potential of tissue-specific stem cells.
Acknowledgments
We thank Ray Reeves for his important contributions to the field and our many other esteemed colleagues and collaborators. We also thank our superlative prior mentors, especially Barbara Migeon, Chi Dang, and the late Donald Coffey and Daniel Nathans. We apologize for omitting many important references due to space limitations.
L. Resar recognizes support from the NIDDK (1R01 DK102943), NCI (R21 CA187495, R03 CA164677, R03 CA182679, R21 CA149559), Alex’s Lemonade Stand Foundation, the Maryland Stem Cell Research Fund, the RALLY Foundation, the Rosencrans Fund, the Patrick C. Walsh Prostate Cancer Fund, AVON, the Sol Goldman Pancreatic Research Fund, and the American Lung Cancer Association. L. Xian also recognizes the Maryland Stem Cell Research Fund and the Hopkins Digestive Diseases Basic Research Core Center.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Nelson WG, Pienta KJ, Barrack ER, Coffey DS. The role of the nuclear matrix in the organization and function of DNA. Ann Rev Biopys Chem 1986;15:457–75. [DOI] [PubMed] [Google Scholar]
- 2.Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer 2014;14:747–53. [DOI] [PubMed] [Google Scholar]
- 3.Bustin M Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol 1999;19:5237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim Biophys Acta 2001; 1519:13–29. [DOI] [PubMed] [Google Scholar]
- 5.Hock R, Furusawa T, Ueda T, Bustin M. HMG chromosomal proteins in development and disease. Trends Cell Biol 2007;17:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sumter TF, Xian L, Huso T, Koo M, Chang YT, Almasri TN, et al. The high mobility group A1 (HMGA1) transcriptome in cancer and development. Curr Mol Med 2016;16:353–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin GH, Sanders C, Johns EW. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem 1973;38:14–9. [DOI] [PubMed] [Google Scholar]
- 8.Goodwin G, Walker JH, Johns EW. The high mobility group (HMG) nonhistone chromosomal proteins. The cell nucleus New York: Academic Press; 1978;181–219. [Google Scholar]
- 9.Lund T, Holtlund J, Fredriksen M, Laland SG. On the presence of two new high mobility group-like proteins in Hela S3 cells. FEBS Lett 1983;152: 163–7. [DOI] [PubMed] [Google Scholar]
- 10.Lund T, Holtlund J, Laland SG. On the phosphorylation of low molecular mass HMG (high mobility group) proteins in Ehrlich ascites cells. FEBS Lett 1985;180:275–9. [DOI] [PubMed] [Google Scholar]
- 11.Krech AB, Wulff D, Grasser KD, Feix G. Plant chromosomal HMGI/Y proteins and histone H1 exhibit a protein domain of common origin. Gene 1999;230:1–5. [DOI] [PubMed] [Google Scholar]
- 12.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Aspects Med 2014;41:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aird K, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 2016;215:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catez F, Brown DT, Misteli T, Bustin M. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep 2002; 3:760–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim J-H, West KL, Rubinstein Y, Berget M, Postnikov YV, Bustin M. Chromosomal protein HMGN enhances acetylation of lysine 14 in histone H3. EMBO J 2005;24:3038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet 2014;46:618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rattner BP, Yusufzai T, Kadonaga JT. HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol Cell 2009;34:620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Resar LM. The high mobility group A1 gene: transforming inflammatory signals into cancer? Cancer Res 2010;70:436–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah SN, Resar LM. High mobility group A1 and cancer: Potential biomarker and therapeutic target. Histol Histopathol 2012;27:567–79. [DOI] [PubMed] [Google Scholar]
- 20.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer 2007;7:899–910. [DOI] [PubMed] [Google Scholar]
- 21.Zhao K, Kas E, Gonzalez E, Laemmli UK. SAR-dependent mobilization of histone H1 by HMG-I/Y in vitro: Hmg-I/Y is enriched in H1-depleted chromatin. Embo J 1993;12:3237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saitoh Y, Laemmli UK. Metaphase chromosome structure: Bands arise from a differential folding path of the highly AT-rich scaffold. Cell 1994;76:609–22. [DOI] [PubMed] [Google Scholar]
- 23.Huth JR, Bewley CA, Nissen MS, Evans JNS, Reeves R, Gronenborn AM, et al. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding protein. Nat Struct Biol 1997; 4657–65. [DOI] [PubMed]
- 24.Geierstanger BH, Volkman BF, Kremer W, Wemmer DE. Short peptide fragments derived from HMG-I/Y proteins bind specifically to the minor groove of DNA. Biochemistry 1994;33:5347–55. [DOI] [PubMed] [Google Scholar]
- 25.Maher JF, Nathans D. Multivalent DNA-binding properties of the HMG-1 proteins. Proc Natl Acad Sci USA 1996;93:6716–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banks GC, Mohr B, Reeves R. The HMG-I(Y) AT-hook peptide motif confers DNA-binding specificity to a structured chimeric protein. J Biol Chem 1999;274:16536–44. [DOI] [PubMed] [Google Scholar]
- 27.Du W, Maniatis T. The high mobility group protein HMG I(Y) can stimulate or inhibit DNA binding of distinct transcription factor ATF-2 isoforms. Proc Natl Acad Sci USA 1994;91:11318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-kappa b-dependent virus induction of the human IFN-beta gene. Cell 1992;71:777–89. [DOI] [PubMed] [Google Scholar]
- 29.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 1995;83: 1091–100. [DOI] [PubMed] [Google Scholar]
- 30.Falvo JV, Thanos D, Maniatis T. Reversal of intrinsic DNA bends in the IFN beta gene enhancer by transcription factors and the architectural protein HMG I(Y). Cell 1995;83:1101–11. [DOI] [PubMed] [Google Scholar]
- 31.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/ p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell 1998;1:277–87. [DOI] [PubMed] [Google Scholar]
- 32.Munshi N, Merika M, Yie J, Senger K, Chen G, Thanos D. Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol Cell 1998;2:457–67. [DOI] [PubMed] [Google Scholar]
- 33.Yie J, Merika M, Munshi N, Chen G, Thanos D. The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. Embo J 1999;18: 3074–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science 2001;293:1133–6. [DOI] [PubMed] [Google Scholar]
- 35.Dragan AI, Carrillo R, Gerasimova TI, Privalov PL. Assembling the human IFN-beta enhanceosome in solution. J Mol Biol 2008;384:335–48. [DOI] [PubMed] [Google Scholar]
- 36.Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat Genet 1995;10:436–44. [DOI] [PubMed] [Google Scholar]
- 37.Rogalla P, Drechsler K, Frey G, Hennig Y, Helmke B, Bonk U, et al. HMGI-C expression patterns in human tissues: implications for the genesis of frequent mesenchymal tumors. Am J Pathol 1996;149:775–9. [PMC free article] [PubMed] [Google Scholar]
- 38.Hess JL. Chromosomal translocations in benign tumors: the HMGI proteins. Am J Clin Pathol 1998;109:251–61. [DOI] [PubMed] [Google Scholar]
- 39.Sarhadi VK, Wikman H, Salmenkivi K, Kuosma E, Sioris T, Salo J, et al. Increased expression of high mobility group A proteins in lung cancer. J Pathol 2006;209:206–12. [DOI] [PubMed] [Google Scholar]
- 40.Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, Schuldenfrei A, et al. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res 2008;6:743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hristov A, Cope L, Delos Reyes M, Singh M, Iacobuzio-Donahue C, Maitra A, et al. HMGA2 protein expression correlates with lymph node metastasis and increased tumor grade in pancreatic ductal adenocarcinoma. Mod Pathol 2009;22:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, et al. HMGA2 is a driver of tumor metastasis. Cancer Res 2013;73:4289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002;415:436–42. [DOI] [PubMed] [Google Scholar]
- 44.Roy S, Di Cello F, Kowalski J, Hristov AC, Tsai HL, Bhojwani D, et al. Hmga1 overexpression correlates with relapse in childhood B-lineage acute lymphoblastic leukemia. Leuk Lymphoma 2013;54:2565–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996;13:2439–46. [PubMed] [Google Scholar]
- 46.Shah SN, Kerr C, Cope L, Zambidis E, Liu C, Hillion J, et al. HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PLoS ONE 2012;7:e48533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morshedi A, Ren Z, Li J, Droge P. Probing into biologic processes influenced by ESC factor and oncoprotein HMGA2 using iPSCS. Stem Cell Rev and Rep 2013;9:514–522. [DOI] [PubMed] [Google Scholar]
- 48.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou BK, Mali P, Huang X, Ye Z, Dowey SN, Resar LM, et al. Efficient human IPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res 2011;21:518–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent +`4’ cell+ markers. Embo J 2012;31:3079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xian L, Georgess D, Huso T, Cope L, Belton A, Chang YT, et al. Hmga1 amplifies Wnt Signaling and expands the intestinal stem cell compartment and Paneth cell niche. Nature Comm 2017;8:15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillion J, Wood LJ, Mukherjee M, Bhattacharya R, Di Cello F, Kowalski J, et al. Upregulation of MMP-2 by HMGA1 promotes transformation in undifferentiated, large-cell lung cancer. Mol Cancer Res 2009;7: 1803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hristov AC, Cope L, Di Cello F, Reyes MD, Singh M, Hillion JA, et al. HMGA1 correlates with advanced tumor grade and decreased survival in pancreatic ductal adenocarcinoma. Mod Pathol 2010;23:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liau SS, Jazag A, Whang EE. HMGA1 is a determinant of cellular invasiveness and in vivo metastatic potential in pancreatic adenocarcinoma. Cancer Res 2006;66:11613–22. [DOI] [PubMed] [Google Scholar]
- 55.Liau SS, Rocha F, Matros E, Redston M, Whang E. High mobility group at-hook 1 (HMGA1) is an independent prognostic factor and novel therapeutic target in pancreatic adenocarcinoma. Cancer 2008;113: 302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah SN, Cope L, Poh W, Belton A, Roy S, Talbot CC Jr, et al. Hmga1: a master regulator of tumor progression in triple-negative breast cancer cells. PLoS One 2013;8:e63419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belton A, Gabrovsky A, Bae YK, Reeves R, Iacobuzio-Donahue C, Huso DL, et al. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One 2012;7:e30034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madison BB, Liu Q, Zhong X, Hahn CM, Lin N, Emmett MJ, et al. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev 2013;27:2233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A, Cuatrecasas M, et al. Let-7 represses carcinogenesis and a stem cell phenotype in the intestine via regulation of Hmga2. PLoS Genet 2015;11:e1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lanahan A, Williams JB, Sanders LK, Nathans D. Growth factor-induced delayed early response genes. Mol Cell Biol 1992;12:3919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holth LT, Thorlacius AE, Reeves R. Effects of epidermal growth factor and estrogen on the regulation of the HMG-I/Y gene in human mammary epithelial cell lines. DNA Cell Biol 1997;16:1299–309. [DOI] [PubMed] [Google Scholar]
- 62.Wood LJ, Mukherjee M, Dolde CE, Xu Y, Maher JF, Bunton TE, et al. HMG-I/Y, a new cmyc target gene and potential oncogene. Mol Cell Biol 2000;20:5490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giannini G, Cerignoli F, Mellone M, Massimi I, Ambrosi C, Rinaldi C, et al. High mobility group A1 is a molecular target for MYCN in human neuroblastoma. Cancer Res 2005;65:8308–16. [DOI] [PubMed] [Google Scholar]
- 64.Dhar A, Hu J, Reeves R, Resar LM, Colburn NH. Dominant-negative c-jun (tam67) target genes: HMGA1 is required for tumor promoter-induced transformation. Oncogene 2004;23:4466–76. [DOI] [PubMed] [Google Scholar]
- 65.Hommura F, Katabami M, Leaner VD, Donninger H, Sumter TF, Resar LM, et al. HMG-I/Y is a c-jun/activator protein-1 target gene and is necessary for c-jun-induced anchorage-independent growth in rat1a cells. Mol Cancer Res 2004;2:305–14. [PubMed] [Google Scholar]
- 66.Grade M, Hormann P, Becker S, Hummon AB, Wangsa D, Varma S, et al. Gene expression profiling reveals a massive, aneuploidy-dependent transcriptional deregulation and distinct differences between lymph nodenegative and lymph node-positive colon carcinomas. Cancer Res 2007;67:41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen X, Lechago J, Ertan A, Ergun G, Verm R, Bridges M, et al. Expression of the high mobility group proteins HMGI(Y) correlates with malignant progression in Barrett’s metaplasia. Cancer Epidemiol Biomarkers Prev 2004;13:30–3. [DOI] [PubMed] [Google Scholar]
- 68.Bandiera A, Bonifacio D, Manfioletti G, Mantovani F, Rustighi A, Zanconati F, et al. Expression of HMGI(Y) proteins in squamous intraepithelial and invasive lesions of the uterine cervix. Cancer Res 1998;58:426–31. [PubMed] [Google Scholar]
- 69.Hillion J, Dhara S, Sumter TF, Mukherjee M, Di Cello F, Belton A, et al. The high-mobility group A1a/signal transducer and activator of transcription3 axis: An Achilles heel for hematopoietic malignancies? Cancer Res 2008;68:10121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belton B, Xian L, Huso T, Koo M, Luo LZ, Turkson J, et al. STAT3 inhibitor has potent anti-tumor activity in B-lineage acute lymphoblastic leukemia cells overexpressing the High Mobility Group A1 (HMGA1)-STAT3 path- way. Leuk Lymphoma 2016;57:2681–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tesfaye A, Di Cello F, Hillion J, Ronnett BM, Elbahloul O, Ashfaq R, et al. The high-mobility group A1 gene up-regulates cyclooxygenase 2 expression in uterine tumorigenesis. Cancer Res 2007;67:3998–4004. [DOI] [PubMed] [Google Scholar]
- 72.Di Cello F, Hillion J, Kowalski J, Ronnett BM, Aderinto A, Huso DL, et al. Cyclooxygenase inhibitors block uterine tumorigenesis in HMGA1a transgenic mice and human xenografts. Mol Cancer Ther 2008;7: 2090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ji YS, Xu Q, Schmedtje JF Jr. Hypoxia induces high-mobility-group protein I(Y) and transcription of the cyclooxygenase-2 gene in human vascular endothelium. Circ Res 1998;83:295–304. [DOI] [PubMed] [Google Scholar]
- 74.Kazmierczak B, Meyer-Bolte K, Tran KH, Wo€ckel W, Breightman I, Rosigkeit J, et al. A high frequency of tumors with rearrangements of genes of the HMGI(Y) family in a series of 191 pulmonary chondroid hamartomas. Genes Chromosomes Cancer 1999;26:125–33. [PubMed] [Google Scholar]
- 75.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007;131: 1109–23 [DOI] [PubMed] [Google Scholar]
- 76.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007;315:1576–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeitels LR, Acharya A, Shi G, Chivukula D, Chivukula RR, Anandam JL, et al. Tumor suppression by miR-26 overrides potential oncogenic activity in intestinal tumorigenesis. Genes Dev 2014;28:2585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wood LJ, Maher JF, Bunton TE, Resar LM. The oncogenic properties of the HMG-I gene family. Cancer Res 2000;60:4256–61. [PubMed] [Google Scholar]
- 79.Reeves R, Edberg DD, Li Y. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol 2001;21:575–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu Y, Sumter TF, Bhattacharya R, Tesfaye A, Fuchs EJ, Wood LJ, et al. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res 2004;64:3371–5. [DOI] [PubMed] [Google Scholar]
- 81.Schuldenfrei A, Belton A, Kowalski J, Talbot CC Jr, Di Cello F, Poh W, et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics 2011;12: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto AJ, Kenyon L, et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005;24:3427–35. [DOI] [PubMed] [Google Scholar]
- 83.Battista S, Fidanza V, Fedele M, Klein-Szanto AJ, Outwater E, Brunner H, et al. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res 1999;59(19):4793–7. [PubMed] [Google Scholar]
- 84.Arlotta P, Tai AK, Manfioletti G, Clifford C, Jay G, Ono SJ. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas. J Biol Chem 2000;275:14394–400. [DOI] [PubMed] [Google Scholar]
- 85.Kugel S, Sebasti´an C, Fitamant J, Ross KN, Saha SK, Jain E, et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 2016;165: 1401–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anand A, Chada K In vivo modulation of Hmgic reduces obesity. Nat Genet 2000;24:377–380. [DOI] [PubMed] [Google Scholar]
- 87.Copley MR, Babovic S, Benz C, Knapp DJ, Beer PA, Kent DG, et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 2013;15:916–25. [DOI] [PubMed] [Google Scholar]
- 88.Liu J, Schiltz JF, Ashar HR, Chada KK. Hmga1 is required for normal sperm development. Mol Reprod Dev 2003;66:81–9. [DOI] [PubMed] [Google Scholar]
- 89.Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa F, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med 2005;11:765–73. [DOI] [PubMed] [Google Scholar]
- 90.Fedele M, Fidanza V, Battista S, Pentimalli F, Klein-Szanto AJ, Visone R, et al. Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res 2006;66: 2536–2543. [DOI] [PubMed] [Google Scholar]
- 91.Resar L, Xian L, Huso T, Belton A, Cope L, Huso D. High Mobility Group A1 chromatin remodeling protein drives self-renewal, niche formation, and regenerative function in adult stem cells through Wnt/b-catenin signaling. Blood 2016;128:2647. [Google Scholar]
- 92.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–7. [DOI] [PubMed] [Google Scholar]
- 93.Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 2007; 178:635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007;133:539–46. [DOI] [PubMed] [Google Scholar]
- 95.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 96.Kubo H, Shimizu M, Taya Y, Kawamoto T, Michida M, Kaneko E, et al. Identification of mesenchymal stem cell (MSC)-transcription factors by microarray and knockdown analyses, and signature molecule-marked MSC in bone marrow by immunohistochemistry. Genes to Cells 2009;14:407–424. [DOI] [PubMed] [Google Scholar]
- 97.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A 2005;102:9194–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prall WC, Czibere A, J€ager M, Spentzos D, Libermann TA, Gattermann N, et al. Age-related transcription levels of KU70, MGST1 and BIK in CD34+ hematopoietic stem and progenitor cells. Mech Ageing Dev 2007;128: 503–10. [DOI] [PubMed] [Google Scholar]
- 99.Bush BM, Brock AT, Deng JA, Nelson RA, Sumter TF. The Wnt/betacatenin/T-cell factor 4 pathway up-regulates high-mobility group A1 expression in colon cancer. Cell Biochem Funct 2013;31:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Akaboshi S, Watanabe S, Hino Y, Sekita Y, Xi Y, Araki K, et al. HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am J Pathol 2009;175:1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol 2013;183:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mukherjee S The Emperor of All Maladies 2010. Toronto: Simon and Schuster Canada. [Google Scholar]