

Abstract
West Nile virus (WNV), a member of the Flavivirus genus, is a leading cause of viral encephalitis in the United States1. The development of neutralizing antibodies against the flavivirus envelope (E) protein is critical for immunity and vaccine protection2. Previously identified candidate therapeutic mouse and human neutralizing monoclonal antibodies (mAbs) target epitopes within the E domain III lateral ridge and the domain I-II hinge region, respectively3. To explore the neutralizing antibody repertoire elicited by WNV infection for potential therapeutic application, we isolated 10 mAbs from WNV-infected individuals. MAb WNV-86 neutralized WNV with a 50% inhibitory concentration (IC50) of 2 ng/mL, one of the most potently neutralizing flavivirus-specific antibodies ever isolated. WNV-86 targets an epitope in E domain II, and preferentially recognizes mature virions lacking an uncleaved form of the chaperone protein prM, unlike most flavivirus-specific antibodies4. In vitro selection experiments revealed a neutralization escape mechanism involving a glycan addition to E domain II. Finally, a single dose of WNV-86 administered two days post-infection protected mice from lethal WNV challenge. This study identifies a highly potent human neutralizing mAb with therapeutic potential that targets an epitope preferentially displayed on mature virions.
Flaviviruses are arthropod-borne, enveloped, positive-stranded RNA viruses that include clinically significant pathogens such as WNV, dengue virus (DENV), yellow fever virus (YFV), and Zika virus (ZIKV). The development of neutralizing antibodies (NAbs) is a surrogate of protection for most licensed flavivirus vaccines3. For emerging flaviviruses such as WNV and ZIKV, licensed vaccines or therapeutic agents for use in humans are lacking.
The main target of flavivirus NAbs is the E protein, which mediates entry into cells, and consists of three structural domains (DI, DII, DIII), a helical stem, and two antiparallel transmembrane helices. Although cryo-electron microscopic reconstructions of mature flavivirus particles reveal a smooth surface densely covered with 90 E dimers that lie flat against the viral membrane3, flaviviruses are structurally heterogeneous, owing partly to an inefficient cleavage of the viral chaperone protein prM during virion maturation5. Unlike the smooth surfaces of mature particles, immature particles incorporate prM-E heterotrimeric spikes, whereas partially mature particles contain structural features of both immature and mature particles5. Compared to mature virions containing little or no uncleaved prM, partially mature virions that retain prM are generally more sensitive to neutralization by antibodies targeting poorly exposed epitopes4,6–8.
Several groups have investigated the therapeutic potential of humanized or human mAbs9,10 and antibody fragments against WNV11. For example, the humanized murine mAb E1612 demonstrates therapeutic efficacy9 and recognizes an accessible epitope in the lateral ridge of E DIII (DIII-LR)13, a common target of potently neutralizing murine mAbs3. However, for some flaviviruses, DIII-LR is not a major target of potently neutralizing human NAbs, which often bind quaternary surfaces comprised of multiple E proteins3. Here, we identified a potently neutralizing WNV-specific human mAb that targets a epitope in DII, preferentially recognizes mature particles lacking prM, and prevents mortality in mice when administered as a single dose post-infection with WNV.
We obtained 13 matched serum and blood cell samples from individuals in Dallas, TX with prior laboratory-confirmed symptomatic WNV infection that occurred during the 2012 outbreak14. We screened serum samples for neutralization of WNV reporter virus particles (RVPs)15 (Figs. 1a-b) and estimated the reciprocal serum dilution that inhibited infectivity by 50% (NT50). These samples displayed a range of neutralizing activities (median NT50 of 1,504), with 9 of 13 samples displaying potent neutralization (NT50 > 1,000). To identify antibodies that mediated serum neutralization, we selected B cells from three donors with potent serum neutralizing activity for the generation of human hybridomas secreting WNV-specific mAbs that were screened for reactivity to WNV E protein. Ten WNV-reactive antibody-secreting hybridoma clones were recovered (Supplementary Table 1). Of these, three mAbs (WNV-61, WNV-39, WNV-18) lacked neutralizing activity, and another three (WNV-6, WNV-13, WNV-15) had modest inhibitory activity, with a large fraction (30% to 50%) of RVPs remaining infectious at the highest mAb concentration tested (10 μg/mL, Figs. 1c and e). The remaining four mAbs (WNV-10, WNV-57, WNV-62, WNV-86) strongly neutralized WNV RVPs and fully infectious WNV in a flavivirus type-specific pattern, although complete inhibition of WNV was only observed with WNV-86 (Figs. 1c-f; Supplementary Figs. 1-2). WNV-86 was particularly potent, neutralizing 50% of virus infectivity at a concentration (IC50) of 2 ng/mL, which is lower than that of the therapeutic mAb E169,12. Like murine E16 (mE16), WNV-86 inhibited infection as a Fab fragment (Supplementary Fig. 3) and was capable of blocking infection at a post-attachment step (Supplementary Fig. 4). Binding studies using recombinant E or DIII fragment protein demonstrated that the epitope recognized by these mAbs was located in DI or DII and therefore distinct from the DIII-LR epitope of mAb E16, and did not require a quaternary arrangement of E proteins on the virion surface (Supplementary Fig. 5).
Figure 1. Serum and mAb neutralization of WNV.
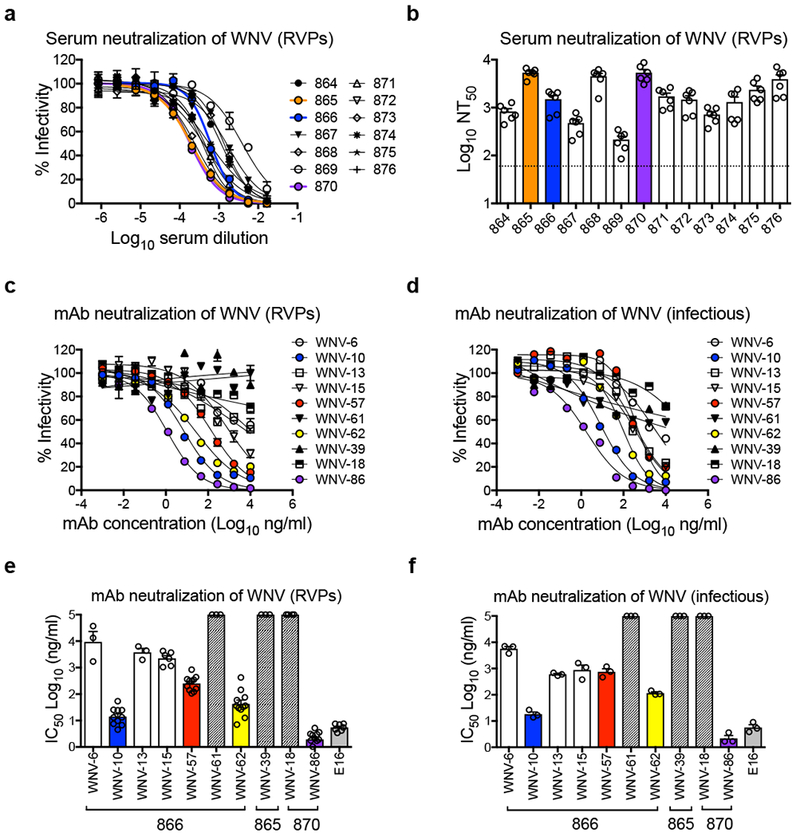
(a) Dose-response neutralization curves of WNV RVPs against 13 WNV convalescent human serum samples representative of five independent experiments with similar results. Infectivity levels were normalized to those observed in the absence of antibody. Data points and error bars indicate the mean and range of duplicate infections, respectively. Reciprocal neutralization titers were obtained by non-linear regression analysis constrained at the bottom to no infection (zero). (b) Mean values of the reciprocal serum dilution required to inhibit infection by 50% (NT50) obtained from five experiments, as indicated by open circles. Error bars represent the standard error of the mean (SEM). The dotted horizontal line shows the lowest serum dilution tested. Dose-response neutralization curves of (c) WNV RVPs or (d) fully infectious WNV against mAbs isolated from donors 866, 865, and 870 are representative of at least three independent experiments (see panels e and f) with similar results. Data points and error bars indicate the mean and range of duplicate infections, respectively. Mean values of mAb concentration required to inhibit infectivity by 50% (IC50) for (e) WNV RVPs or (f) fully infectious WNV were calculated from 3 independent experiments for all mAbs except WNV-10, WNV-57, WNV-62, WNV-86, E16 in panel e (8 independent experiments), as indicated by open circles. Error bars represent SEM. Hashed bars indicate that neutralization was not observed at the highest mAb concentration tested (10 μg/mL). The donor from which mAbs were isolated is indicated below the x-axis in panels e and f. Murine mAb E16 was included as a control in panels e and f.
To determine whether virion maturation state affected the activity of the mAbs, we compared their ability to neutralize prM- or prM+ WNV RVPs. As previously shown, the DIII-LR-specific murine mAb E16 neutralized these particles with similar potency4 (Fig. 2a), whereas murine mAb E53, which targets the cryptic DII fusion loop (DII-FL) epitope, neutralized prM+ RVPs but not prM- RVPs4,6 (Fig. 2b). As seen with most WNV antibodies4,7,8, three (WNV-10, WNV-57, or WNV-62) of the four human mAbs displayed increased neutralization potency (7 to 27-fold) against prM+ RVPs relative to prM- RVPs (Figs. 2c-e and g). In contrast, the IC50 of WNV-86 was 4-fold lower against prM- RVPs compared to prM+ RVPs (Figs. 2f-g), suggesting that this mAb recognizes an epitope preferentially displayed on prM- mature virions.
Figure 2. Effect of virion maturation on mAb neutralization.
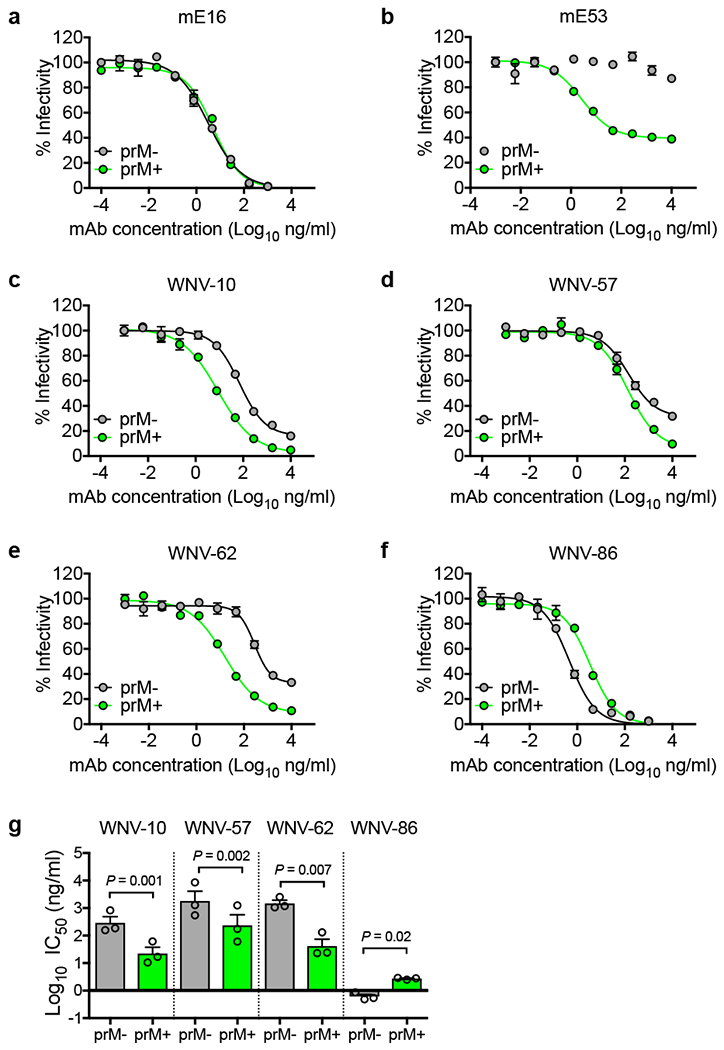
WNV RVPs prepared in the presence of overexpressed furin (prM-) or ammonium chloride (prM+) to increase or decrease the efficiency of virion maturation, respectively, were tested for sensitivity to neutralization by murine mAbs (a) mE16 and (b) mE53, or human mAbs (c) WNV-10, (d) WNV-57, (e) WNV-62, and (f) WNV-86. Shown are dose-response curves with infectivity normalized to levels observed in the absence of antibody representative of three independent experiments with similar results. Data points and error bars indicate the mean and range of duplicate infections, respectively. Mean IC50 values for each mAb (g) were obtained from three experiments. Error bars indicate SEM. The indicated P values were obtained from two-tailed paired t-tests. The mean difference between groups in panels g (and the 95% confidence interval of this difference) were as follows: WNV-10 [Log10 −1.11 (−1.29 to −0.94)]; WNV-57 [Log10 −0.89 (−1.05 to −0.73)]; WNV-62 [Log10 −1.5 (−2.11 to −0.99)]; WNV-86 [Log10 0.65 (0.23 to 1.06)].
To investigate the epitope targeted by WNV-86, we selected for neutralization escape mutations. After three serial passages in Vero cells under mAb selection pressure, WNV accumulated to high titres (~105 IU/mL) in the presence of WNV-86, indicating neutralization escape (Fig 3a and Supplementary Fig. 6a-b). Sequencing of escape variants from two independent selection experiments revealed a single nucleotide change corresponding to an amino acid substitution at E DII residue 64 (T64N), resulting in the addition of a potential N-linked glycosylation site (Fig 3c). WNV T64N replicated to near wild-type levels in mammalian and mosquito cell lines (Supplementary Fig. 6e-f). We confirmed the presence of an additional N-linked glycan on the T64N variant by SDS-PAGE and western blotting of RVP lysates with or without PNGaseF treatment (Supplementary Fig. 7a). We next evaluated the contribution of the additional N-linked sugar to neutralization resistance by creating a panel of 13 additional RVP variants incorporating amino acid substitutions that do not introduce an extra glycan at E residue 64. Of 14 total variants, 11 (excluding T64A, T64S, T64P) resulted in a 20-fold or greater reduction in WNV-86 potency (Supplementary Figs. 7b and d) but minimally impacted sensitivity to neutralization by mE16 (Supplementary Figs. 7c and e). These findings suggest that mutation at residue 64 directly affected WNV-86 neutralization potency, regardless of glycan occupancy.
Figure 3. In vitro selection of WNV-86 escape variants.
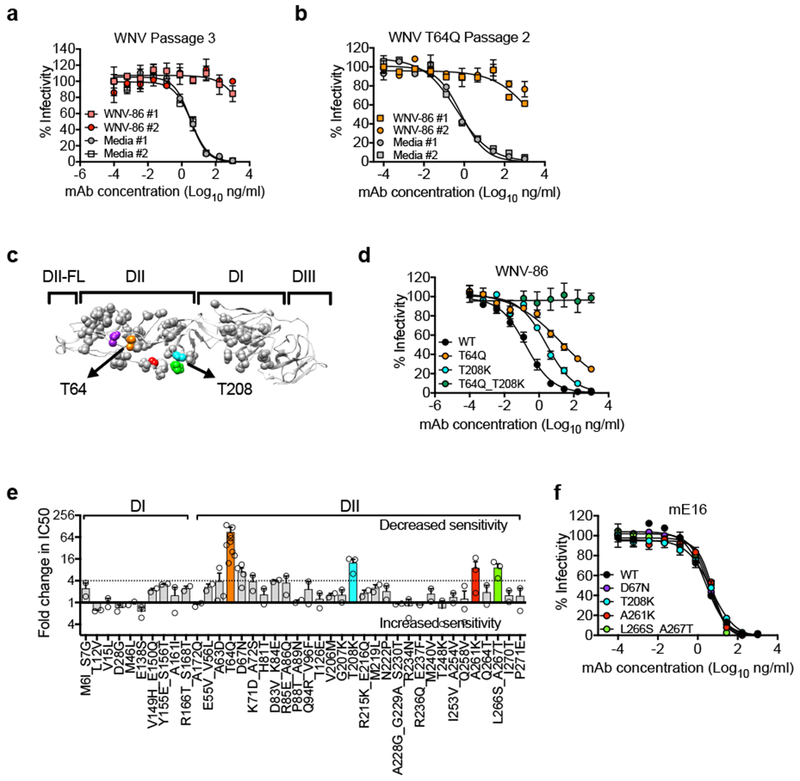
Vero cells were infected with WNV WT or T64Q in duplicate in the presence of mAb WNV-86 or media. (a) Passage 3 WNV WT and (b) passage 2 WNV T64Q virus supernatants obtained from duplicate wells of WNV-86 or medium only selection were tested independently for neutralization sensitivity against WNV-86. Data points and error bars indicate the mean and range of duplicate infections, respectively. Dose response curves are representative of duplicate infections from two independent experiments in panel a, and duplicate infections from one experiment in panel b. (c) Crystal structure of the WNV E protein monomer (PDB 2HG0) with DI, DII, DIII, and DII-FL indicated above the structure. Gray spheres indicate mutated residues selected for epitope mapping studies. Colored spheres indicate mutations at residues that reduce WNV-86 neutralization potency by > 4-fold relative to WT. Arrows indicate the location of mutation at residues T64 (orange spheres) and T208 (cyan spheres), respectively. (d) Representative dose-response neutralization curves of WNV WT, T64Q, T208K, and T64Q_T208K RVPs against WNV-86. Data points and error bars indicate the mean and range of duplicate infections, respectively. Data are representative of two experiments. (e) Average fold change in WNV-86 IC50 values against WNV E variants containing mutations in DI and DII relative to WT. Average values were obtained from two independent experiments for most variants, except A63D, T208K, A261K, L266S_A267T (n = 3), D67N (n = 4), and T64Q (n = 8), as shown by the open circles. Error bars indicate SEM. Colored bars represent residues highlighted in color on the crystal structure in panel c. (f) RVP variants encoding mutations resulting in a > 4-fold reduction in WNV-86 neutralization potency were tested for sensitivity to neutralization against murine mAb E16. Data points and error bars indicate the mean and range of duplicate infections, respectively. Data are representative of two experiments.
To identify additional E residues that contribute to WNV-86 recognition, a second neutralization escape study was performed in which a WNV T64Q infectious clone variant, which displayed an intermediate sensitivity to neutralization by WNV-86 (Supplementary Figs. 7b and d), was passaged in the presence of antibody. Two serial passages under WNV-86 selection pressure were sufficient to select a neutralization escape virus that grew to wild-type levels in the presence of neutralizing antibody (Fig. 3b and Supplementary Figs. 6c-d). Comparison of bulk viral RNA sequences isolated from WNV T64Q passaged in the presence or absence of mAb WNV-86 identified the same single nucleotide change that resulted in a second amino acid substitution at E DII residue 208 (T208K, Fig. 3c). As observed with WNV T64Q RVPs, when tested individually, WNV RVPs containing a T208K mutation reduced, but did not eliminate sensitivity to neutralization by WNV-86 (Fig. 3d). However, in combination, these two mutations abrogated WNV-86 neutralization, suggesting that mutation at both residues is required for neutralization escape from WNV-86.
To further define the epitope targeted by WNV-86, we screened 40 RVP variants encoding single, double, or triple mutations at a total of 56 solvent-accessible residues throughout E DI and DII, including DII residues 64 and 208 identified in our in vitro selection experiments. Of 56 total mutations tested, 29 displayed minimal effects on neutralization sensitivity (< 2-fold change in IC50), 19 modestly decreased neutralization sensitivity (2- to 4-fold increase in IC50), and 6 reduced sensitivity by > 4-fold (Fig. 3e). These 6 mutations are clustered in DII and, with the exception of D67N, are bounded by residues T64 and T208 (Figs 3c), suggesting that the binding footprint of WNV-86 lies within this region and may partially overlap with the binding site of prM of the E protein16. These mutations did not alter antigenicity non-specifically, as their introduction did not affect sensitivity to neutralization by mE16 (Fig. 3f).
To evaluate the therapeutic potential of human neutralizing mAbs, five-week old C57BL/6J mice were inoculated with WNV, treated with a single dose (100 μg; 5 mg/kg) of mAb WNV-86 or WNV-10 two days post-inoculation, and monitored for survival. These mAbs represented the two most potently neutralizing mAbs in our screen (Fig 1), target non-competing epitopes, and selected for different mutations during neutralization escape studies (Supplementary Fig. 8). As seen with mice treated with the therapeutic humanized murine mAb E16 (hE16)9, mice treated with WNV-86 were protected completely from mortality (Fig. 4a). In contrast, treatment with WNV-10 afforded partial protection, with 5 out of 10 mice succumbing to infection by day 19. Thus, the potent neutralizing activity of WNV-86 in vitro correlated with therapeutic efficacy in vivo. Although the in vitro neutralization potency of WNV-10 was similar to that of E16 (~10 ng/mL, Figs. 1e-f)12, only the latter afforded complete protection from lethal infection, suggesting that additional properties determine mAb therapeutic efficacy in vivo. Notably, administration of a LALA variant of WNV-86 that is incapable of engaging Fc receptor displayed significant albeit slightly reduced protection (Fig 4b). Moreover, both wild-type and LALA versions of WNV-86 significantly reduced viral burden in the spinal cord and brain of infected animals (Figs. 4c-d), suggesting that the therapeutic mechanism of WNV-86 is largely due to an ability to directly inhibit virus infection and dissemination.
Figure 4. Therapeutic efficacy of mAbs.
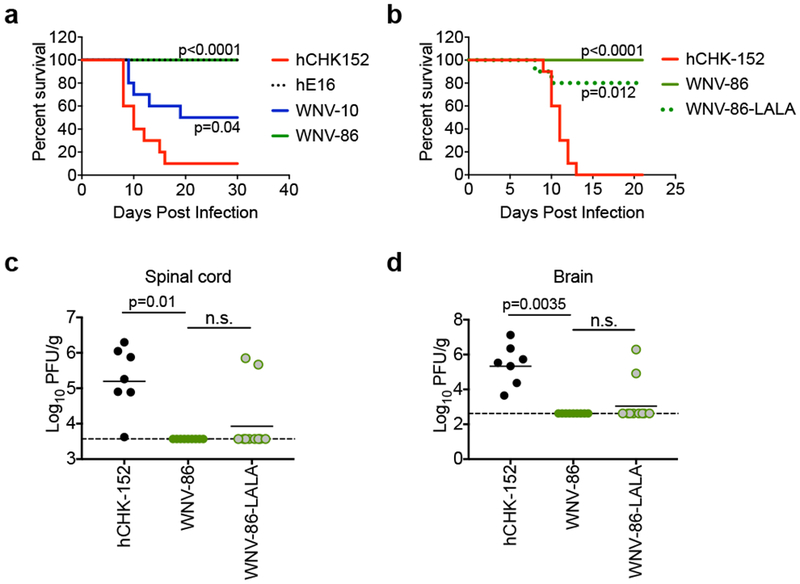
(a) Five-week old C57/BL6J mice were inoculated with 102 FFU of WNV, treated with 100 μg of mAb WNV-10 or WNV-86 two days following inoculation, and monitored for survival. Mice administered WNV-specific mAb hE16 or isotype-control mAb hCHK152 were used as a positive or negative control, respectively. Data were pooled from two experiments (n = 10 per group). The indicated p-value was obtained from a one-sided log-rank test. (b) To address the contribution of effector functions in protection in mice following the administration of WNV-86, a LALA variant of this antibody was administered to 5-week old C57BL/6 mice (100 μg per mouse, n = 10) two days following inoculation, and monitored for survival. The indicated p-values were obtained from a one-sided log-rank test. (c-d) At day 8 post-infection, spinal cords and brains were harvested after extensive perfusion, and WNV levels were titrated by plaque assay on Vero cells. Dashed line indicates limit of detection, and bars indicate the median. Data were pooled from two independent experiments for WNV-86 (n = 9), WNV-86 LALA (n = 10), and hCHK-152 (n = 7; 3 mice died in this group prior to tissue harvest), and analyzed using an ANOVA with Dunn’s post-test.
In summary, we have isolated a potently neutralizing WNV-specific human mAb with therapeutic potential. WNV-86 neutralized WNV with an IC50 that was approximately 3-fold lower than mAb E16, which has been shown previously to prevent mortality in WNV-infected mice9. Unlike E16 and most potently neutralizing mouse antibodies, WNV-86 did not recognize an epitope within DIII, consistent with the recognition pattern of recently characterized potently neutralizing human mAbs for other flaviviruses3. WNV-86 bound a recombinant soluble E protein, suggesting its epitope does not require a complex quaternary arrangement, which contrasts with other human neutralizing mAbs3. Despite its potency, competition and neutralization studies suggest that antibodies that bind this epitope are neither common nor contribute significantly to the neutralizing activity of sera (Supplementary Fig. 9).
WNV-86 neutralized mature virions lacking uncleaved prM better than those retaining prM due to incomplete maturation, in contrast to many WNV antibodies characterized to date. The increased sensitivity of prM-containing virions to neutralization by many antibodies may be explained by improved epitope accessibility on E proteins arranged as heterotrimeric spikes relative to that on the E homodimers of mature virions5. An exception to this model may apply to antibodies targeting quaternary epitopes that span multiple E proteins3; these antibodies should in theory preferentially neutralize mature virions over those that retain prM17. Despite our observation that WNV-86 recognition did not require a quaternary structure, structural studies are required to determine whether the accessibility of its epitope is improved on E proteins assembled as dimers on the mature virus particle, or whether residues on the opposing E protein within the dimer may contribute to WNV-86 interactions.
In vitro selection of neutralization escape virus variants and epitope mapping studies revealed a cluster of DII residues that likely comprise the WNV-86 epitope. Although the exact binding footprint of WNV-86 awaits structural studies, the DII epitope identified by our mapping studies appears distinct from that within the DI-DII hinge recognized by some strongly neutralizing human mAbs18–20. Although E protein residue 64, which strongly impacted WNV-86 recognition, is highly conserved among WNV strains21, we observed strain-dependent neutralization sensitivity, with the more distantly related African lineage II WNV strain WN 956 D117 3B displaying reduced sensitivity (Supplementary Figure 10). Notably, the sequence of this and other lineage II strains differs at both E protein positions 64 and 208 shown here to be important for WNV-86 recognition.
As seen with mAb E16, the in vitro neutralization potency of WNV-86 correlated with in vivo therapeutic efficacy. In contrast, despite its potent neutralizing activity in vitro, when administered as a single dose post-exposure, WNV-10 prevented mortality in only 50% of mice. Reduced in vivo efficacy of WNV-10 may reflect the existence of a measurable fraction of virions resistant to neutralization or an inability to neutralize at a post-attachment step (Supplementary Figs 1 and 4). Other factors may uniquely contribute to in vivo protection. For example, although administration of WNV-86 resulted in significant protection independently of Fc-mediated functions, the protective efficacy of E16 was diminished in mice lacking Fcγ receptors9. Understanding whether the cellular mechanism of antibody neutralization contributes to therapeutic efficacy awaits further studies.
METHODS
Human subjects
Blood samples were obtained in 2014 from adult subjects with history of symptomatic laboratory-confirmed WNV infection during the 2012 West Nile encephalitis outbreak in Dallas, TX. The studies were approved by the Institutional Review Board of Vanderbilt University Medical Center; samples were collected after written informed consent was obtained by the Vanderbilt Clinical Trials Center.
Generation of anti-E mAbs
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood by gradient centrifugation after layering on Ficoll Histopaque, and cryopreserved in the vapor phase of liquid nitrogen until use. B cells from donors 865, 866, and 870 were transformed by infection with Epstein Barr virus (EBV) obtained from the supernatant of cultured B95.8 cotton-top tamarin lymphoblastoid line (ATCC). The transformation medium contained 2.5 μg/mL TLR agonist CpG (phosphorothioate-modified oligodeoxynucleotide ZOEZOEZZZZZOEEZOEZZZT, Life Technologies), 10 μM Chk2 inhibitor [Chk2i] (Sigma), and 10 μg/mL cyclosporine A (Sigma). B cells were plated in 384-well culture plates and cultured for seven days and then expanded into four 96-well culture plates containing CpG, Chk2i and irradiated heterologous human PBMCs to serve as feeder layers for the growth of lymphoblastoid cell line (LCL) clusters. After an additional three days of culture, the supernatants were screened for binding to recombinant soluble WNV E protein by ELISA. Approximately 5 μL of supernatant from each well of transformed B cell cultures (in a total assay volume of 50 μL) were added to wells coated with 2 μg/mL of recombinant WNV E protein. The bound antibodies were detected using alkaline phosphatase conjugated goat anti-human Ig (γ-chain specific) (Southern Biotech). The frequency of WNV E protein-specific EBV-transformed B cells was similar for the three donors (0.9% for 865; 1.1% for 866; and 0.7% for 870).
Cells from wells positive for binding to WNV E were subjected to electrofusion with HMMA2.5 myeloma cells22. The fused cells then were cultured in a selective medium containing 100 μM hypoxanthine, 0.4 μM aminopterin, 16 μM thymidine (HAT Media Supplement, Sigma HO262), and 7 μg/mL ouabain (Sigma O3125) and incubated for 14 – 18 days before screening hybridomas for antibody production by ELISA using recombinant WNV E protein. Cells from positive wells were cloned by sorting live unlabelled single cells into 384-well plates using a FACSAria III fluorescence-activated cell sorter (Becton Dickinson), cultured for about 14 days and screened for specific antibody production by ELISA using WNV E protein. For expression of antibodies from hybridoma clones, cells were cultured in serum-free medium, Hybridoma SFM (Life Technologies), for 21 days. Antibodies were harvested from the supernatants by affinity chromatography on HiTrap MabSelect SuRe columns (Life Technologies) according to the manufacturer’s instructions. Antibodies eluted from affinity columns were concentrated using Amicon centrifugal filters (Millipore). Antibody heavy and light chain variable genes for one hybridoma clone (WNV-86) were amplified by RT-PCR from hybridoma cell RNA and then sequenced by Sanger sequencing. cDNAs encoding the WNV-86 antibody variable genes were synthesized and cloned into expression vectors for 1) Fab, 2) WT IgG1 or 3) IgG1 with a LALA variant Fc region, and recombinant antibodies expressed in 293F cells.
Cells
Raji B lymphoblast cells (ATCC) engineered to stably express DC-SIGNR (Raji-DCSIGNR)23 were cultured in RPMI 1640 medium containing Glutamax (Invitrogen) supplemented with 7% fetal bovine serum (FBS; Invitrogen) and 100 U/mL penicillin-streptomycin (P/S; Invitrogen). HEK-293T (ATCC), Vero (ATCC), and C6/36 (ATCC) cells were maintained in Dulbecco’s Modified Eagle medium (DMEM) containing 25 mM HEPES (Invitrogen) supplemented with 7% FBS and 100 U/mL P/S. Raji-DCSIGNR, HEK-293T, and Vero cells were maintained at 37°C in the presence of 7% CO2. C6/36 cells were maintained at 28°C in the presence of 7% CO2. Cell lines used in this study were not authenticated nor tested for mycoplasma contamination prior to use in these studies. Cell banks from which these cells were grown were shown to be mycoplasma-free using the Universal Mycoplasma Detection Kit (ATCC). DCSIGNR expression on stable Raji cells was confirmed using mouse anti-human DC-SIGNR/CD299 (R&D Systems MAB162 clone 120604; RBO1; lot: DZV0215051; 10 μg/ml).
Generation of plasmids encoding E protein variants
We used a previously described expression vector encoding the structural genes (C-prM-E) of the WNV NY99 strain15 as a template for site-directed mutagenesis using the Pfu Ultra DNA polymerase system (Agilent Technologies). PCR cycling parameters were: 1 cycle of 95°C for 1 m; 18 cycles of 95°C for 50 s, 60°C for 50 s, and 68°C for 9 m; and 1 cycle of 68°C for 7 m. Following digestion with DpnI (New England Biolabs) for 3 h at 37°C, PCR products were used to transform Stbl2 cells (Invitrogen) and propagated at 30°C. After confirming the presence of the desired mutation by sequencing, the entire C-prM-E region was sequenced to ensure that no additional mutations were present.
Production of RVPs
RVPs were produced by co-transfection of a plasmid expressing a WNV subgenomic replicon in which the structural genes have been replaced with GFP, and a plasmid encoding the structural genes, as described previously15,24,25. To prepare mature RVPs with increased efficiency of prM cleavage, RVPs were produced by co-transfecting plasmids encoding the replicon, structural genes, and human furin at a 1:3:1 ratio by mass. Immature RVPs with decreased prM cleavage efficiency were produced in cells treated with 20 mM NH4Cl, as described previously4.
Production of fully infectious WNV
A DNA fragment encoding WT WNV structural genes or WNV structural genes encoding a mutation at E residue 64 was ligated into a GFP-expressing WNV “backbone” replicon plasmid (pWNV-GFP-backbone V3), and transfected directly into HEK-293T cells to generate infectious virus particles, as described previously26. Cells were incubated at 37°C in the presence of 7% CO2. Viral supernatant was harvested at three days post-transfection, filtered using a 0.22 μm filter (Millipore), and stored at −80°C.
Determination of virus titer
Virus-containing supernatant was serially diluted 2-fold in a total volume of 100 μL and used to infect 5 × 104 Raji-DCSIGNR cells in an equal volume at 37°C. Cells were fixed in 1.8% paraformaldehyde at 48 h or 16 h following infection by RVPs or fully infectious virus, respectively, and GFP-positive cells were enumerated using flow cytometry (BD FACSCalibur). Infectious titer was calculated using the linear portion of the resulting dose-response infectivity curve using the following formula: Infectious units (IU) per virus volume = (% GFP-positive cells) × (dilution factor) × (number of cells).
Neutralization assays
RVP or fully infectious virus stocks were diluted to ~5-10% infectivity and incubated with serial dilutions of mAbs for 1 h at room temperature before the addition of Raji-DCSIGNR cells. All infections were performed in duplicate at 37°C. At 48 h (RVP) or 16 h (fully infectious virus) post-infection, infectivity was measured as a percentage of GFP-positive cells by flow cytometry (BD FACSCalibur). Antibody dose-response curves were analysed using non-linear regression with a variable slope (GraphPad Prism v 6.0g, GraphPad Software Inc.) to estimate the reciprocal serum dilution (NT50) or concentration of antibody (IC50) required to inhibit infection by 50%.
RVP binding assays
Standard preparation WNV RVPs were incubated with six-fold serial dilutions of mAbs for 1 h at 37°C to allow binding to reach equilibrium. Raji-DCSIGNR cells were added and incubated with the RVP-antibody complexes for 3.5 h at 37°C. As a negative control, Raji-DCSIGNR cells were incubated with anti-DCSIGNR antibody (detailed above) at 30°C for 30 m prior to addition. After incubation cells were washed to remove unbound RVPs. Half of the cells were moved to a new 96-well plate in fresh media and incubated at 37°C for 48 h. Infectivity was measured by flow cytometry as described previously. The remaining cells were lysed using the QIAshredder kit (Qiagen) and total RNA was isolated using the RNeasy Mini kit (Qiagen), in accordance with the manufacturer’s instructions. The relative amount of WNV RNA was determined by quantitative reverse-transcriptase PCR using known copy numbers of the WNV replicon plasmid to generate a standard curve. Additional experiments (not shown) were performed to ensure that at 3.5 h, genomic RNA replication in infected cells does not impact WNV RNA copy number. The data was analyzed using non-linear regression with a variable slope (GraphPad Prism v 6.0g, GraphPad Software Inc.).
Selection of neutralization escape variants
Fully infectious GFP-expressing WNV26 was incubated with mAb WNV-86 at a concentration that was approximately 200-fold greater than its IC50 (0.6 μg/mL for WT; 2 μg/mL for WNV T64Q) in a total volume of 2 mL for 30 m at 37°C, followed by duplicate infection of pre-plated Vero cells (8.5 × 105 /well) in a 6-well dish at a MOI of 0.1. After 3-4 days of infection at 37°C, continuous viral replication and antibody selection pressure was maintained by serial passaging of virus supernatant diluted 1:10 in medium with or without WNV-86 in a total volume of 2 mL and incubated for 30 m at 37°C prior to addition to fresh Vero cells. The remaining volume of virus supernatant was aliquoted and stored at −80°C until further use. Replication of antibody escape viral variants was monitored by visually inspecting cells for GFP expression and confirmed by inoculating Raji-DCSIGNR cells with an aliquot of the virus supernatant (serially diluted 2-fold) in the presence or absence of a neutralizing concentration (2 μg/mL) of WNV-86, and by neutralization assays as described above. Following confirmation of mAb escape, viral RNA was isolated from a 50 μL aliquot of viral supernatant (adjusted to 140 μL in RNase-free water) using the QIAamp viral RNA mini kit (Qiagen) according to the manufacturer’s instructions. cDNA encoding WNV structural genes was amplified using the SuperScript III One-Step RT-PCR system (Invitrogen). The structural genes were sequenced directly from the gel-purified PCR product and were compared to the structural gene sequence of WNV passaged in parallel in the absence of antibody to identify mAb-induced escape mutations.
MAb binding to recombinant E proteins
MAb binding to WNV was measured by ELISA, as described previously27. Briefly, recombinant WNV E ectodomain or DIII fragment was diluted to 5 μg/mL in 0.1 M sodium carbonate buffer (pH 9.3) and adsorbed on 96-well Nunc MaxiSorp microtitre plates (ThermoFisher Scientific) overnight at 4°C. After blocking with phosphate-buffered saline (PBS) containing 2% bovine serum albumin and 0.05% Tween 20 (PBS-BT) for 1 h at 37°C, three-fold serial dilutions of antibody in PBS-BT were incubated for 1 h at room temperature. Plates were washed with PBS plus 0.05% Tween 20 and incubated with biotin-conjugated goat anti-mouse IgG (1 μg/mL; Sigma-Aldrich SAB4600004) for 1 h at room temperature. After washing, all plates were incubated with streptavidin-horseradish peroxidase (2 μg/mL; Zymed) for 1 h at room temperature and developed with tetramethylbenzidine substrate (Dako). After the addition of 1 N H2SO4, the optical density at 450 nm was measured. Best-fit lines were fit using GraphPad Prism v 6.0g (GraphPad Software, Inc).
Determining E protein glycosylation status
RVP-containing supernatant (1.5 mL) from transfected cells in a 6-well plate was concentrated and partially purified by microcentrifugation (14,000 × g overnight at 4°C) through a 20% sucrose cushion (0.25 mL per 1.5 mL RVP). The resulting RVP pellet was re-suspended and lysed in TNE buffer (50 mM Tris, 140 mM NaCl, 5 mM EDTA, pH adjusted to 7.4) containing 1% Triton-X100 followed by digestion with PNGase F (New England Biolabs) for 3 h at 37°C. Undigested and PNGase F-digested E proteins in RVP lysates were detected by SDS-PAGE and Western blot analysis using mouse mAb 4G2 (1 μg/mL; Novus Biologicals NBP2-52666 clone D1-4G2-4-15). IRDye 800CW goat-anti mouse IgG (LI-COR Biosciences 925-32210) diluted 1:2,500 was used as a secondary antibody. Protein bands were visualized using the Odyssey infrared imaging system (LI-COR Biosciences).
Mouse experiments
Five-week old C57BL/6J male mice (Jackson Laboratories, 000644) were inoculated subcutaneously with 102 focus forming units (FFU) of WNV NY99 after anaesthetization with xylazine and ketamine. Two days post-infection, mice were given 100 μg of mAb (~5 mg/kg) via intraperitoneal injection and monitored for survival for 30 days. Sample size choice (as indicated in figure legends) was estimated based on power calculations. We did not perform randomization nor blinding. Mouse experiments were approved and performed according to the guidelines of the Washington University School of Medicine Institutional Animal Care and Use Committee (IACUC) (Assurance Number: A3381-01).
Statistical analysis
All data were analysed in GraphPad Prism v 6.0g (GraphPad Software Inc.) and expressed as mean or median values and their range (two data points) or standard error (SEM) (three or more data points). P-values were calculated using paired t-tests, ANOVA, or the log-rank test, with post-hoc corrections for multiple comparisons, as indicated in the figure legends.
Supplementary Material
ACKNOWLEDGEMENTS
This study was funded by NIH grants R01 AI073755 (to M.S.D. and J.E.C.) and HHSN272201400018C (to M.S.D.); and by the intramural research program of the Division of Intramural Research, National Institutes of Allergy and Infectious Diseases (T.C.P.). Flow cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The project was supported by CTSA award No. UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Footnotes
DATA AVAILABILITY
Additional datasets generated and/or analysed during the current study are available from the corresponding authors on reasonable request.
COMPETING INTERESTS
M.S.D. is a consultant for Inbios and Sanofi-Pasteur and is on the Scientific Advisory Board of Moderna. J.E.C. has served as a consultant for Takeda Vaccines, Sanofi Pasteur, Pfizer, and Novavax, is on the Scientific Advisory Boards of CompuVax, GigaGen, Meissa Vaccines, PaxVax, and is Founder of IDBiologics.
ETHICAL COMPLIANCE
We have complied with all relevant ethical regulations for research animals and human research participants.
REFERENCES
- 1.Reimann CA et al. Epidemiology of neuroinvasive arboviral disease in the United States, 1999-2007. Am J Trop Med Hyg 79, 974–979 (2008). [PubMed] [Google Scholar]
- 2.Rey FA, Stiasny K, Vaney MC, Dellarole M & Heinz FX The bright and the dark side of human antibody responses to flaviviruses: lessons for vaccine design. EMBO Rep 19, 206–224, doi: 10.15252/embr.201745302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.VanBlargan LA, Goo L & Pierson TC Deconstructing the Antiviral Neutralizing-Antibody Response: Implications for Vaccine Development and Immunity. Microbiol Mol Biol Rev 80, 989–1010, doi: 10.1128/MMBR.00024-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson S et al. Maturation of West Nile virus modulates sensitivity to antibody-mediated neutralization. PLoS Pathog 4, e1000060, doi: 10.1371/journal.ppat.1000060 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierson TC & Diamond MS Degrees of maturity: the complex structure and biology of flaviviruses. Curr Opin Virol 2, 168–175, doi: 10.1016/j.coviro.2012.02.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherrier MV et al. Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J 28, 3269–3276, doi: 10.1038/emboj.2009.245 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guirakhoo F, Bolin RA & Roehrig JT The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology 191, 921–931 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinz FX et al. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 198, 109–117, doi: 10.1006/viro.1994.1013 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Oliphant T et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med 11, 522–530, doi: 10.1038/nm1240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Throsby M et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J Virol 80, 6982–6992, doi: 10.1128/JVI.00551-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gould LH et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol 79, 14606–14613, doi: 10.1128/JVI.79.23.14606-14613.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pierson TC et al. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1, 135–145, doi: 10.1016/j.chom.2007.03.002 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nybakken GE et al. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 437, 764–769, doi: 10.1038/nature03956 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung WM et al. The 2012 West Nile encephalitis epidemic in Dallas, Texas. JAMA 310, 297–307, doi: 10.1001/jama.2013.8267 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Pierson TC et al. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346, 53–65, doi: 10.1016/j.virol.2005.10.030 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Li L et al. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319, 1830–1834, doi: 10.1126/science.1153263 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Dejnirattisai W et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat Immunol 16, 170–177, doi: 10.1038/ni.3058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufmann B et al. Neutralization of West Nile virus by cross-linking of its surface proteins with Fab fragments of the human monoclonal antibody CR4354. Proc Natl Acad Sci U S A 107, 18950–18955, doi: 10.1073/pnas.1011036107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teoh EP et al. The structural basis for serotype-specific neutralization of dengue virus by a human antibody. Sci Transl Med 4, 139ra183, doi: 10.1126/scitranslmed.3003888 (2012). [DOI] [PubMed] [Google Scholar]
- 20.de Alwis R et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc Natl Acad Sci U S A 109, 7439–7444, doi: 10.1073/pnas.1200566109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatcher EL et al. Virus Variation Resource - improved response to emergent viral outbreaks. Nucleic Acids Res 45, D482–D490, doi: 10.1093/nar/gkw1065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu X, McGraw PA, House FS & Crowe JE Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods 336, 142–151, doi: 10.1016/j.jim.2008.04.008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis CW et al. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol 80, 1290–1301, doi: 10.1128/JVI.80.3.1290-1301.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS & Pierson TC Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 381, 67–74, doi: 10.1016/j.virol.2008.08.021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowd KA et al. Broadly Neutralizing Activity of Zika Virus-Immune Sera Identifies a Single Viral Serotype. Cell Rep 16, 1485–1491, doi: 10.1016/j.celrep.2016.07.049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin TY et al. A novel approach for the rapid mutagenesis and directed evolution of the structural genes of west nile virus. J Virol 86, 3501–3512, doi: 10.1128/JVI.06435-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliphant T et al. Induction of epitope-specific neutralizing antibodies against West Nile virus. J Virol 81, 11828–11839, doi: 10.1128/JVI.00643-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.