

SUMMARY
Metagenomic sequencing is a promising approach for identifying and characterizing organisms and their functional characteristics in complex, polymicrobial infections, such as airway infections in people with cystic fibrosis. These analyses are often hampered, however, by overwhelming quantities of human DNA, yielding only a small proportion of microbial reads for analysis. In addition, many abundant microbes in respiratory samples can produce large quantities of extracellular bacterial DNA originating either from biofilms or dead cells. We describe a method for simultaneously depleting DNA from intact human cells and extracellular DNA (human and bacterial) in sputum, using selective lysis of eukaryotic cells and endonuclease digestion. We show that this method increases microbial sequencing depth and, consequently, both the number of taxa detected and coverage of individual genes such as those involved in antibiotic resistance. This finding underscores the substantial impact of DNA from sources other than live bacteria in micro-biological analyses of complex, chronic infection specimens.
Graphical Abstract
In Brief
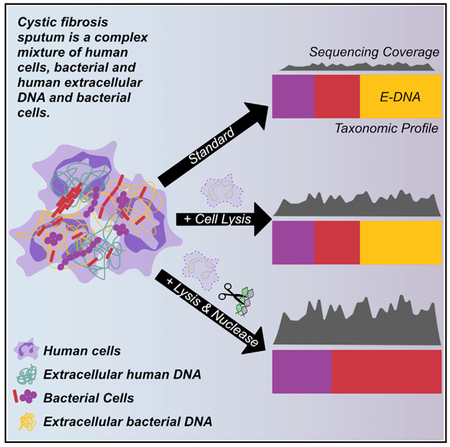
Nelson et al. describe a method for reducing both human cellular DNA and extracellular DNA (human and bacterial) in a complex respiratory sample using hypotonic lysis and endonuclease digestion. This method increases effective microbial sequencing depth and minimizes bias introduced into subsequent phylogenetic analysis by bacterial extracellular DNA.
INTRODUCTION
Sequencing-based microbiome methods have greatly improved our understanding of the microbial inhabitants of the human body in both health and disease and have been particularly instrumental in expanding our view of microbes in polymicrobial communities and infections. Polymicrobial lung infections in individuals with cystic fibrosis (CF) serve as a paradigm for studying many chronic, complex human infections. CF is a genetic disorder that is characterized by aberrant ion and fluid balances at multiple body sites. These defects result in lifelong multiorgan disease, with the respiratory tract most prominently affected. The resulting buildup of thick mucus in the airways is associated with chronic infections and progressive respiratory disease, the leading cause of morbidity and mortality in people with CF (Cystic Fibrosis Foundation, 2015; Emerson et al., 2002; Gibson et al., 2003). Historically, CF respiratory infections have been characterized, diagnosed, and treated using culture methods that are optimized for detecting species readily grown under routine clinical laboratory conditions, including Pseudomonas aeruginosa and Staphylococcus aureus (Cystic Fibrosis Foundation, 2015; Saiman et al., 2014). The declining cost of high-throughput, next-generation sequencing (NGS) technology has permitted culture-free analysis of CF sputum, a respiratory specimen that variably samples secretions from the mouth to the lower airways, most often by sequencing the bacterial 16S ribosomal RNA gene (16S amplicon sequencing). These culture-free methods have shown the microbiota (the full complement of bacterial taxa present) in CF respiratory samples to be more diverse than previously thought, often comprising species not detected by routine clinical culture (Cox et al., 2010; Rogers et al., 2004; Rudkjøbing et al., 2011).
Despite a growing body of work characterizing CF respiratory microbiota, the determinants of clinical decline and microbial persistence remain incompletely understood, as is the case for many chronic, polymicrobial infections. Current therapies in CF generally target culture-identifiable organisms, but CF lungs remain persistently infected with these “standard pathogens” throughout patients’ lifetimes despite frequent antibiotic treatments. CF sputum microbial communities are resilient to therapy, typically rebounding to pre-exacerbation profiles regardless of antibiotic treatment (Carmody et al., 2015; Fodor et al., 2012; Price et al., 2013; Stressmann et al., 2011; Zhao et al., 2012a). Furthermore, microbial communities in CF sputum can differ dramatically between individuals with similar clinical characteristics (Kramer et al., 2015). These observations, together with the diagnostic imprecision of routine clinical culture, make it difficult to infer which taxa are the most responsible for clinical status or response to treatment. Thus, a deeper understanding of sputum microbial community constituency and function than that provided by current methods could determine mechanisms by which microorganisms persist, and how these infections may be more effectively treated.
Although bioinformatic pipelines exist to infer the functional capacity of a community from 16S amplicon sequencing (Langille et al., 2013), these methods can only use what is available in annotated bacterial genomic databases and can miss differences in accessory genomes across strains. Sequence analysis of the “metagenome,” the total complement of genes present in a community, can provide insight into not only the taxonomic composition of the microbiota but also its functional capacity directly from sequencing data (Yatsunenko et al., 2012). Metagenomic analysis has been used in fecal samples (Lloyd-Price et al., 2017) and, to a limited extent, in respiratory samples (Feigelman et al., 2017; Lim et al., 2013; Moran Losada et al., 2016) and has the potential to identify functional traits that are required for persistence in chronic infections like those of the CF lung. Unlike some complex microbiota communities, such as those in fecal samples from the healthy GI tract and in soil, many complex clinical samples include large quantities of immune cells and comparatively low microbial loads. As a result, metagenomic sequencing of sputum and other respiratory samples can be hindered by the overabundant proportions of human DNA, relative to microbial DNA; for example, approximately 95% of metagenomic sequencing reads from CF sputum samples are annotated as human (Feigelman et al., 2017; Moran Losada et al., 2016). High ratios of human-to-microbial DNA are a barrier shared by many complex, human-associated microbial communities, such as those found in healthy oral (Horz et al., 2010), skin (Ferretti et al., 2017), vaginal (Goltsman et al., 2018), blood (Bright et al., 2012), middle ear, and nasopharyngeal samples (Jervis-Bardy et al., 2015). Human-to-microbial DNA ratios may be even higher in samples from inflamed and/or infected sites due to the influx of immune cells (Bhatt et al., 2014; Chiodini et al., 2013). The cost of metagenomic analysis of samples with high human-to-microbial DNA ratios can thus become prohibitive when considering the depths required for the thorough investigation of the microbiome in a large number of such complex samples. For this reason, functional characterization of sputum and other samples containing relatively high ratios of human-to-microbial DNA has lagged behind studies investigating stool and other microbial communities that contain a high microbial abundance.
A related problem affecting metagenomic analyses of infected tissues is the presence of extracellular DNA derived from human cells and/or microbes. Extracellular DNA in complex infections can be produced as a result of the processes that occur during and in response to such infections, including cellular turnover accelerated by antibiotic therapy, inter-bacterial competition, and host defense, as well as from the production of microbial biofilms (DNA is an abundant component of these extracellular matrices for certain microbes). Failing to address the impact of extracellular DNA on molecular microbial analyses has the potential for systematic bias when the intended focus is on viable microbial cells. For example, proportions of extracellular microbial DNA may vary when collected before and after antibiotic treatment (Rogers et al., 2010a) and may bias the characterization of live bacteria persisting after antibiotic treatment if not accounted for during processing. Therefore, methods to more accurately characterize the functional capacity of infecting communities using metagenomic sequencing require not only human DNA depletion but also enrichment for the cellular DNA fraction.
A number of studies have detailed techniques for depleting human DNA from microbial communities (Hasan et al., 2016; Horz et al., 2010; Lim et al., 2014a; Marotz et al., 2018; Thoendel et al., 2016; Zhou and Pollard, 2012). Few have evaluated the efficiency of these different methods for depleting human DNA from inflammatory and (relatively) low-abundance infections, their ability to remove extracellular DNA of either human or microbial origin, or the bias introduced by these methods to the phylogenetic composition of these samples. The aim of this study is to compare the results of different methods designed to deplete human DNA extracted from complex clinical specimens, focusing in particular on (1) the relative effect of each method on the calculated taxonomic constituency of the community and (2) how accurately the results of each method reflect the viable microbial fraction. We quantified human DNA depletion via both NGS and qPCR, demonstrating that a combination of hypotonic lysis and nuclease digestion most effectively reduces human and extracellular microbial DNA. Using phylogenetic analysis of metagenomic and 16S amplicon sequencing data, we compared the effects of this and other methods on microbial sequence read depth and on the detection of rare taxa, as well as the contribution of extracellular bacterial DNA to apparent community composition, in clinical sputum samples. We then compared the accuracy of these methods for measuring the constituency of mock communities containing cultured bacterial species common in CF sputum. Finally, we demonstrate that the increased read depth afforded by this method enhanced sensitivity for detecting genes of potential clinical importance. Our ultimate goal is to apply this optimized method to a large number of clinical samples to determine the relative importance of this viable microbial fraction as a correlate of clinical outcomes.
RESULTS
Efficiency of Human DNA Depletion
We first compared 4 methods, each used in published studies (Hasan et al., 2016; Horz et al., 2010; Lim et al., 2014a; Marotz et al., 2018; Thoendel et al., 2016; Zhou and Pollard, 2012) or available as commercial kits specific for human DNA depletion, for their ability to deplete human DNA from CF sputum samples. “Antibody depletion” uses immunoglobulins against methylated epitopes specific to eukaryotic DNA; “cell lysis” uses selective eukaryotic cell lysis with trypsin-EDTA and Tween-20; “ MolYsis “ uses the chaotropic lysis of human cells followed by the endonuclease digestion of extracellular DNA; and “benzonase1” uses the hypotonic lysis of human cells followed by the endonuclease digestion of extracellular DNA. Each of these methods was used to supplement a standard phenyl:chloroform-based DNA extraction method, which was also tested without pre-processing (“standard extraction”).
Sputum samples from 8 CF patients (a collection we refer to here as “test set 1”) were processed using each method for the selective depletion of human DNA followed by standard DNA extraction or standard extraction alone. To assess the degree of selective human DNA reduction, we calculated the proportion of human to total (human and bacterial) DNA in terms of genome equivalents (GEs) per microliter identified in respective broad-range qPCR reactions (Figure 1A), for all samples and extraction methods. In parallel, we performed metagenomic sequencing of all of the DNA extracts and then calculated the proportion of human to total sequence reads by mapping reads to the human genome (Figure 1B). By both sequencing and qPCR measures, non-depleted samples contained 96% human DNA on average, which is consistent with previous reports (Feigelman et al., 2017; Lim et al., 2014b). In the present study, the benzonase1 method was the most efficient for selective human DNA removal from test set 1; on average, 68% of the reads from benzonase1-treated samples mapped to the human genome. This reduction in human reads resulted in a 5-fold increase in microbial reads on average and increased microbial sequencing depth (Figure 1C). Reduction in human DNA did not correspond to a decrease in the total bacterial load after any extraction method, indicating that increased microbial sequencing depth could be achieved (Figure 1C) without significant loss of bacterial DNA (Figure 1D).
Figure 1. Comparison of 4 Methods for Selective Depletion of Human DNA from CF Sputum Samples.

DNA was extracted from 8 CF sputum samples with standard extraction and 4 different methods for selectively depleting human DNA.
(A) Proportion of human DNA to total calculated DNA as determined by qPCR of both human and bacterial DNA.
(B) Proportion of human to total reads, calculated by mapping all metagenomic sequencing reads to the human genome.
(C) Ratio of microbial metagenomic sequencing reads yielded by each extraction method compared to standard extraction.
(D) Total bacterial load yielded by each extraction method, calculated by qPCR targeting the 16S rRNA gene. The colored lines connect data points from the same sample, and the dotted line indicates the limit of detection.
Boxes represent the interquartile region, and black lines within boxes indicate the median value. Pairwise Wilcoxon signed rank tests were used to compare results from selective human DNA depletion methods to standard extraction, with a Benjamini-Hochberg correction for multiple comparisons with all of the comparisons combined (A–D). No comparison reached the level of significance (p < 0.05).
Effect of Human DNA Depletion on Apparent Microbial Community Composition
We next assessed the effect of the different depletion methods on apparent microbial community structure for test set 1. We hypothesized that selectively depleting human DNA in these samples would result in higher effective microbial sequence read depth and would improve the detection of low-abundance taxa in these samples. MetaPhlAn2 (Truong et al., 2015) was used to define the taxonomic composition of test set 1 metagenomes, and the effects of different depletion methods on the resulting calculated sputum microbial taxonomic profiles were compared. We identified more microbial taxa (Figure 2A), as well as an apparent shift in the relative abundance of a number of taxa, in samples processed by either nuclease-based method (MolYsis and benzonase1; Figures 2B and S1). Although this difference between each depletion method and standard DNA extraction was not statistically significant, we did observe a large decrease in P. aeruginosa relative abundance, an important CF pathogen that is known to exude extracellular DNA (Mulcahy et al., 2008; van Tilburg Bernardes et al., 2017), with both nuclease-based processing methods. Because of this pronounced effect of the nuclease-based processing methods on P. aeruginosa relative abundance (Figures 2B and S1) and because of the importance of this pathogen in CF, we wondered whether our sample collection (which routinely includes freezing at −80°C) or processing methods could have affected the viability of P. aeruginosa. First, we compared DNA extracted from 4 test set 1 samples both before and after freezing and showed no appreciable alteration in microbiota profiles (Figure S2). Second, we tested whether the observed impact of nuclease-based extraction methods was due to the premature lysis of bacterial cells during initial processing with EDTA, which is used to inactivate the endonuclease used in benzonase1 extraction and has been demonstrated to kill P. aeruginosa growing planktonically (Haque and Russell, 1974) and in biofilms (Banin et al., 2006). We tested the effect of hypotonic lysis followed by nuclease treatment (benzonase1) on viable counts of 6 clinical P. aeruginosa isolates cultured from test set 1. We also modified the benzonase1 protocol by adding EDTA later during processing (referred to here as “benzonase2”) and tested this procedural change on the viability of P. aeruginosa. These experiments demonstrated that, while benzonase1 extraction resulted in an average 1.3 log10 reduction in P. aeruginosa colony-forming units (CFUs), the benzonase2 method had no detectable impact on viable count (Figure 2C). We did not observe a similar effect with other taxa commonly identified in CF sputum (Figure S3). We also enumerated viable counts of P. aeruginosa directly from a single CF sputum sample (309) before and after benzonase2 processing, similarly demonstrating no difference in viable counts (data not shown).
Figure 2. Effect of Extraction Method on Metagenomic Sequencing Taxonomic Profile and Viable Counts of the 8 Test Set 1 CF Sputum Samples.

(A) Number of distinct genera detected by metagenomic sequencing from DNA prepared with each extraction method. Results from extractions, including selective depletion of human and extracellular DNA, were compared to standard extraction conditions. Boxes represent the interquartile region, and black lines indicate the median value. Pairwise Wilcoxon signed rank tests were performed comparing each extraction method to standard extraction with a Benjamini-Hochberg correction for multiple comparisons, with no comparison reaching the level of significance (p < 0.05).
(B) Difference in relative abundance identified by each extraction method (compared with standard extraction) for the 10 most abundant taxa. Each data point indicates an individual sample.
(C) Effect of benzonase treatment on viable P. aeruginosa counts. Cultures of 6 separate P. aeruginosa clinical isolates were subjected to both benzonase1 and benzonase2 processing methods, which differ only in the order in which EDTA is added. Viable counts were measured after the hypotonic, host-cell lysis step and after enzyme inactivation, as indicated.
See also Figures S1–S4.
Effect of Adapted Benzonase2 Protocol on Metagenomic and 16S Amplicon Sequencing
To assess the effect of the benzonase2 method on taxonomic profiles from metagenomic sequencing, we constructed a smaller set of 4 sputum samples (“test set 2”), 1 from test set 1 (186), and 1 sample each from 3 additional CF patients. Each sample was treated with the mucolytic agent dithiothreitol (DTT) (Burns and Rolain, 2014) and then mixed to fully homogenize, thus limiting bias from potential regional differences among the different sputum aliquots used for extraction. DNA was extracted with standard extraction and benzonase1 and benzonase2 methods, and taxonomic profiles were defined by metagenomic sequencing followed by MetaPhlAn2 analysis. As before, we observed a decrease in the relative abundance of P. aeruginosa and an increased detection of lower-abundance taxa with both benzonase methods compared with standard extraction (Figure 3, top).
Figure 3. Sequence-Based Phylogenetic Composition of DTT-Treated Test Set 2 Samples after Standard and Benzonase Extraction.

DNA extracted from each of 4 DTT-treated, homogenized CF sputum samples was analyzed using both metagenomic sequencing (MetaPhlAn2, top row) and 16S amplicon sequencing (16S Amplicon, bottom row).
See also Tables S1–S4.
This observed decrease in the relative abundance of P. aeruginosa may be due solely to an increase in reads from other microbes after benzonase processing, perhaps as a result of increased microbial sequencing depth, rather than reduction of the absolute abundance of Pseudomonas. Because taxonomic coverage in 16S amplicon sequencing is less affected by large quantities of human DNA due to the amplification of a bacterial-specific gene, we addressed this possibility by analyzing each sample and extraction method using 16S amplicon sequencing (Figure 3, bottom). It is important to note that although large quantities of human DNA can inhibit amplification of the 16S gene (Glassing et al., 2015), we saw similar total 16S amplicon read numbers and proportions of amplicon sequencing reads annotated as human across different extraction methods when applied to test set 1, suggesting that this effect was minimal (Table S1). 16S amplicon sequencing demonstrated decreases in the relative abundance of P. aeruginosa after benzonase1 and benzonase2 extractions that were similar to those demonstrated by metagenomic sequencing, although of a lower magnitude. On average, 97% of metagenomic sequencing reads from DNA prepared with the standard extraction and 60% of reads in the benzonase-treated extracts mapped to the human genome (Figures 4A and 4B). Microbial reads increased 15-fold following benzonase1 treatment and 14-fold following benzonase2 treatment, on average, compared with standard extraction (Figure 4C). As before, the total bacterial load was similar between extraction methods, indicating a minimal effect of these processing methods on microbial DNA extraction efficiency (Figure 4D). Pretreatment with DTT alone did not lead to an appreciable difference in community structure (Figure S4), but our data indicate a further decrease in the proportion of human reads and increase in microbial reads when comparing standard extraction to benzonase treatment in test set 1 versus test set 2 (68% versus 60%, Figures 1B and 1C versus Figures 4B and 4C), suggesting that DTT-based homogenization may increase the efficiency of nuclease-mediated reduction in host DNA.
Figure 4. Effect of the Refined Benzonase2 Extraction Method on Selective Human DNA Depletion and Microbial Sequencing Depth.

Total DNA from the 4 test set 2 sputum samples was extracted using standard, benzonase1, and benzonase2 extraction methods and analyzed to show the following.
(A) Proportion of human DNA relative to total DNA as determined by qPCR.
(B) Proportion of human to total reads calculated by mapping all of the metagenomic sequencing reads to a reference human genome.
(C) Ratio of microbial shotgun sequencing reads yielded by each extraction method compared to the standard extraction.
(D) Total bacterial load (genome equivalents [GEs]) yielded from each extraction method, as determined by qPCR targeting the 16S rRNA gene. Boxes represent the interquartile region, and black lines indicate the median value.
(E and F) Number of genera detected in each extract using (E) metagenomic sequencing or (F) 16S amplicon sequencing. Each color represents a different sample.
Results from each extraction method were compared to the standard extraction conditions using pairwise, 2-sided t tests with a Benjamini-Hochberg correction for multiple comparisons, identifying no significant differences.
Finally, the number of genera detected after both benzonase treatments compared to standard extraction alone was increased in metagenomic sequencing but not 16S amplicon sequencing (Figures 4E and 4F). Notably, for 3 of the 4 samples analyzed, a similar number of genera were detected by metagenomic sequencing and 16S amplicon sequencing following either benzonase treatment, suggesting that nuclease-based methods provide similar microbial sequencing depths via both metagenomic sequencing and amplification of a bacterial-specific gene.
Nuclease-Based Extraction Better Reflects True, Viable Diversity in the Sputum Microbiota Than Other Tested Methods
Because benzonase treatment depletes human reads by digesting extracellular DNA, it remained possible that the taxonomic differences found with benzonase processing compared with standard extraction (Figure 3) were due primarily to the degradation of extracellular microbial DNA rather than improved microbial sequence read depth. Extracellular DNA is also excluded from sequencing by another processing method, treatment with propidium monoazide (PMA), a chemical that cross-links extracellular DNA and selectively prevents its amplification (Nocker et al., 2007). PMA has been used in clinical samples to specifically focus on viable microbial cells (Bellehumeur et al., 2015; Exterkate et al., 2015) (similar to the intended use of benzonase processing) and has been used in CF sputum 16S amplicon sequencing studies for this reason (Rogers et al., 2008; van Tilburg Bernardes et al., 2017). PMA has also been used to deplete human DNA from human-associated microbiota (Marotz et al., 2018). However, these prior studies focused on complex clinical samples, as in our experiments, and thus did not determine the relative effects on the exclusion of microbial versus human DNA. Therefore, to more rigorously define and compare the effects of benzonase and PMA processing on microbial community structure, we constructed an in vitro bacterial mock community containing cultured cells of taxa commonly present in CF respiratory samples. DNA was extracted from these communities using 6 methods: standard extraction, standard extraction with additional cell lysis, MolYsis, benzonase1, benzonase2, or PMA treatment. We cultured the mock community to determine the viable cell counts of each community member and to confirm viable relative abundances. We were also able to detect extra-cellular DNA in the supernatant of these cultured taxa using qPCR (Figure S5). We then compared phylogenetic composition identified by 16S amplicon sequencing and metagenomic sequencing to culture-validated input community composition (Figure 5). We predicted that the benzonase2 taxonomic profile from 16S amplicon sequencing would most closely resemble PMA (reflecting depletion of extracellular bacterial DNA), and that the benzonase2 taxonomic profile from metagenomic sequencing (which is less subject to the amplification bias of 16S amplicon sequencing [Jovel et al., 2016]) would most closely resemble calculated input. As predicted, the taxonomic composition identified by metagenomic sequencing after benzonase2 processing was most similar to culture-validated input (Figure 5, left), indicating that DNA extracted after benzonase2 processing most closely resembles the DNA of the viable bacterial community, whereas relatively low concordance was observed between cultured results and the PMA-treated DNA extracts. In contrast, community composition identified by 16S amplicon sequencing after MolYsis and benzonase2 processing was most similar to PMA-treated communities (rather than calculated input), consistent with the ability of PMA to prevent the amplification of extra-cellular DNA (Figure 5, right). MolYsis, which differs from the benzonase methods in both the nuclease and eukaryotic cell lysis method used, produced a community structure similar to that produced by benzonase2 as identified by both metagenomic sequencing and 16S amplicon sequencing, further underscoring the effect of endonuclease digestion of extracellular DNA on apparent phylogenetic composition. Therefore, nuclease-based processing before DNA extraction performed at least as well (16S amplicon sequencing) or better (metagenomic sequencing) than PMA at identifying the viable bacterial constituency of polymicrobial mixtures.
Figure 5. Effect of Extraction Method on Sequencing-Based Taxonomic Profile of a Bacterial Mock Community.

Data represent a single mock community extracted in parallel with each extraction method. Phylogenetic tree constructed using Bray-Curtis dissimilarity (“Tree”) and the corresponding phylogenetic composition (“Community Composition”) determined via (left) metagenomic sequencing and (right) 16S amplicon sequencing. Input refers to the relative abundance of the mock community before extraction based on quantitative culture. Viable counts were corrected for 16S copy number in the analysis of 16S amplicon sequence data.
See also Figure S5.
Selective Depletion of Human DNA Increases Coverage of Microbial Genes in Metagenomic Sequencing
To assess the effect of selective human DNA depletion on sequence coverage of microbial genomes, we computationally constructed contigs from the metagenomic sequencing dataset from all 4 test set 2 sputum samples using standard, benzonase1, and benzonase2 processing. We then mapped all of the microbial reads from the dataset to these contigs, quantifying the mean coverage (average sequencing depth at each base pair) of each DNA extract as a measure of microbial sequence read depth. For both benzonase methods, mean coverage increased across all of the contigs compared to standard extraction (Figure 6A), despite similar raw read counts between extraction methods (Table S2), indicating that higher microbial sequencing coverage was achieved by benzonase processing. Subsampling metagenomic sequencing reads followed by MetaPhlan2 analysis suggested that this increased microbial sequencing depth was adequate to profile community structure (Figure S6A). For both benzonase extractions, the graph of species richness versus number of sequences reached a plateau for 3 of 4 samples at 1–5 million reads, with sample 186 beginning to plateau at 7 million reads. These data concur with the results presented in Figures 4E and 4F, which demonstrated that benzonase extraction results in taxonomic richness that is similar to 16S amplicon sequencing. It is interesting to note that although the rarefaction curves for standard extraction plateau for all of the samples, fewer species were detected with standard than with benzonase extraction, further revealing the improved microbial detection afforded by benzonase-based extraction (Figure S6A). Furthermore, Figure 3 indicates that the microbiota identified by metagenomic sequencing only resembles that of 16S amplicon sequencing when the latter was performed after nuclease-based extraction, again suggesting that the species richness calculated after benzonase-based extraction more closely resembles the “true” CF sputum microbiota, at least as determined by the sequencing method used most often in this field (16S amplicon sequencing).
Figure 6. Increase in Microbial Sequence Coverage after Human DNA Depletion of the 4 Test Set 2 Sputum Samples.

(A) Contigs were assembled using the same 4 sputum samples and processing methods as in Figures 3 and 4, and all of the reads were subsequently mapped back onto these contigs. Each radial line represents a single contig ordered by Euclidean distance based on sequence content. The height of each bar of darker shading represents the average coverage across that contig (average of the sequence coverage of each nucleotide across a given contig) in a given sample from 0 to 10×. Radial black lines in the outermost ring indicate contigs annotated as human.
(B) Average coverage of 145 identified antibiotic resistance genes in each sample as a proportion of 10× coverage. Each radial line represents an individual antibiotic resistance gene. Three genes described in the text are indicated. The height of the bars represents mean coverage for each gene, from 0 to 10×. See also Figures S6 and S7 and Methods S1–S3.
We next assessed the effects of benzonase processing on the detection and characterization of individual microbial genes in the sputum metagenome data. We focused our analysis on antibiotic resistance genes due to their potential clinical importance. All of the reads from all of the samples were mapped against the Comprehensive Antibiotic Resistance Database (CARD), an annotated database of antibiotic resistance genes and their associated proteins and phenotypes (McArthur et al., 2013). Of the 2,239 genes in this collection, 145 were detected in 4 samples (at least 1 read mapping to a given gene in at least 1 sample). Across all 4 samples, an average of 67% of the antibiotic resistance genes detected from extracts prepared with either benzonase method were not detected from DNA prepared using the standard extraction. No resistance genes were detected solely in standard extraction samples. For benzonase2 extracted samples, the mean coverage for detected resistance genes was 11.0×, 17.8×, 6.5×, and 35.4× in samples 186, 205, 309, and 312, respectively. In contrast, the highest mean coverage across all standard extractions was 0.4× (Figure 6B). This increased coverage was seemingly independent of the degree to which a given taxon was detected by metagenomic sequencing or 16S amplicon sequencing. For example, sequencing coverage for the chromosomal aminoglycoside acetyltransferase from Stenotrophomonas, AAC(6′)-IZ, was <1× after standard extraction but 303 after benzonase2 (Figures 6B and S7A), despite this genus being detected at a similar relative abundance after standard or benzonase extractions (Figure 3). Metagenomic sequencing also detected differences in species-specific antibiotic resistance gene profiles among different samples with that species. For example, both mecA, a gene that confers methicillin-resistant S. aureus (MRSA) status to S. aureus, and mecI, encoding an inhibitor of mecA, were detected in sample 312, whereas only mecA was detected in sample 309, despite a similar relative abundance of S. aureus in both samples (Figure 3). Standard extraction afforded <1× average coverage for both of these genes (Figures 6B, S7B, and S7C). It is particularly useful to note that that we used the lowest limit of detection possible (1 read mapping to a given gene), and it is likely that higher stringency criteria required to confidently detect antibiotic resistance genes using metagenomic sequencing would have further limited the sensitivity of standard extraction. Finally, subsampling metagenomic sequencing reads followed by mapping these rarefied reads against the same antibiotic resistance gene database indicated that the increased microbial sequencing depth provided by benzonase extraction was sufficient to characterize the inferred antibiotic resistance profile of our communities (Figures S6B–S6D).
DISCUSSION
As the price of NGS technology continues to drop, the prospect of metagenomic analysis is becoming a realistic goal for a wide variety of clinical samples. Metagenomic sequencing offers particularly enticing advantages for complex samples, including from chronic, inflammatory infections of the lung, nose, sinuses, skin, and other surfaces. Current methods for analyzing these polymicrobial infections are insufficient to handle samples with overwhelming quantities of human DNA relative to microbial DNA. For example, metagenomic sequencing of sputum from people with chronic airway infections due to CF results in a majority of human reads that must be computationally identified and removed before downstream analyses, a process that commonly results in inadequate microbial sequence read depth. Bacteria in the CF lung can undergo frequent cellular turnover due to interspecies competition, antibiotic therapy, and host defense, resulting in large quantities of extracellular bacterial DNA that can persist in infected tissues and secretions. Copious amounts of extracellular DNA can lead to inaccurate estimates of viable bacterial cell abundance and can undermine the ability of metagenomic sequencing to analyze the functional capacity of a microbial community by masking functions or taxa in a community, especially those persisting after antibiotic therapy.
Here, we describe an optimized method for DNA extraction from complex sputum microbiomes that enriches for microbial DNA from viable cells by (1) selective lysis of human cells with hypotonic treatment and (2) endonuclease digestion of both human and microbial extracellular DNA. Our analysis demonstrated that this approach has a superior ability to deplete human DNA from CF sputum compared with 4 other published or commercially available methods, resulting in a 14-fold increase in microbial reads without affecting the total bacterial load measured by qPCR.
The benzonase2 method also shifted the community structure compared with the standard extraction method, most notably by reducing the relative abundance of the canonical CF pathogen P. aeruginosa. A similar decrease was observed for a related Gram-negative taxon, Achromobacter, which is also an opportunist and biofilm-former common in CF infections (Chmiel et al., 2014; Nielsen et al., 2016), in one test set 1 sample (Figure S1). There are several potential reasons for these observations. For example, it is possible that heterogeneity in microbial distribution within sputum samples led repeatedly to these differences occurring by chance; however, we consider this to be unlikely, as we observed similar differences between standard and benzonase extraction for all of the samples with Pseudomonas. While it was also possible that treatment with benzonase selectively lyses Pseudomonas and Achromobacter cells, we identified no change in the viable counts of either taxon after benzonase2 treatment (Figures 2C and S3). Instead, the analysis of mock communities suggests that the reduction of extracellular bacterial DNA by nuclease treatment explained the relative abundance shifts that were observed for sputum samples. Thus, nuclease treatment likely enables a more optimized characterization of viable bacterial communities than standard DNA extraction methods. Furthermore, the increased sequence read depth afforded by the digestion of all extracellular DNA enables higher microbial gene coverage and, thus, may allow improved detection of taxa present at low relative abundance. This increased sensitivity, in turn, allows a more in-depth study of the contribution of these less-well-studied taxa to disease status and response to therapy.
There are a number of lines of evidence supporting this interpretation. For example, we were able to culture taxa from 5/8 test set 1 samples that were detected only by sequencing after nuclease processing (both MolYsis and benzonase1, results not shown), confirming their presence in the samples. Although this does not provide conclusive evidence that those taxa detected after benzonase2 extraction are solely those that were alive in CF sputum, it does indicate that standard extraction methods would fail to detect potentially important, viable species in CF respiratory samples and that benzonase2 extraction provides a more focused reflection of the viable microbial community in these samples. In addition, several taxa detected by metagenomic sequencing only after nuclease processing were detected via 16S amplicon sequencing after all of the extraction methods. These findings suggest that benzonase treatment achieved metagenomic coverage to determine microbial community composition reflecting that of a method less biased by sequence read depth. These results indicate that the increased microbial sequence read depth provided by nuclease processing improves the detection of lower abundance taxa by metagenomic sequencing. Nevertheless, the relative abundances of Pseudomonas and Achromobacter yielded by the 2 nuclease methods (benzonase and MolYsis) were similar, despite differences in human DNA depletion, indicating that the depletion of human DNA alone was not responsible for this effect. Pseudomonas is known to extrude extracellular DNA into its environment, particularly when forming biofilms (Jakubovics et al., 2013), further supporting the idea that the taxonomic differences observed after benzonase2 extraction were due to the depletion of extracellular bacterial DNA.
Using mock bacterial communities, we further demonstrated that endonuclease treatment results in a more accurate representation of viable community diversity compared with standard DNA extraction. When analyzed by metagenomic sequencing, mock community profiles generated from DNA prepared with benzonase2 processing most closely resembled the input community structure, again suggesting that extracellular DNA may influence sequencing-based taxonomic profiling, even with in vitro cultured bacteria. The larger reduction in the relative abundance of Pseudomonas that we observed for some samples after benzonase1 processing, which differs from the benzonase2 protocol only in the timing of EDTA treatment, reflected the loss of viable counts seen with this extraction method, providing further support for the close relation between results from sequencing and culture. Results after extraction with the MolYsis method, another nuclease-based processing method that does not use EDTA to inhibit enzymatic digestion of extra-cellular DNA, also closely resembled the input mock community, further underscoring the effect of extracellular DNA depletion on phylogenetic composition defined by sequencing.
When mock communities were analyzed via 16S amplicon sequencing, results from both benzonase2 and MolYsis processing, which involve nuclease treatment, closely resembled those from PMA extraction (a method used commonly to prevent the amplification of extracellular DNA), further underscoring the negative impact that extracellular DNA can have on the sequence-based characterization of viable community structure. PMA treatment creates double-strand breaks and DNA aggregates only in DNA that it can access (e.g., extracellular) (Emerson et al., 2017; Soejima et al., 2007), limiting the amplification of longer targets (e.g., amplicon sequencing). Because metagenomic sequencing principally uses smaller fragments and involves little amplification, it is less likely to be affected by this depletion method, a prediction that is supported by the differences that we found between the results from PMA and nuclease methods by metagenomic versus 16S sequencing (Figure 5). Two previous studies used PMA to exclude extracellular DNA before metagenomic sequencing (Erkus et al., 2016; Thoendel et al., 2016), but both used an additional whole-genome amplification step before sequencing. Our results indicate that PMA is not ideal with limited-amplification sequencing methods currently in use (such as in the present study). Marotz et al. (2018) successfully used PMA to deplete human DNA from saliva for metagenomic sequencing, although they did not specifically explore its effects on extracellular bacterial DNA. It is unclear why PMA was not as useful in reflecting viable input than were nuclease methods in our mock communities by metagenomic sequencing. It is possible that PMA efficacy is limited by the physical properties of either extracellular DNA or sample chemistry. For example, in the limited number of sputum samples tested, we did not see an effect of PMA on the proportion of human DNA (data not shown). The complexity and viscosity of these CF sputum samples compared to saliva may explain this effect.
We found that benzonase processing substantially increased metagenomic sequencing coverage of microbial genes in CF sputum compared with standard extraction. While people with CF are frequently treated with antibiotics, the microbial determinants of response (or lack of response) to these treatments are poorly understood. Metagenomic sequencing offers a promising approach to this problem; for example, these methods can detect, classify, and quantify longitudinal changes in antibiotic resistance genes in infectious bacterial communities during treatment. However, the sequencing read depth provided by standard extraction is insufficient for confident detection or classification of many of these genes and gene variants, which will limit the utility of metagenomic sequencing to infer functionality. These limitations can be overcome through the increased microbial coverage yielded by benzonase2 processing. Furthermore, metagenomic sequencing with benzonase2 processing identified differences in antibiotic resistance gene profiles among samples with similar relative abundances of a given taxon; by contrast, functional inferences from 16S amplicon sequencing of the same samples would not indicate any differences, highlighting the potential of metagenomic sequencing and this extraction method for infectious disease studies. Comparison of sequencing results from mock communities revealed the potential bias introduced by extracellular DNA in defining viable community structures in infections, which are continuously perturbed and remodeled by host immune activity, antibiotics and other treatments, nutrient limitation, interspecies interactions, and natural cell turnover, and during biofilm production. This creates the potential for the extracellular DNA load to be systematically larger among subjects in the treatment arms of antibiotic therapy trials, which would bias the comparison of metagenomes between sample groups if extracellular DNA was not excluded. We found that the nuclease-based processing technique benzonase2 resulted in the most accurate representation of viable community structure among the tested methods. While MolYsis also provided an accurate picture of the viable bacterial community, this method was not as effective in enriching for bacterial reads in our samples.
Many of the methods used in this comparative study have been used individually in CF respiratory microbiome studies. For example, PMA treatment has been used in advance of 16S amplicon sequencing (Rogers et al., 2010b, 2013) and metagenomic sequencing (Marotz et al., 2018). Regarding nuclease treatment, Lim et al. (2013) used DNaseI to reduce human DNA in sputum samples before metagenomic sequencing and demonstrated a reduction in human genome equivalents, but did not define the impact of this processing on microbial reads or coverage. Furthermore, 25 mM EDTA was used to inactivate the enzyme in these studies; here, we show that EDTA concentrations as low as 5 mM can lyse P. aeruginosa, with unclear effects on community profiles. Leo et al. used the MolYsis method to treat CF bronchoalveolar lavage fluid (BALF) (Leo et al., 2017), a method that resulted in 72% human DNA after depletion. By comparison, we found this method to be less efficient at reducing human DNA in CF sputum, perhaps due to the relatively high viscosity and complexity of sputum compared to BALF. In addition, we are aware of a commercially available kit for the depletion of human DNA that uses detergent-mediated lysis of human cells followed by benzonase digestion of extracellular DNA, but differs from the methods described here in the use of proteinase K to inactivate the endonuclease. Although not tested here, we believe that this method may not be optimal for analyzing CF sputa or perhaps other clinical samples, which often require enzymatic digestion steps after extracellular DNA removal to efficiently extract DNA from common taxa, such as S. aureus (Zhao et al., 2012b).
There are several important considerations from this work that may extend beyond the samples tested in this study. While benzonase2 was shown here to work well for metagenomic studies of CF sputum, this method is likely equally suited for other types of samples from inflamed, infected tissues with overwhelming amounts of human DNA such as wound, vaginal, blood, oral, sinus, or upper airway respiratory specimens that sample more directly from the lower airways (e.g., bronchoalveolar lavage), or other chronic infection samples. It is also easy to append benzonase2 to a variety of DNA extraction protocols, whether custom or kit based. We also found little change in calculated taxonomic profiles in samples extracted before versus after freezing, indicating utility for previously banked samples. Nevertheless, it is important to consider that the mock communities tested here were constructed to reflect the microbiota found in CF sputum, but contained neither added human DNA nor the complex matrix of proteins, glycans, and cellular debris that binds extracellular DNA in vivo. We did construct bacterial mock communities with exogenous human DNA (constituting approximately 95% of total DNA), but found that even standard extraction efficiently removed >99% of this host DNA, highlighting the differences between purulent sputum and liquid culture. Therefore, to demonstrate generalizable results, this method should be tested and optimized on samples and mock communities based on other sample types. Furthermore, we did observe taxonomic changes, occasionally marked, with benzonase2 as compared to standard extraction. Although we present a number of lines of evidence that this revised method, coupled with metagenomic sequencing, results in a microbial profile that more closely reflects the viable bacterial constituencies in both mock communities and CF sputum samples in comparison to 16S amplicon sequencing, this analysis does not rigorously quantify the bias introduced by benzonase2-aided metagenomic sequencing compared to more standard 16S amplicon sequencing. We should also note that 16S amplicon sequencing carries its own biases and limitations: differences in the amplification efficiency of the 16S gene between taxa and the inability to directly infer the genetic capacity of the community.
There are also logistical considerations for benzonase2 extraction. Because nuclease-based methods such as benzonase2 decrease the total DNA yield from samples, sometimes by 1–2 logs (Table S2), these techniques often require extraction from larger sample volumes to make a sequencing library compared with standard extraction methods, an issue that likely would be compounded for samples with lower microbial abundance compared to sputum. This issue may improve with newer library-construction technologies that require lower amounts of DNA input. Benzonase2 is also a lengthy protocol that is not commercially available as a kit, raising the potential for both contamination and errors, particularly when processing large sample sets.
Regardless, this method represents an important step toward extending metagenomic sequencing, a powerful and promising technology for analyzing complex clinical samples such as those from chronic infections. While this method affords both enhanced sequencing coverage adequate for functional metagenomics and a sharper focus on viable bacterial cells, it is not yet known whether microbiome analyses focused on viable bacterial cells would correlate more closely with clinical outcomes than would the results from other microbiological approaches. This study involved an in-depth comparative analysis of a relatively small sample set, precluding clinical correlation. We therefore plan to apply this refined extraction method to a large sample set of CF sputa collected before, during, and after antibiotic treatment, with matched clinical data, and compare with standard extraction. We hope that such a large, more refined sample set will allow us to further define the association of the viable bacterial community and their functional characteristics and clinical outcomes.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lucas R. Hoffman (lhoffm@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Sputum Samples
This study was approved by the Seattle Children’s Hospital (SCH) Institutional Review Board. Sputum samples were collected from children diagnosed with CF who presented to Seattle Children’s Hospital as part of regular clinical care, were willing to provide samples and provide informed consent, and were able to expectorate at least 1mL of total sputum (so that we would have enough sputa to split each sample into multiple aliquots). Participants were selected to reflect a range of prior sputum culture results (both in terms of dominant culturable species and abundances), and they were on a mix of inhaled, oral and/or IV antibiotics and presented with a range of clinical statuses (stable, exacerbation, in treatment). Finally, samples 183 and 189 were from the same individual, collected on different days. Test Set 1 samples were homogenized by passing through a 1mL syringe approximately 10 times, aliquoted, and either processed immediately or frozen at −80°C prior to extraction. Test Set 2 samples were diluted 1:1 with 10% Sputolysin (EMD Millepore, 56-000-010ML), aliquoted evenly and frozen prior to extraction.
Bacteria
All bacterial isolates used in this study were derived from the sputum samples detailed above, see Method Details.
METHOD DETAILS
Mock Community Construction
Seven individual taxa isolated from CF sputa (Pseudomonas aeruginosa, Staphylococcus aureus, Neisseria sp. unclassified, Achromobacter xylosoxidans, Streptococcus salivarius, Stenotrophomonas maltophila and Rothia mucilaginosa), were grown in liquid culture in to mid-log phase in Brain Heart Infusion broth (Neisseria, R. mucilaginosa, S. salivarius) or Tryptic soy broth (P. aeruginosa, S. aureus, A. xylosoxidans, S. maltophila) and taxonomy confirmed by Sanger sequencing of the 16S rRNA gene. Specified volumes of each culture were mixed by gently vortexing and inverting and were then diluted to generate a mock community. Serial dilutions of each taxa were simultaneously grown on 5% Sheep’s Blood Agar for viable counts to retrospectively determine viable community composition, resulting in the following calculated relative abundances: 63.8% Pseudomonas, 29.5% Staphylococcus, 2.89% Neisseria, 0.398% Achromobacter, 1.02% Streptococcus, 2.04% Stenotrophomonas and 0.370% Rothia. For comparison with 16S amplicon sequencing, we adjusted relative abundances by 16S rRNA copy number, resulting in 60.0% Pseudomonas, 34.3% Staphylococcus, 2.89% Neisseria, 0.398% Achromobacter, 1.02% Streptococcus, 2.04% Stenotrophomonas and 0.370% Rothia. DNA extraction was performed immediately after mock community construction as detailed below.
To quantify extracellular DNA in in vitro culture supernatants, three separate isolates of A. xylosoxidans, three of P. aeruginosa, two of R. dentocariosa, two of S. aureus and one of R. aeria were cultured as indicated above and 1mL of culture was removed. Bacterial cells were pelleted by spinning at 10,000 g for 3 min, supernatant was collected and syringe filtered using a 0.22 μm PES filter. DNA was extracted using Standard extraction as detailed below, beginning at the Proteinase K step. Total bacterial load was determined via 16S qPCR as detailed below.
DNA Extraction
For all samples and depletion methods, Standard DNA extraction was performed as follows: approximately 200 mg of sputum or 200 μL of mock community was suspended in 1 mL of PBS and centrifuged at 13,000 g for 3 min. The pellet was suspended in 400 μL TE. A mixture of 1 mm and 0.1 mm silica:zirconia beads and a single tungsten-carbide bead was added to TE solution and followed by bead-beating for 1 min in a BioSpec MiniBeadBeater. The resulting solution was boiled for 5 min at 95°C. Lysozyme (Sigma L6876, 3 mg/mL final) and lysostaphin (Ambi LSPN, 0.14 mg/mL final) were added, and the sample was incubated for 1 h at 37°C. Proteinase K (Invitrogen 25530049, 1.4 mg/mL final) and SDS (1.8% final) were added, and the resulting solution incubated at 56°C for 30 min before cooling to room temperature. The solution was removed to a separate tube and 5 M NaCl was added (2 M final) before adding phenol:chloroform:isoamylalcohol (25:24:1) at a 1:1 volume. The solution was then incubated for 20 min at room temperature, centrifuged at 13,000 g for 20 min and the top aqueous layer was collected. 0.133 volume equivalent of 7.5 M ammonium acetate was added to the aqueous layer, and the resulting solution diluted 1:1 with cold 100% ethanol to precipitate DNA. DNA product was cleaned with a spin column.
For the “Antibody Depletion” method, 1 μg of total extracted DNA was processed with the NEBNext Microbiome DNA Enrichment Kit (NEB E2612S) according to the manufacturer’s instructions. For the “Cell Lysis” method (Hunter et al., 2011), prior to Standard DNA extraction sputum was suspended in 1 mL PBS, vortexed and centrifuged at 13,000 g for 2 min. The resulting pellet was suspended in 1 mL TrypZean (Sigma T3449) and 0.05% Tween-20 (Sigma P9416) and incubated at 37°C for 60 min. The solution was then vortexed to mix and centrifuged at 5,000 g for 2 min to pellet eukaryotic cells. The supernatant was then removed and centrifuged at 13,000 g for 10 min to pellet any prokaryotic cells and suspended in PBS before proceeding with Standard DNA extraction. For the “Molysis” method, prior to Standard DNA extraction sputum was initially processed with Molzym’s Molysis Human DNA removal kit (D-300–050) according to manufacturer’s instructions, pausing before the “Buglysis” step. Cells were washed once with PBS and suspended in TE before proceeding with Standard DNA extraction. For the “Benzonase1” method (Hunter et al., 2011), sputum was suspended in 7 mL dH2O and incubated at room temperature for 1 h with gentle agitation. 10x strength Benzonase buffer (200 mM Tris-HCl, 10 mM MgCl2) to a final 1X and 250U Benzonase (Sigma E-1014) was added and the sample was incubated at 37°C for 2 h with gentle agitation. The Benzonase reaction was quenched by adding EDTA (5 mM final) and NaCl (150 mM final). The resulting solution was spun at 8000 g for 10 min and the pellet washed once with PBS, then suspended in 400 μL TE before proceeding with Standard DNA extraction. The “Benzonase2” method differed from Benzonase1 in the moving of one step: after the two-hour nuclease incubation and before EDTA inhibition, the bacteria were pelleted by centrifugation at 8000 g for 10 min and washed once in PBS, then suspended in 400 μL TE, at which point EDTA (5 mM final) was added to inactivate the endonuclease before proceeding directly to Standard extraction. For the “PMA” method, 300uL PBS was added to 200uL of the bacterial mock community, 1 μL propidium monoazide (20mM in water) was added and the solution incubated at room temperature for 5 min. The solution was then incubated under 160 LED white light for 15 min with gentle agitation. The bacteria were pelleted by centrifugation at 13,000 g for 10min, washed once in PBS and then suspended in 400 μL TE before proceeding with Standard extraction. Reagent blanks consisting of PBS alone were processed for each extraction method and sequenced via 16S amplicon sequencing.
Quantitative PCR
Total bacterial load was determined using quantitative PCR with PowerUp SYBR Green Master mix (Applied Biosystems A25742) and previously published primers and reaction conditions (Nadkarni et al., 2002). Total human DNA load was determined using primers targeting the beta-globin gene, as previously described (Handschur et al., 2009). Data were analyzed with the Bio-Rad CFX Manager 3.1 software, using software-defined Cq thresholds. Proportion of human DNA was determined by calculating quantity of human and bacterial DNA from genome equivalents (GE), using a genome size of 6.5×109 bp for human and 5×106 bp for the average microbial genome. GEs for human cells and bacterial cells were multiplied by their respective genome sizes to calculate number of base pairs per microliter. “Total DNA” per microliter was calculated by adding the total bacterial and human base pairs. The proportion of human DNA was calculated by dividing total human base pairs per microliter by total base pairs per microliter. Based on the following commonly used definition of limit of detection (LOD) for qPCR (Burns and Valdivia, 2007; Bustin et al., 2009), “the lowest copy number associated with the serial dilution that gave a positive PCR response on 95% of occasions,” the LOD of our qPCR assay is approximately 8.24 Genome Equivalents (GE) per uL.
Effect of Benzonase processing on bacterial viability
Six separate isolates of P. aeruginosa, three separate isolates of S. aureus, two separate isolates of Rothia dentocariosa and one isolate each of Streptococcus salivarius, Rothia aeria and Achromobacter xylosoxidans cultured from Test Set 1 samples were grown in liquid culture to mid-log phase. Cells were washed in PBS and plated for viable counts on Brain Heart Infusion agar or Tryptic Soy Agar. Cells were then subjected to Benzonase1 and Benzonase2 extraction as described above, with aliquots plated for viable counts at input, after hypotonic lysis and after enzyme inactivation.
Phylogenetic composition from metagenomic shotgun sequencing
Next generation sequencing libraries were prepared for all samples using the Nextera DNA Sample Prep Kit (Illumina FC-121–1031) following manufacturer’s instructions. Three of the Test Set 1 samples (183–185) were sequenced on the Illumina MiSeq platform, producing an average of 2.55 × 106 reads per sample. The remaining samples (186–190, 205, 309 and 312) were sequenced on the Illumina HiSeq platform, producing an average of 1.55 × 107 reads per sample in our first set of 8 (Test Set 1) sputum samples and 3.48 × 107 reads in Test Set 2. Sequencing data from all samples were de-duplicated using SeqUniq (version 0.1) https://github.com/standage/sequniq) and quality filtered using KneadData (version 0.6.1) and Trimmomatic (version 0.33) (Bolger et al., 2014). Human reads were identified and removed with BMTagger (version 3.101) and community phylogenetic composition was determined using MetaPhlAn2 (Thompson et al., 2017; Truong et al., 2015) (version 2.2.0) to produce a MetaPhlAn2 taxa table. All commands were executed with default settings, with the exception of KneadData, which was used with the “–run-bmtagger” flag. For our mock communities, the Achromobacter genus represents reads originally annotated either as Achromobacter or Bordetella, the latter of which we believe represents misidentified Achromobacter reads. We have found MetaPhlAn2 consistently splits this taxon into Achromobacter and Bordetella even when this later taxon was not added to mock communities and was not detected in 16S amplicon sequencing of the same samples.
Phylogenetic composition from 16S amplicon sequencing
The V4 region of the 16S rRNA gene was amplified using primers from the Earth Microbiome Project (Thompson et al., 2017) and barcodes adapted from Kozich et al. (Kozich et al., 2013). (detailed at https://github.com/SchlossLab/MiSeq_WetLab_SOP/blob/master/MiSeq_WetLab_SOP.md). 16S amplicons were made under the following conditions: 94°C for 3min, 30 cycles of the following sequence: [94°C for 45 s, 50°C for 60 s, 72°C for 90 s], and then 72°C for 10min. Libraries were constructed by pooling equimolar amounts of each sample or of each blank at the volume of the least concentrated sample. Libraries were sequenced on the Illumina MiSeq platform producing paired 300 bp reads.
16S amplicon sequencing data were analyzed using the denoising program DADA2 (Wang et al., 2007) (version 1.6.0) and the complete code is listed in Methods S1. Briefly, we computationally trimmed 10bp off the beginning of the both the forward and reverse reads and truncated the forward read to 200bp and the reverse read to 100bp. We used our entire dataset to define an error rate at each base pair and then denoised all sequences. Forward and reverse reads were merged and any pair without perfect overlap was removed. Finally, chimeric sequences were removed. This program produces a list of “Amplicon Sequence Variants” (ASV) analogous to OTUs generated with a 97% clustering method. Each ASV was annotated with the RDP Bayesian classifier (Wang et al., 2007) against the SILVA database (Quast et al., 2013) to produce a 16S amplicon taxa table. ASVs identified as Pseudomonas, Staphylococcus and Achromobacter were analyzed with BLASTn to determine species identity. Code is detailed further in Methods S1.
Relative Abundance Analysis
After quality filtering, sputum samples produced an average of 56,347 reads per sample, mock communities produced an average of 18,687 reads, extraction blanks produced an average of 101 reads and amplicon blanks produced an average of 8 reads. Most reads in the blanks were taxa in common among samples with similar sequencing barcodes. As the absolute abundance of these reads was 2–3 logs lower in blanks than in neighboring samples during sequencing, we found it unlikely they contributed to the taxonomic profiles of our samples, but that these reads were more likely due to errors in barcode reading (Kircher et al., 2012; Sinclair et al., 2015). The remaining taxa were those noted by Salter et al. to be common reagent contaminants (Salter et al., 2014) and were found at < 1% relative abundance in samples. We did not observe any increase in those taxa identified in extraction blanks with any extraction method compared to standard, suggesting that background contamination was not the source of the increase in taxonomic richness we observed with Benzonase extraction. As the number of reads from blanks were substantially outnumbered by those detected in sputum samples, we did not analyze the reads from these controls further (raw taxonomic tables from 16S amplicon sequencing are in Table S3).
Analysis of community composition was performed in R (R Core Team, 2017) (version 3.4.2) and visualized using ggplot2 (Wick-ham, 2009) (version 2.2.1). Taxa tables from 16S amplicon sequencing and MetaPhlAn2 output were merged. For sputum samples, all taxa below 1% relative abundance in all samples or above 5% relative abundance solely in extraction blanks, were pooled into the “Other” category (these genera are listed in Table S4). For mock communities, we excluded two detected taxa which were not added to our mock community (Pusillimonas and Caulobacter), which both comprised < 0.03% of the community.
Phylogenetic trees were constructed using the Vegan (Oksanen et al., 2017) and Ape packages (Paradis et al., 2004) (versions 2.4.4 and 5.0 respectively) using the bray-curtis dissimilarity metric and the “Unweighted Pair Group Method with Arithmetic Mean” agglomeration method for hierarchical clustering. Tree was visualized using the ggtree package (Yu et al., 2016) (version 1.10.0).
Microbial read depth analysis
Contigs from metagenomic sequencing data were assembled with Megahit (Version 1.1.2) (Li et al., 2015), producing a total of 162,842. All reads were then mapped back to assembled contigs with Bowtie2 (Version 2.2.6) (Langmead and Salzberg, 2012). Taxonomy was determined by predicting open reading frames with Prodigal (Version 2.6.3) (Hyatt et al., 2010). Contigs of human origin were identified with Centrifuge (Kim et al., 2016) based on these ORFs. The “nucleotide protein homolog model” collection from the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013) was used to detect antibiotic resistance genes. This model comprises 2,239 unique genes for which presence is sufficient to confer resistance (i.e., not including ubiquitous genes that only confer resistance when mutated such as DNA gyrase). All reads were mapped to this database with Bowtie2 as above. The complete code for this analysis is listed in Methods S2. Mapping results were processed and visualized with Anvi’o (Version 4) (Eren et al., 2015) and figures were finalized with Inkscape (https://inkscape.org). Code is detailed in Methods S2.
For subsampled analysis, metagenomic sequencing reads after quality filtering and human read removal were subsampled to indicated depths and analysis was repeated as detailed above. Code is detailed in Methods S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed in R. For all comparisons between standard extraction and processing methods for human DNA removal, p values were calculated using a pairwise, Wilcoxon signed rank tests with a Benjamini-Hochberg correction for multiple comparisons. p values smaller than 0.05 were considered significant.
DATA AND SOFTWARE AVAILABILITY
The accession number for all metagenomic sequencing and 16S amplicon sequencing data reported in this paper is NCBI Bioproject: PRJNA516442.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Pseudomonas aeruginosa | This paper: Isolated from samples 184 (mock community), 185 and 187 | N/A |
| Staphylococcus aureus | This paper: Isolated from samples 183, 188 (mock community) and 189 | N/A |
| Neisseria sp. unclassified | This paper: Isolated from sample 188 | N/A |
| Achromobacter xylosoxidans | This paper: Isolated from samples 183 and 189 (mock community) | N/A |
| Streptococcus salivarius | This paper: Isolated from sample 190 | N/A |
| Stenotrophomonas maltophila | This paper: Isolated from sample 188 | N/A |
| Rothia mucilaginosa | This paper: Isolated from samples 188 (mock community) | N/A |
| Rothia dentocariosa | This paper: Isolated from samples 183 and 190 | N/A |
| Rothia aeria | This paper: Isolated from sample 188 | N/A |
| Biological Samples | ||
| Sputum sample 183 | This paper | N/A |
| Sputum sample 184 | This paper | N/A |
| Sputum sample 185 | This paper | N/A |
| Sputum sample 186 | This paper | N/A |
| Sputum sample 187 | This paper | N/A |
| Sputum sample 188 | This paper | N/A |
| Sputum sample 189 | This paper | N/A |
| Sputum sample 190 | This paper | N/A |
| Sputum sample 205 | This paper | N/A |
| Sputum sample 309 | This paper | N/A |
| Sputum sample 312 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phosphate Buffered Saline (PBS) | Sigma | D8537–500ML |
| Tris-EDTA, pH 8.0 | Fisher Scientific | AM9849 |
| 0.1mm silica:zirconia beads | Fisher Scientific | 11079101z |
| 1mm silica:zirconia beads | Fisher Scientific | NC9847287 |
| Tungsten-carbide beads | QIAGEN | 69997 |
| Lysozyme | Sigma | L6876 |
| Lysostaphin | Ambi | LSPN |
| Proteinase K | Fisher Scientific | 25-530-049 |
| 10% Sodium Dodecyl Sulfate | Fisher Scientific | 15-553-027 |
| 5M Sodium chloride | Fisher Scientific | AM9759 |
| Pheno:Chloroform:lsoamyl alcohol | Sigma | P2069–400ML |
| 3M Ammonium Acetate | Fisher Scientific | FERR1181 |
| 200 proof Ethanol | Fisher Scientific | 07-678-003 |
| TrypZean | Sigma | T3449 |
| Tween-20 | Sigma | P9416 |
| Molecular grade water | WVR | M46000 |
| 1M Tris-HCI, pH8.0 | Fisher Scientific | 15-568-025 |
| 1M Magnesium Chloride | Fisher Scientific | AM9530G |
| Benzonase | Sigma | E-1014 |
| 0.5M EDTA | Fisher Scientific | 15-575-020 |
| Propidium monoazide | VWR | 89139–068 |
| PowerUp SYBR Green Master mix | Applied Biosystems | A25742 |
| 5 Primer Hot Master Mix | VWR | 2200410 |
| Sputolysin (DTT) | Fisher Scientific | 56-000-010mL |
| Critical Commercial Assays | ||
| EZ-10 Spin Column Bacterial Genomic DNA Mini-prep Kit | Biobasic | BS423 |
| Deposited Data | ||
| Metagenomic sequencing and 16S amplicon sequencing data | This paper | NCBI Bioproject: PRJNA516442 |
| Oligonucleotides | ||
| GGGCAACGTGCTGGTCTG | Handschur et al., 2009 | Human-betaG-F |
| AGGCAGCCTGCACTGGT | Handschur et al., 2009 | Human-betaG-R |
| TCCTACGGGAGGCAGCAGT | Nadkarni et al., 2002 | Universal16S_F |
| GGACTACCAGGGTATCTAATCCTGTT | Nadkarni et al., 2002 | Universal16S_R |
| Software and Algorithms | ||
| Bio-Rad CFX Manager 3.1 | Biorad | 1845000 |
| SeqUniq |
https://github.com/standage/sequniq |
Version 0.1 |
| KneadData | https://bitbucket.org/biobakery/kneaddata/wiki/Home | Version 0.6.1 |
| Trimmomatic | Bolger et al., 2014 | RRID:SCR_011848; Version 0.33 |
| BMTagger | https://bioconda.github.io/recipes/bmtagger/README.html; Human Microbiom Project | RRID:SCR_014619; Version 3.101 |
| MetaPhlAn2 | Thompson et al., 2017; Truong et al., 2015 | RRID:SCR_004915; Version 2.2.0 |
| DADA2 | Wang et al., 2007 | Version 1.6.0 |
| RDP Bayesian classifier | Wang et al., 2007 | Implemented through DADA2 (Version 1.6.0) |
| R | R Core Team, 2017 | RRID:SCR_001905; Version 3.4.2 |
| ggplot2 | Wickham, 2009 | RRID:SCR_014601; Version 2.2.1 |
| Vegan | Oksanen et al., 2017 | RRID:SCR_011950; Version 2.4.4 |
| Ape | Paradis et al., 2004 | Version 5.0 |
| ggtree | Yu et al., 2016 | Version 1.10.0 |
| Megahit | Li et al., 2015 | Version 1.1.2 |
| Bowtie2 | Langmead and Salzberg, 2012 | RRID:SCR_016368; Version 2.2.6 |
| Prodigal | Hyatt et al., 2010 | RRID:SCR_011936; Version 2.6.3 |
| Centrifuge | Kim et al., 2016 | RRID:SCR_016665; Version 1.0.3-beta |
| Anvi’o | Eren et al., 2015 | Version 4 |
| Inkscape | https://inkscape.org | RRID:SCR_014479; Version 0.92.3 |
Highlights.
Human and extracellular bacterial DNA can bias metagenomic sequencing
Hypotonic lysis, endonuclease digestion limit human and extracellular bacterial DNA
Reducing extracellular DNA optimizes metagenomic sequencing of viable bacterial cells
Increased microbial sequencing coverage improves detection of important genes
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (DK089507), the Cystic Fibrosis Foundation (SINGH15R0), an institutional training grant (T32AI55396), and an unrestricted grant from Novartis. We thank Sharon McNamara, Laura Nay, and Cheryl Majors for recruiting participants and collecting sputum samples; Angshita Dutta for assistance with the graphical abstract; and Pradeep Singh for constructive discussion. Finally, we thank all of the patients who participated in this study.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, four tables, and three Supplemental Methods and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.091.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Banin E, Brady KM, and Greenberg EP (2006). Chelator-induced dispersal and killing of Pseudomonas aeruginosa cells in a biofilm. Appl. Environ. Microbiol 72, 2064–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellehumeur C, Boyle B, Charette SJ, Harel J, L’Homme Y, Masson L, and Gagnon CA (2015). Propidium monoazide (PMA) and ethidium bromide monoazide (EMA) improve DNA array and high-throughput sequencing of porcine reproductive and respiratory syndrome virus identification. J. Virol. Methods 222, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt AS, Manzo VE, Pedamallu CS, Duke F, Cai D, Bienfang DC, Padera RF, Meyerson M, and Docken WP (2014). In search of a candidate pathogen for giant cell arteritis: sequencing-based characterization of the giant cell arteritis microbiome. Arthritis Rheumatol. 66, 1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright AT, Tewhey R, Abeles S, Chuquiyauri R, Llanos-Cuentas A, Ferreira MU, Schork NJ, Vinetz JM, and Winzeler EA (2012). Whole genome sequencing analysis of Plasmodium vivax using whole genome capture. BMC Genomics 13, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JL, and Rolain J-M (2014). Culture-based diagnostic microbiology in cystic fibrosis: can we simplify the complexity? J. Cyst. Fibros 13, 1–9. [DOI] [PubMed] [Google Scholar]
- Burns M, and Valdivia H (2007). Modelling the limit of detection in real-time quantitative PCR. Eur. Food Res. Technol 226, 1513–1524. [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Carmody LA, Zhao J, Kalikin LM, LeBar W, Simon RH, Venkataraman A, Schmidt TM, Abdo Z, Schloss PD, and LiPuma JJ (2015). The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodini RJ, Dowd SE, Davis B, Galandiuk S, Chamberlin WM, Kuenstner JT, McCallum RW, and Zhang J (2013). Crohn’s disease may be differentiated into 2 distinct biotypes based on the detection of bacterial genomic sequences and virulence genes within submucosal tissues. J. Clin. Gastroenterol 47, 612–620. [DOI] [PubMed] [Google Scholar]
- Chmiel JF, Aksamit TR, Chotirmall SH, Dasenbrook EC, Elborn JS, LiPuma JJ, Ranganathan SC, Waters VJ, and Ratjen FA (2014). Antibiotic management of lung infections in cystic fibrosis. I. The microbiome, methicillin-resistant Staphylococcus aureus, gram-negative bacteria, and multiple infections. Ann. Am. Thorac. Soc 11, 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, et al. (2010). Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One 5, e11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cystic Fibrosis Foundation (2015). Patient Registry 2015 Annual Data Report. https://www.cff.org/Our-Research/CF-Patient-Registry/2015-Patient-Registry-Annual-Data-Report.pdf.
- Emerson J, Rosenfeld M, McNamara S, Ramsey B, and Gibson RL (2002). Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 34, 91–100. [DOI] [PubMed] [Google Scholar]
- Emerson JB, Adams RI, Román CMB, Brooks B, Coil DA, Dahlhausen K, Ganz HH, Hartmann EM, Hsu T, Justice NB, et al. (2017). Schrödinger’s microbes: tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 5, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Esen ö.C., Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO, and van Gulik W (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkus O, de Jager VCL, Geene RTCM, van Alen-Boerrigter I, Hazel-wood L, van Hijum SAFT, Kleerebezem M, and Smid EJ (2016). Use of propidium monoazide for selective profiling of viable microbial cells during Gouda cheese ripening. Int. J. Food Microbiol. 228, 1–9. [DOI] [PubMed] [Google Scholar]
- Exterkate RAM, Zaura E, Brandt BW, Buijs MJ, Koopman JE, Crielaard W, and Ten Cate JM (2015). The effect of propidium monoazide treatment on the measured bacterial composition of clinical samples after the use of a mouthwash. Clin. Oral Investig. 19, 813–822. [DOI] [PubMed] [Google Scholar]
- Feigelman R, Kahlert CR, Baty F, Rassouli F, Kleiner RL, Kohler P, Brutsche MH, and von Mering C (2017). Sputum DNA sequencing in cystic fibrosis: non-invasive access to the lung microbiome and to pathogen details. Microbiome 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti P, Farina S, Cristofolini M, Girolomoni G, Tett A, and Segata N (2017). Experimental metagenomics and ribosomal profiling of the human skin microbiome. Exp. Dermatol 26, 211–219. [DOI] [PubMed] [Google Scholar]
- Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, and Wolfgang MC (2012). The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 7, e45001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson RL, Burns JL, and Ramsey BW (2003). Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med 168, 918–951. [DOI] [PubMed] [Google Scholar]
- Glassing A, Dowd SE, Galandiuk S, Davis B, Jorden JR, and Chiodini RJ (2015). Changes in 16s RNA Gene Microbial Community Profiling by Concentration of Prokaryotic DNA. J. Microbiol. Methods 119, 239–242. [DOI] [PubMed] [Google Scholar]
- Goltsman DSA, Sun CL, Proctor DM, DiGiulio DB, Robaczewska A, Thomas BC, Shaw GM, Stevenson DK, Holmes SP, Banfield JF, and Relman DA (2018). Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res. 28, 1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschur M, Karlic H, Hertel C, Pfeilstöcker M, and Haslberger AG (2009). Preanalytic removal of human DNA eliminates false signals in general 16S rDNA PCR monitoring of bacterial pathogens in blood. Comp. Immunol. Microbiol. Infect. Dis 32, 207–219. [DOI] [PubMed] [Google Scholar]
- Haque H, and Russell AD (1974). Effect of ethylenediaminetetraacetic acid and related chelating agents on whole cells of gram-negative bacteria. Antimicrob. Agents Chemother. 5, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MR, Rawat A, Tang P, Jithesh PV, Thomas E, Tan R, and Tilley P (2016). Depletion of Human DNA in Spiked Clinical Specimens for Improvement of Sensitivity of Pathogen Detection by Next-Generation Sequencing. J. Clin. Microbiol 54, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horz H-P, Scheer S, Vianna ME, and Conrads G (2010). New methods for selective isolation of bacterial DNA from human clinical specimens. Anaerobe 16, 47–53. [DOI] [PubMed] [Google Scholar]
- Hunter SJ, Easton S, Booth V, Henderson B, Wade WG, and Ward JM (2011). Selective removal of human DNA from metagenomic DNA samples extracted from dental plaque. J. Basic Microbiol 51, 442–446. [DOI] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubovics NS, Shields RC, Rajarajan N, and Burgess JG (2013). Life after death: the critical role of extracellular DNA in microbial biofilms. Lett. Appl. Microbiol 57, 467–475. [DOI] [PubMed] [Google Scholar]
- Jervis-Bardy J, Leong LEX, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, Nosworthy E, Morris PS, O’Leary S, Rogers GB, and Marsh RL (2015). Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T, Perry T, Kao D, Mason AL, Madsen KL, and Wong GK (2016). Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol 7, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song L, Breitwieser FP, and Salzberg SL (2016). Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Sawyer S, and Meyer M (2012). Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R, Sauer-Heilborn A, Welte T, Jauregui R, Brettar I, Guzman CA, and Höfle MG (2015). High individuality of respiratory bacterial communities in a large cohort of adult cystic fibrosis patients under continuous antibiotic treatment. PLoS One 10, e0117436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol 31, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo S, Gaïa N, Ruppé E, Emonet S, Girard M, Lazarevic V, and Schrenzel J (2017). Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing. Int. J. Mol. Sci 18, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu C-M, Luo R, Sadakane K, and Lam T-W (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. [DOI] [PubMed] [Google Scholar]
- Lim YW, Schmieder R, Haynes M, Willner D, Furlan M, Youle M, Abbott K, Edwards R, Evangelista J, Conrad D, and Rohwer F (2013). Metagenomics and metatranscriptomics: windows on CF-associated viral and microbial communities. J. Cyst. Fibros 12, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Haynes M, Furlan M, Robertson CE, Harris JK, and Rohwer F (2014a). Purifying the impure: sequencing metagenomes and metatranscriptomes from complex animal-associated samples. J. Vis. Exp (94) 10.3791/52117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Evangelista JS 3rd, Schmieder R, Bailey B, Haynes M, Furlan M, Maughan H, Edwards R, Rohwer F, and Conrad D (2014b). Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. J. Clin. Microbiol 52, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, and Zengler K (2018). Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother 57, 3348–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Losada P, Chouvarine P, Dorda M, Hedtfeld S, Mielke S, Schulz A, Wiehlmann L, and Tümmler B (2016). The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2, 00096–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy H, Charron-Mazenod L, and Lewenza S (2008). Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4, e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadkarni MA, Martin FE, Jacques NA, and Hunter N (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148, 257–266. [DOI] [PubMed] [Google Scholar]
- Nielsen SM, Nørskov-Lauritsen N, Bjarnsholt T, and Meyer RL (2016). Achromobacter Species Isolated from Cystic Fibrosis Patients Reveal Distinctly Different Biofilm Morphotypes. Microorganisms 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocker A, Sossa KE, and Camper AK (2007). Molecular monitoring of disinfection efficacy using propidium monoazide in combination with quantitative PCR. J. Microbiol. Methods 70, 252–260. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, et al. (2017). vegan: Community Ecology Package. https://rdrr.io/cran/vegan/.
- Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Price KE, Hampton TH, Gifford AH, Dolben EL, Hogan DA, Morrison HG, Sogin ML, and O’Toole GA (2013). Unique microbial communities persist in individual cystic fibrosis patients throughout a clinical exacerbation. Microbiome 1, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A Language and Environment for Statistical Computing. https://www.R-project.org/.
- Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G, and Bruce KD (2004). characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16s ribosomal DNA terminal restriction fragment length polymorphism profiling. J. Clin. Microbiol 42, 5176–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GB, Stressmann FA, Koller G, Daniels T, Carroll MP, and Bruce KD (2008). Assessing the diagnostic importance of nonviable bacterial cells in respiratory infections. Diagn. Microbiol. Infect. Dis 62, 133–141. [DOI] [PubMed] [Google Scholar]
- Rogers GB, Hoffman LR, Whiteley M, Daniels TWV, Carroll MP, and Bruce KD (2010a). Revealing the dynamics of polymicrobial infections: implications for antibiotic therapy. Trends Microbiol 18, 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GB, Marsh P, Stressmann AF, Allen CE, Daniels TVW, Carroll MP, and Bruce KD (2010b). The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin. Microbiol. Infect 16, 1656–1658. [DOI] [PubMed] [Google Scholar]
- Rogers GB, Cuthbertson L, Hoffman LR, Wing PAC, Pope C, Hooftman DAP, Lilley AK, Oliver A, Carroll MP, Bruce KD, and van der Gast CJ (2013). Reducing bias in bacterial community analysis of lower respiratory infections. ISME J. 7, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkjøbing VB, Thomsen TR, Alhede M, Kragh KN, Nielsen PH, Johansen UR, Givskov M, Høiby N, and Bjarnsholt T (2011). True microbiota involved in chronic lung infection of cystic fibrosis patients found by culturing and 16S rRNA gene analysis. J. Clin. Microbiol 49, 4352–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiman L, Siegel JD, LiPuma JJ, Brown RF, Bryson EA, Chambers MJ, Downer VS, Fliege J, Hazle LA, Jain M, et al. ; Cystic Fibrous Foundation; Society for Healthcare Epidemiology of America (2014). Infection prevention and control guideline for cystic fibrosis: 2013 update. Infect. Control Hosp. Epidemiol 35 (Suppl 1), S1–S67. [DOI] [PubMed] [Google Scholar]
- Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, and Walker AW (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair L, Osman OA, Bertilsson S, and Eiler A (2015). Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform. PLoS One 10, e0116955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima T, Iida K, Qin T, Taniai H, Seki M, Takade A, and Yoshida S (2007). Photoactivated ethidium monoazide directly cleaves bacterial DNA and is applied to PCR for discrimination of live and dead bacteria. Microbiol. Immunol 51, 763–775. [DOI] [PubMed] [Google Scholar]
- Stressmann FA, Rogers GB, Marsh P, Lilley AK, Daniels TWV, Carroll MP, Hoffman LR, Jones G, Allen CE, Patel N, et al. (2011). Does bacterial density in cystic fibrosis sputum increase prior to pulmonary exacerbation? J. Cyst. Fibros 10, 357–365. [DOI] [PubMed] [Google Scholar]
- Thoendel M, Jeraldo PR, Greenwood-Quaintance KE, Yao JZ, Chia N, Hanssen AD, Abdel MP, and Patel R (2016). Comparison of microbial DNA enrichment tools for metagenomic whole genome sequencing. J. Microbiol. Methods 127, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, et al. ; Earth Microbiome Project Consortium (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- van Tilburg Bernardes E, Charron-Mazenod L, Reading DJ, Reckseidler-Zenteno SL, and Lewenza S (2017). Exopolysaccharide-Repressing Small Molecules with Antibiofilm and Antivirulence Activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother 61, e01997–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis (Springer). [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Smith DK, Zhu H, Guan Y, and Lam TT-Y (2016). ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol 8, 28–36. [Google Scholar]
- Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, et al. (2012a). Decade-long bacterial community dynamics in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 109, 5809–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Carmody LA, Kalikin LM, Li J, Petrosino JF, Schloss PD, Young VB, and LiPuma JJ (2012b). Impact of enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS One 7, e33127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, and Pollard AJ (2012). A novel method of selective removal of human DNA improves PCR sensitivity for detection of Salmonella Typhi in blood samples. BMC Infect. Dis 12, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for all metagenomic sequencing and 16S amplicon sequencing data reported in this paper is NCBI Bioproject: PRJNA516442.