

Abstract
INTRODUCTION:
We sought to examine the genetic overlap between vascular pathologies and Alzheimer’s disease (AD) dementia, and the potential mediating role of vascular pathologies between AD-related genetic variants and late-life cognition.
METHODS:
For 2,907 stroke-free older individuals, we examined the association of polygenic risk scores for AD dementia (ADPRS) with vascular pathologies and with cognition. Mediation analyses addressed whether association between ADPRS and cognition was mediated by a vascular pathology.
RESULTS:
ADPRS was associated with lobar cerebral microbleeds (CMB), white matter lesion load (WML) and coronary artery calcification (CAC), mostly explained by SNPs in the 19q13 region. The effect of ADPRS on cognition was partially but significantly mediated by CMB, WML, and CAC.
DISCUSSION:
Our findings provide evidence for genetic overlap, mostly due to APOE, between vascular pathologies and AD dementia. The association between AD polygenic risk and late-life cognition is mediated in part via effects on vascular pathologies.
Keywords: Alzheimer’s dementia, Polygenic risk score, Cerebral microbleeds, White matter lesions, Coronary calcification, Cognitive impairment, Causal mediation
1. BACKGROUND
The etiology of Alzheimer’s disease (AD) dementia is complex and multifactorial. AD dementia refers to the clinical diagnosis of dementia considered likely to be due to underlying AD pathology, the accumulation of amyloid plaques and neurofibrillary tangles, which may lead to neurodegeneration and neuronal cell death. However, it is well-established that a large fraction of those with a diagnosis of AD dementia also have cerebrovascular pathology [1]. Systematically collected cohort-based autopsy data have shown that vascular pathology often coexists with AD pathology, adds to the likelihood of cognitive impairment, and lowers the threshold of AD pathology for the development of clinically diagnosed AD dementia [2].
A variety of cerebral small vessel diseases (SVD) have been associated with AD dementia. Cerebral microbleeds (CMB) are more prevalent in individuals with dementia [3–5]. The presence of CMB in deep and infratentorial regions is generally ascribed to hypertensive vasculopathy, while a lobar distribution of CMB is associated with cerebral amyloid angiopathy (CAA) [6], which has been considered a major contributor to the pathogenesis of AD dementia [7]. White matter lesions (WML), an imaging marker of cerebral SVD, may also play a role in the development of AD dementia [8, 9]. A metaanalysis found that WML predicted an increased risk of AD and other dementia [10]. Retinal venular diameter (RVD), an indicator to visualize microcirculation in vivo, has been related to WML, brain atrophy, and increased risk of dementia [11–13].
Research efforts have also been devoted to the association between large vessel disease (LVD) and AD dementia. Possible mechanisms linking large-vessel atherosclerosis to AD dementia include shared etiology and brain hypoperfusion [14]. Several longitudinal studies suggest that carotid intima-media thickness (CIMT), a marker of atherosclerosis, is associated with brain atrophy [15] and a later incidence of AD dementia [16, 17]. Atherosclerotic coronary artery calcification (CAC) is another marker of LVD. Although there have been few reports on the relation between CAC and AD , current evidence suggests that larger volume of CAC is associated with brain atrophy, worse cognitive function, and all-cause dementia [18–20].
Genetic studies may provide clues to the biological link of AD dementia with cerebro- and cardio-vascular disease (collectively “CVD”). Apolipoprotein E (APOE), the major susceptibility gene for AD [21], has been reported to be a risk factor for hyperlipidemia, lobar CMB, WML, ischemic stroke, and coronary heart disease [22–25]. In addition to APOE, genome-wide association studies (GWAS) of AD dementia have identified single nucleotide polymorphisms (SNPs) with known or hypothesized relationships to lipid metabolism, such as CLU, ABCA7, and SORL1 [26]. Recent studies using large-scale GWAS data suggest that AD dementia may be genetically correlated with levels of biomarkers for CVD risk (plasma lipids and C-reactive protein) [27] and small vessel stroke [28]. A gene-based pathway approach to GWAS data has also identified shared genetic pathways between CVD and AD dementia [29].
A recent study found that the effect of APOE-e4 on late-life cognition was partially mediated by cerebrovascular pathologies [30]. In the present study, we expand to additional vascular pathologies beyond the brain and full genome data to more fully understand the relationship of AD genes and vascular pathology in the development of cognitive impairment. We generated genome-wide polygenic risk scores for AD dementia (GW-ADPRS) to examine the polygenic overlap between AD dementia and each of the following vascular pathologies: lobar CMB, WML, RVD, CIMT, and CAC. We also generated two partitioned ADPRS, estimating genetic risk for AD dementia contributed separately by the 19q13 region that includes APOE and SNPs in linkage disequilibrium (LD) with APOE, and all other SNPs outside of the APOE-linkage region. We tested each ADPRS separately for association with cognition scores and with each vascular pathology. For vascular markers observed to be genetically correlated with AD dementia, we performed mediation analyses to explore the causal relationship among ADPRS, vascular pathology, and cognitive function.
2. METHODS
2.1. Study Sample
The analyses were performed in data from the Age, Gene/Environment Susceptibility— Reykjavik Study (AGES-Reykjavik), a population-based cohort in Iceland [31] (see Supplementary Methods). For our phenotypic analyses, from the full AGES-Reykjavik sample of 5,764 participants, we excluded those with a history of stroke or vascular dementia, leaving 5,161. Of these participants, the 2,907 with clean genotype data available constituted the sample for our genetic analyses (see Supplementary Figure S1). Genotyping was performed using the Illumina HumanCNV370-Duo (Illumina Inc.; San Diego, CA, USA). Rigorous quality control procedures were performed on the genotyped markers and individuals. Non-genotyped markers were imputed using the 1000 Genomes-V3-phase I reference panel (see Supplementary Methods).
2.2. Vascular Pathology Markers
Markers of vascular pathologies were measured with standard protocols and assessed by well-trained raters (see Supplementary Methods). We systematically examined cerebrovascular and cardiovascular markers in our analyses, including lobar CMB (binary; multiple [>=2] versus non-multiple [0 or 1]), WML load (binary; the highest quartile versus the lower three), RVD (continuous), CIMT (continuous; log-transformed), and CAC (continuous; log-transformed).
2.3. Measures of Cognitive Function
Participants received a comprehensive cognitive assessment battery including tests of memory, executive function, and processing speed. Based on the scores of domain-specific cognitive tests, we calculated the Z-score of the composite memory score and the Z-score of the composite global cognition score, as the main cognitive outcomes for our analyses (see Supplementary Methods and Supplementary Figure S2).
2.4. Other Covariates
Other covariates used in the analyses included age, sex, education, smoking status, midlife physical activity, diet quality, prevalent diabetes, hypertension, high LDL level, and obesity (see Supplementary Methods).
2.5. Polygenic Risk Scores for AD Dementia (ADPRS)
2.5.1. Genome-wide ADPRS (GW-ADPRS)
We used the summary statistics from the Alzheimer’s Disease Genetics Consortium (ADGC) GWAS (8,309 AD cases and 7,366 controls of European ancestry) [32] as the discovery dataset to calculate GW-ADPRS in our study sample. We applied an LD clumping procedure to the discovery datasets, retaining the SNP with smallest P-value in each 250kb window and removed all those in LD (r2> 0.2) with this SNP. We used three association P-value thresholds (PTs), 0.0001, 0.001, and 0.01, to select index SNPs from the clumped independent SNPs for generating the PRSs. For each individual, and each PT, we calculated GW-PRS by summing the risk allele counts of the index SNPs, weighted by the log of their association odds ratios estimated from the ADGC GWAS results.
2.5.2. 19q13-ADPRS and non-19q13-ADPRS
Because APOE is the strongest risk gene for AD dementia, we further partitioned the GW-ADPRS into an APOE region score and a non-APOE region score to separately assess the polygenic effects of SNPs in the APOE-linkage region 19q13 (ch19:4500000–4580000) and all other SNPs. We followed the same steps as for the calculation of the GW-ADPRS to generate a 19q13-ADPRS (the summation of log-odds-ratio weighted risk allele counts of the index SNPs in the 19q13 region) and a non-19q13-ADPRS (the summation of log-odds-ratio weighted risk allele counts of the index SNPs across whole genome except 19q13) for each individual.
2.6. Data Analysis
2.6.1. Phenotypic associations of vascular markers with cognition
We used univariate and multivariate linear regressions to assess the associations of each vascular marker with the cognitive outcomes. Multivariate models adjusted for age, sex, education, diabetes, hypertension, high LDL level, obesity, physical activity, diet quality, and smoking status.
2.6.2. Association of ADPRS with vascular and cognitive phenotypes
We examined if any of the PTs generates an ADPRS significantly associated with each of the cognitive outcomes and vascular pathologies. We used linear (for continuous phenotypes) or logistic (for binary phenotypes) regression models to test the association of each phenotype with each of GW-ADPRS, 19q13-ADPRS, and non-19q13-ADPRS, adjusting for age and sex. The Wald test P-value for each association test was reported, and squared semi-partial correlations (R2) were calculated to estimate the proportion of variance explained by the PRSs. We used Bonferroni correction to adjust for multiple testing (see Supplementary Methods).
2.6.3. Causal Mediation Analyses
We performed mediation regression analyses [33], based on the counterfactual framework for causal inference [34], to examine how much of the effect of an ADPRS on cognition score was mediated by a vascular pathology observed to be genetically correlated with AD dementia.
For each ADPRS (GW-ADPRS, 19q13-ADPRS, or non-19q13-ADPRS) as the predictor, we estimated the direct and indirect (mediated) effects of each vascular pathology as the mediator, and Z-score of the composite memory or global cognition score as the outcome. In order to gain more statistical power, the ADPRS predictors used in the mediation analyses were those with the PT that showed the highest association with each cognitive outcome. We adjusted for potential mediator-outcome confounders, including age, sex, smoking status, midlife physical activity, diet quality, and other genetic risk scores if necessary. A counterfactual outcome variable denotes the outcome that would have been observed had a predictor been set to a particular value. In order to compare high and low values of each ADPRS in our estimates of the direct and the indirect effects, we chose to compare the 75th percentile and the 25th percentile of each.
Finally, we conducted sensitivity analyses of unmeasured confounding and the choice of 75th versus 25th percentile comparison (see Supplementary Methods).
All the mediation analyses were performed by using the PARAMED module in STATA [35]. We used bootstrap procedures with 200 replications to compute a 95% bias-corrected bootstrap confidence interval (95% BCCI) for the direct and indirect effects.
3. RESULTS
3.1. Sample Characteristics
Table 1 presents descriptive statistics for the AGES sample used here. The mean age of all subjects without stroke or vascular dementia (n=5,161) was 76.7 (5.8) years. Vascular pathologies were relatively rare: for example, only 2% had multiple lobar CMB. Subjects with genotype data available (n=2,907) were similar to the full sample but had somewhat lower coronary calcification score (with vs. without genotypes; Mann Whitney U test, P=0.01).
Table 1.
Descriptive statistics of demographic and clinical characteristics
| Characteristics | All subjects (N=5161) |
Subjects with genotype data (N=2907) |
|---|---|---|
| Demoaraphic | ||
| Age at AGES I (years), mean (SD) | 76.73 (5.83) | 76.20 (5.43) |
| Sex | ||
| Female, N (%) | 3022 (58.6) | 1706 (58.7) |
| Education | ||
| Secondary, N (%) | 2406 (49.9) | 1441 (49.7) |
| College, N (%) | 755 (15.7) | 451 (15.6) |
| University, N (%) | 547 (11.4) | 334 (11.5) |
|
Vascular Patholoaies, Baseline Lobar cerebral microbleeds |
||
| Count>=2, N(%) | 110 (2.1) | 69 (2.7) |
| White matter lesion load, median(Q1, Q3) | 1.91 (0.51, 5.64) | 1.92 (0.50, 5.59) |
| Central retinal venular equivalent, mean(SD) | 202.19 (19.56) | 202.14 (19.50) |
| Carotid intima-media thickness, median(Q1,Q3) | 0.97 (0.88, 1.06) | 0.97 (0.88, 1.06) |
| Coronary calcification score, median(Q1, Q3) | 271.23 (43.61, 898.78) | 253.52 (38.94, 841.53) |
|
Other Covariates, Baseline Midlife physical activity |
||
| Intermediate, N(%) | 2166 (46.6) | 1327 (47.5) |
| Poor, N(%) | 909 (19.5) | 524 (18.8) |
| Diet quality | ||
| Intermediate, N(%) | 4011 (84.6) | 2418 (84.8) |
| Poor, N(%) | 354 (7.5) | 205 (7.2) |
| Smoking | ||
| Ever, N(%) | 2111 (43.9) | 1303 (44.8) |
| Current, N(%) | 593 (12.3) | 372 (12.8) |
| Diabetes, N(%) | 640 (12.4) | 324 (11.2) |
| Hypertension | ||
| Prehypertension, N(%) | 758 (14.8) | 445 (15.3) |
| Hypertension, N(%) | 4112 (80.3) | 2318 (79.8) |
| LDL level >=130 mg/dL, N(%) | 2830 (54.9) | 1643 (56.6) |
| BMI>=30, N(%) | 1139 (22.3) | 642 (22.1) |
Subjects with GWAS genotype data available (n=2,907) had lower coronary calcification score than those without genotype data (n=2,254) (Mann-Whitney U test, P=0.01). No significant difference was observed in the distribution of any other characteristic listed in the Table between subjects with and without genotype data available.
3.2. Phenotypic Associations
Table 2 presents phenotypic associations between each vascular pathology and cognitive outcomes. All unadjusted associations were significant. After adjusting for potential confounders, CMB, CAC, and WML were significantly associated with memory score, whereas the former two were significantly associated with global cognition score.
Table 2.
Phenotypic associations between vascular pathologies and late-life cognitive function
| Vascular pathologies | Memory | Global Cognition | ||||||
|---|---|---|---|---|---|---|---|---|
| Unadjusted | Adjusted | Unadjusted | Adjusted | |||||
| Beta (S.E.) | P | Beta (S.E.) | P | Beta (S.E.) | P | Beta (S.E.) | P | |
| Lobar CMB | −0.368 (0.097) | 1.5E-04* | −0.187 (0.086) | 0.03* | −0.443 (0.100) | 8.9E-06* | −0.268 (0.083) | 0.001* |
| WML | −0.193 (0.036) | 1.1E-07* | −0.070 (0.033) | 0.03* | −0.244 (0.037) | 6.0E-11* | −0.060 (0.032) | 0.06 |
| RVD | 0.002 (0.001) | 0.01* | −0.001 (0.001) | 0.53 | 0.003 (0.001) | 0.001* | 0.0004 (0.0007) | 0.56 |
| CIMT | −1.110 (0.105) | 6.3E-26* | −0.107 (0.102) | 0.29 | −1.004 (0.109) | 4.5E-20* | 0.073 (0.099) | 0.46 |
| CAC | −0.086 (0.006) | 1.5E-46* | −0.015 (0.006) | 0.01* | −0.087 (0.006) | 2.4E-45* | −0.023 (0.006) | 7.8E-05* |
Lobar CMB: Multiple lobar cerebral microbleeds; count >=2 vs. 0 or 1.
WML: White matter lesion load; highest quartile vs. other three quartiles of the total volume of white matter lesions. RVD: Retinal venular diameter; represented by central retinal venular equivalent, continuous.
CIMT: Carotid intima-media thickness; log-transformed, continuous.
CAC: Coronary artery calcification score; log-transformed, continuous.
All multivariate models adjusted for age, sex, education, diabetes, hypertension, high LDL level, obesity, physical activity, diet quality, and smoking status.
Asterisk indicates significance at P<0.05.
If all potential confounders remain constant, those with multiple lobar CMB had, on average, 0.19 SD lower memory score and 0.26 SD lower global cognition score than those with 0 or 1 lobar CMB. Individuals in the highest quartile of WML load had 0.07 SD lower memory score than others. For each one-unit increase in log-transformed CAC, the mean memory and global cognition scores declined by 0.015 SD and 0.023 SD, respectively.
3.3. Associations of GW-ADPRS with Cognitive or Vascular Phenotypes
Results are shown in Figure 1 and Table 3. Lower memory score was significantly associated with higher GW-ADPRS with a P-value threshold of 0.0001 (GW-ADPRSPT = 0.0001; P = 0.006, R2 = 0.22%) and higher GW-ADPRS with a P-value threshold of 0.01 (GW-ADPRSPT = 0.01; P = 0.001, R2 = 0.29%), after the Bonferroni correction. We also found nominal associations of lower global cognition score with higher GW-ADPRSPT = 0.0001 and higher GW-ADPRSPT = 001. In terms of the association between the GW-ADPRSs and vascular pathologies, we found that higher GW-ADPRS at all three PT were nominally associated with multiple lobar CMB. There were also nominal associations between higher GW-ADPRSPT = 0.01 and greater WML and between higher GW-ADPRSPT = 0.0001 and higher CAC.
Figure 1. Pair-wise polygenic association analyses between GW-ADPRS and (A) cognition scores, (B) vascular pathologies.
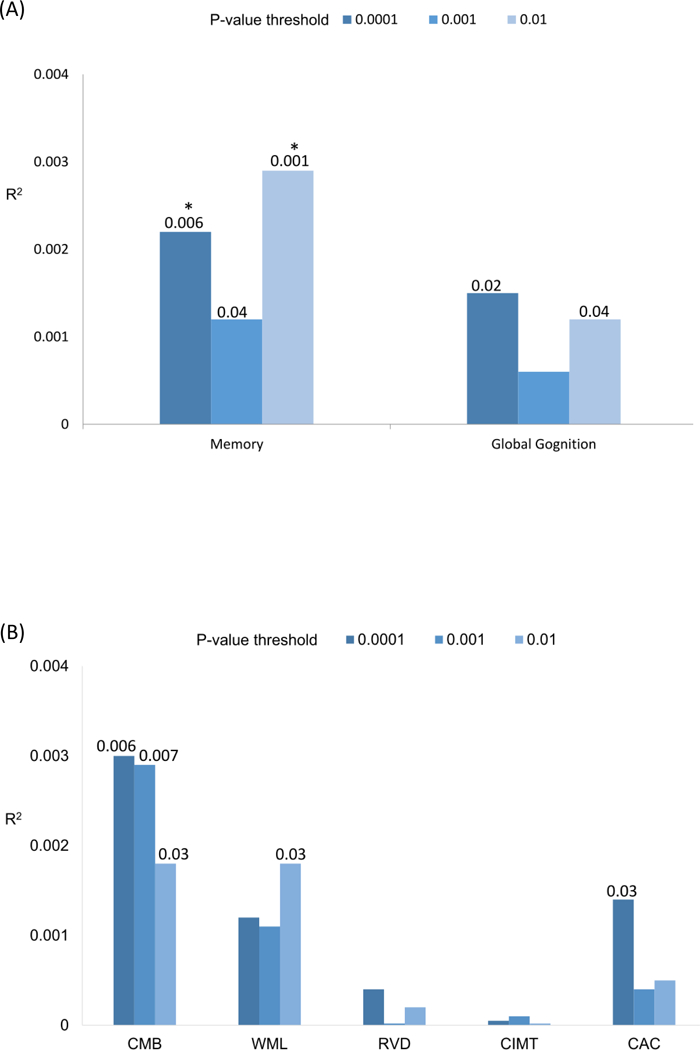
We derived genome-wide PRS for AD dementia using ADGC GWAS as the discovery sample with three different P-value thresholds (PT used to select training set SNPs: 0.0001, 0.001, and 0.01) and apply them to (A) Z-score of the composite memory or global cognition score; and (B) each of the markers of vascular pathologies. Age and sex were included as covariates in the association analyses.
Each pair is shown on the x-axis and the proportion of variance explained for each phenotype (estimated via partial correlation R2) on the y-axis.
Unadjusted P-values are shown on the top of the bars if < 0.05.
An asterisk indicates Bonferroni-corrected P-value < 0.05.
Table 3.
Associations between polygenic risk scores for AD dementia and each target phenotype
| PRS for AD dementia | Cognitive Outcomes | Vascular Marker Mediators | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genomic region | P-threshold | nSNP | Memory (n=2752) |
Global Cognition (n=2582) |
Lobar CMB (n=2562) |
WML (n=2559) |
RVD (n=2682) |
CIMT (n=2777) |
CAC (n=2869) |
| GW-ADPRS | PT < 0.0001 | 190 | R2=0.0022 P=0.006† |
R2=0.0015 P=0.02 |
R2=0.0030 P=0.006* |
R2=0.0012 P=0.07 |
R2=0.0004 P=0.29 |
R2<0.0001 P=0.71 |
R2=0.0014 P=0.03* |
| PT < 0.001 | 1342 | R2=0.0012 P=0.04* |
R2=0.0006 P=0.14 |
R2=0.0029 P=0.007* |
R2=0.0011 P=0.09 |
R2<0.0001 P=0.90 |
R2=0.0001 P=0.61 |
R2=0.0004 P=0.24 |
|
| PT < 0.01 | 8918 | R2=0.0029 P=0.001† |
R2=0.0012 P=0.04* |
R2=0.0018 P=0.03* |
R2=0.0018 P=0.03* |
R2=0.0002 P=0.49 |
R2<0.0001 P=0.99 |
R2=0.0005 P=0.19 |
|
| 19q13-ADPRS | PT < 0.0001 | 40 | R2=0.0015 P=0.02* |
R2=0.0013 P=0.04* |
R2=0.0038 P=0.002† |
R2=0.0014 P=0.05 |
R2=0.0003 P=0.40 |
R2=0.0002 P=0.46 |
R2=0.0012 P=0.04* |
| PT < 0.001 | 54 | R2=0.0015 P=0.02* |
R2=0.0013 P=0.04* |
R2=0.0035 P=0.003† |
R2=0.0018 P=0.03* |
R2=0.0002 P=0.48 |
R2=0.0002 P=0.48 |
R2=0.0015 P=0.02* |
|
| PT < 0.01 | 76 | R2=0.0012 P=0.04* |
R2=0.0011 P=0.06 |
R2=0.0040 P=0.002† |
R2=0.0019 P=0.02* |
R2=0.0002 P=0.46 |
R2=0.0003 P=0.30 |
R2=0.0015 P=0.02* |
|
| Non-19q13-ADPRS | PT < 0.0001 | 150 | R2=0.0008 P=0.10 |
R2=0.0002 P=0.39 |
R2<0.0001 P=0.94 |
R2<0.0001 P=0.82 |
R2=0.0002 P=0.50 |
R2=0.0001 P=0.50 |
R2=0.0001 P=0.49 |
| PT < 0.001 | 1288 | R2=0.0002 P=0.45 |
R2<0.0001 P=0.81 |
R2=0.0004 P=0.30 |
R2=0.0001 P=0.68 |
R2=0.0001 P=0.67 |
R2<0.0001 P=0.94 |
R2<0.0001 P=0.71 |
|
| PT < 0.01 | 8842 | R2=0.0020 P=0.008† |
R2=0.0006 P=0.15 |
R2=0.0005 P=0.27 |
R2=0.0008 P=0.14 |
R2=0.0004 P=0.31 |
R2<0.0001 P=0.70 |
R2=0.0001 P=0.59 |
|
P-threshold (PT): the P-value threshold used in the training dataset to select SNPs for calculating the PRS for AD dementia. nSNP: different number of independent SNPs included for calculating the PRS for AD dementia, which is determined by the selection of PT.
Lobar CMB: lobar cerebral microbleeds (count >=2 vs. 0 or 1).
WML: total brain white matter lesion load (highest quartile vs. other three quartiles of the total volume of white matter lesions).
RVD: the average retinal venular diameter, represented by central retinal venular equivalent (continuous).
CIMT: mean of carotid intima-media thickness (log-transformed, continuous).
CAC: coronary artery calcification score (log-transformed, continuous).
R2: squared semi-partial correlation, the proportion of variance in the target phenotype explained by the PRS for AD dementia.
P: the P-value of the test for association between the PRS and the target phenotype, before multiple testing correction.
Associations were tested using linear (for continuous phenotype) or logistic (for binary phenotype) regression models with age and sex as covariates.
The missing data status of each vascular marker was associated neither with memory / global cognition scores nor with any of the PRS (all P > 0.20).
An asterisk indicates significance at P-value <0.05.
A dagger indicates significance after Bonferroni-correction. We considered a PRS-wise significant threshold for the correction of multiple comparisons (P< 0.008, after Bonferroni correction for the 6 association tests between 2 cognitive outcomes and 3 ADPRS for each genomic region; and P<0.003, Bonferroni correction for the 15 association tests between 5 vascular pathologies and 3 ADPRS).
3.4. Associations of 19q13-ADPRS and non-19q13-ADPRS with Cognitive or Vascular Phenotypes
Our data showed that higher 19q13-ADPRS at all three PTs were significantly associated with having lobar CMB. We also found nominal associations of higher 19q13-ADPRS with greater WML, higher CAC, and poorer performance on both cognitive outcomes (Table 3). For non-19q13-ADPRS, the only association was that between higher non-19q13-ADPRSPT = 0.01 and lower memory score (Table 3).
3.5. Mediation Analyses
The PRS that most associated with each cognitive outcome was selected as the predictor (GW-ADPRSPT = 0.01, 19q13-ADPRSPT = 0.001, and non-19q13-ADPRSPT = 0.01 for memory; GW-ADPRSPT = 0.0001, 19q13-ADPRSPT = 0.001, non-19q13-ADPRSPT = 0.01 for global cognition). Vascular pathologies with a p-value lower than 0.05 for PRS associations with AD dementia were tested as potential mediators (CMB, WML, and CAC).
Results are shown in Table 4. The proportion mediated (PM) was obtained by dividing the estimated indirect effect by the estimated total effect, as an index of the degree of mediation. The total effect of GW-ADPRS on memory score was significantly mediated by multiple lobar CMB and WML load. CMB, WML, and CAC were all identified as significant mediators of the effects of GW-ADPRS on global cognition score. The total effect of 19q13-ADPRS on memory score was significantly mediated by CMB and WML, and its effect on global cognition was mediated by CMB, WML, and CAC. The total effect of non-19q13-ADPRS on both memory and global cognition was mediated by CMB.
Table 4.
Total, direct and indirect effects of PRS for AD dementia on late-life cognitive function mediated by vascular pathologies
| Memory | Global Cognition | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Predictor | Mediator | N | Total effect (95% BCCI) |
Direct effect (95% BCCI) |
Indirect effect (95% BCCI) |
PM | Total effect (95% BCCI) |
Direct effect (95% BCCI) |
Indirect effect (95% BCCI) |
PM |
| GW-ADPRS | CMB | 2308 | −.074 (−.117, −.025) | −.072 (−.115, −.023) | −.002 (−.006, −.001) | 3.2% * | −.038 (−.081, .008) | −.034 (−.076, .013) | −.004 (−.009, −.002) | 10.8% * |
| WML | 2307 | −.078 (−.124, −.033) | −.076 (−.123, −.032) | −.002 (−.007, −.00004) | 2.7% * | −.041 (−.087, .011) | −.039 (−.085, .013) | −.002 (−.006, −.0003) | 4.0% * | |
| CAC | 2577 | −.069 (−.112, −.023) | −.067 (−.110, −.022) | −.001 (−.006, .0001) | 1.6% | −.048 (−.093, −.003) | −.046 (−.092, −.001) | −.002 (−.006, −.0001) | 4.4% * | |
| 19q13-ADPRS | CMB | 2308 | −.038 (−.087, .013) | −.035 (−.085, .017) | −.003 (−.008, −.001) | 7.4% * | −.036 (−.090, .009) | −.032 (−.079, .015) | −.004 (−.013, −.001) | 11.3% * |
| WML | 2307 | −.043 (−.091, .007) | −.041 (−.088, .010) | −.002 (−.008, −.0004) | 5.6% * | −.040 (−.091, .008) | −.038 (−.088, .012) | −.002 (−.007, −.0002) | 4.8% * | |
| CAC | 2577 | −.048 (−.096, −.001) | −.046 (−.093, .002) | −.002 (−.005, .0004) | 3.6% | −.048 (−.100, −.003) | −.045 (−.096, −.0004) | −.002 (−.006, −.0001) | 5.0% * | |
| Non-19q13ADPRS | CMB | 2308 | −.068 (−.118, −.022) | −.067 (−.116, −.020) | −.001 (−.005, −.0001) | 2.1% * | −.028 (−.075, .016) | −.026 (−.074, .019) | −.002 (−.006, −.0001) | 7.5% * |
| WML | 2307 | −.070 (−.115, −.022) | −.069 (−.114, −.021) | −.001 (−.005, .0003) | 2.0% | −.029 (−.075, .017) | −.028 (−.073, .018) | −.001 (−.005, .0003) | 3.7% | |
| CAC | 2577 | −.058 (−.102, −.013) | −.057 (−.101, −.012) | −.001 (−.004, .001) | 0.9% | −.031 (−.078, .015) | −.031 (−.077, .015) | −.001 (−.004, .001) | 2.2% | |
Predictor: GW-ADPRS, 19q13-ADPRS, and non-19q13-ADPRS (the 75th percentile vs. the 25th percentile).
Mediator: CMB (lobar cerebral microbleeds; >=2 vs. 0 or 1), WML (total brain white matter lesion load; highest quartile vs. other three quartiles of the total volume of white matter lesions), or CAC (coronary artery calcification score; log-transformed, continuous).
Outcome: z-score of the memory composite score (left panel) or z-score of the global cognition composite score (right panel).
Values for total, direct and indirect effects indicate changes in each outcome.
BCCI: bias-corrected confidence interval.
PM: proportion mediated=indirect effect beta coefficient/ total effect beta coefficient. An asterisk indicates that the PM is significantly greater than 0. Models for the effects of GW-PRS adjusted for age, sex, smoking status, midlife physical activity, and diet quality.
Models for the effects of 19q13-ADPRS adjusted for age, sex, smoking status, midlife physical activity, diet quality, and non-19q13-ADPRS.
Models for the effects of non-19q13-ADPRS adjusted for age, sex, smoking status, midlife physical activity, diet quality, and 19q13-ADPRS.
An asterisk indicates that the estimated indirect effect is significantly different from zero at the 5% level.
When an interaction between the PRS and the vascular mediator was included in each mediation model, there was very little change in the estimated direct and indirect effects, so we decided not to include the interaction in the mediation models, as suggested by Vanderweele [33].
3.6. Sensitivity Analyses
Sensitivity analyses of unmeasured confounding suggest that under the seemingly more likely scenarios of unmeasured confounders associated with better cognition and less severe vascular pathology, or unmeasured confounders associated with poorer cognition and more severe vascular pathology, our estimated PMs would underestimate the true mediation effects of vascular pathologies (see Figure 2, Supplementary Results, Table S1, and Figure S3). Sensitivity analyses for selection of predictor levels for comparison found that mediation analyses comparing the effects of the 90th percentile and the 10th percentile of each ADPRS yielded very similar PMs as those shown in Table 4 (see Supplementary Table S2).
Figure 2. Sensitivity analyses of unmeasured confounding for the relationship between 19q13-ADPRS and memory score mediated by (A) CMB, (B) WML, or (C) CAC.
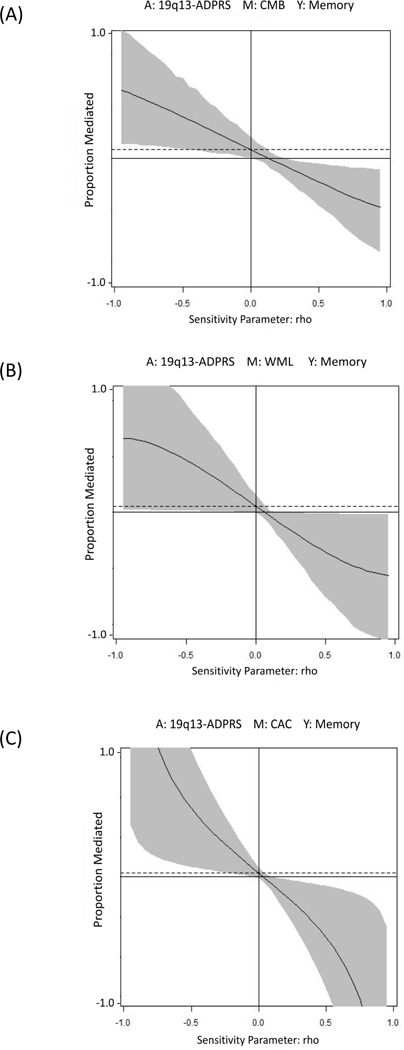
Figure 2 presents how the PM would change if unmeasured confounding of a specified direction and magnitude is allowed for in the mediation relationship between 19q13-ADPRS, vascular markers, and memory score. Detailed results of sensitivity analyses of unmeasured confounding for all other mediation analyses are described in Supplementary Results, Table S1, and Figure S3.
A: predictor; 19q13-ADPRS
M: mediator; (A) CMB, (B) WML, or (C) CAC
Y: outcome; memory score
The y-axis is the proportion mediated The x-axis, denoted by rho, is the degree of hypothetical unmeasured confounding, estimated by the size of the correlation between the residuals in the equation predicting M and the equation predicting Y. The larger the absolute value of rho, the stronger the confounding.
The solid curve shows the estimated proportion mediated for different values of the correlation between the residuals in equations. The shaded part of the plot represents the 95% intervals surrounding the mediated effect. The x-intercept represents the value of rho at which proportion mediated equals to 0.
The horizontal broken line denotes the proportion mediated without considering unmeasured confounding. When rho is equal to zero, the reported proportion mediated is the same as that we estimated in the mediation analysis without considering unmeasured confounding. For other values of rho, the proportion mediated is calculated under different levels of unobserved confounding. When rho<0, which means there is unmeasured confounding associated with better cognition and less severe vascular pathology or unmeasured confounding associated with poorer cognition and more severe vascular pathology (seemly more likely), our estimated PMs would underestimate the true mediation effects of vascular pathologies.
4. DISCUSSION
In a community-based sample of 5,161 stroke-free older individuals, we found that multiple lobar CMB, higher WML load, and greater CAC --but not RVD nor CIMT--were associated with poorer late-life memory and global cognition. In the 2,907 genotyped individuals, we found that a higher genetic risk score for AD dementia, driven primarily by APOE, was associated with these three vascular pathologies and two cognition outcomes. In mediation analyses, we found that the effects of APOE and SNPs near APOE on memory may be partially mediated by CMB and WML, and their effects on global cognition may be partially mediated by CMB, WML, and CAC. With the possible exception of CMB, there was little evidence of an effect of non-APOE AD dementia-associated alleles on either memory or global cognition.
In a relatively large sample of older adults, our phenotypic analyses replicated previously reported phenotypic associations of cerebral SVD [36, 37] and atherosclerosis [18]with cognitive function. We further examined if shared genetic factors contribute to these associations, and found genetic overlap between AD dementia, vascular pathologies, and late-life cognition (Table 3 and Figure 1). Our data showed that lobar CMB, WML load, and CAC score were associated with GW-ADPRS, and were even more strongly associated with 19q13-ADPRS. Our findings of the strongest genetic overlap between AD dementia and CMB are consistent with recently reported genetic correlation between AD and cerebral SVD but not LVD [28]. Lobar CMB may be caused by CAA [38, 39], which is highly prevalent in post-mortem analyses of brains of persons with clinical diagnosis of AD dementia [40]. In addition, the APOE-e4 allele has been associated with the presence of CAA [41, 42]. These previous findings support the possible genetic overlap between lobar CMB and AD dementia observed in our data. Since WML [43] and CAC [44] may share some common risk factors with CMB, and both have been related to dementia (although the evidence is not as strong as that for CMB), a genetic overlap of AD dementia with WML and CAC makes sense. On the other hand, we found no association between ADPRS and RVD, which has been reported as an indicator of cerebral small-vessel pathology [12] and a predictor of dementia [11]. One possible explanation is that the central retinal venular equivalent is observer-dependent and may not accurately reflect the degree of retinal venular dilatation, but there is no indication of even an attenuated signal.
Our results suggest that the APOE gene explains most of the SNP-based genetic overlap of AD dementia with the vascular pathologies (see Supplementary Methods and Results on ‘Conditional Regression Analyses’ and Supplementary Table S6). APOE has been related to cerebrovascular dysfunction by affecting cerebral blood flow, blood-brain-barrier integrity, and neuronal-vascular coupling [45]. As mentioned above, the APOE-e4 allele is a risk factor for CAA [41, 42]. In terms of peripheral vascular disease, APOE has been shown to be an important factor in the development of hyperlipoproteinemia and atherosclerosis [45, 46]. Our data also showed no association between the non-19q13-ADPRS and any of the examined vascular pathologies, despite previously reported associations of non-APOE AD risk genes with inflammation and abnormal lipid metabolism, which are both risk factors for vascular disease [47]. Future research with larger samples are needed to test for association between vascular pathologies and AD dementia-associated alleles outside of the APOE-linkage region.
In our sample, we observed associations of the APOE-ADPRS with both memory and global cognition, whereas the non-APOE-ADPRS was associated with memory, but not global cognition. A previous meta-analysis including 77 studies of the association between APOE and cognitive function suggested that carriers of APOE-e4 performed worse on multiple domains of cognitive tests, including memory, executive functioning, perceptual speed, and overall global cognition [48]. On the other hand, non-APOE-ADPRS calculated by using summary statistics from the International Genomics of Alzheimer’s Project (IGAP) was found to be associated with memory impairment but not executive function in non-demented subjects, with mean age of 75.3 years, in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) [49]. It is possible that impaired memory was more likely to be detected than deficits in other cognitive domains for individuals in the early stage of cognitive decline. However, more research is needed on the relationship between specific genes and different domains of cognitive function.
Having established a SNP-based genetic overlap between AD dementia, vascular pathologies, and late-life cognition, we then sought to identify the causal relationships between ADPRS, vascular pathologies, and cognition scores. Our findings indicate that AD dementia-associated SNPs affect late-life cognition partially through pathways involving vascular pathologies, providing insight into potential pathogenic mechanisms in clinical AD dementia. The results also may lend further support to interventions to reduce vascular pathologies may be of value in the prevention of AD dementia. It is worth noting that we separately examined the mediation effects of CMB, WML, and CAC. Although measures of these vascular pathologies were correlated with each other, their correlations were relatively weak in our sample (Kendall’s tau-b=0.07 for CMB-WML, Point-Biserial correlation coefficient=0.08 for CMB-CAC and 0.12 for WML-CAC). Thus, it is reasonable to believe that a certain proportion of AD dementia-associated SNP effects on cognitive function were mediated by vascular pathologies when considering all vascular mediators together.
The only previous study investigating the mediation role of cerebrovascular imaging markers between genetic variants and cognitive function, which used an overlapping sample from the same cohort (the AGES-Reykjavik), reported that about 9% of the total effect of APOE4 carriership on global cognition was mediated by CMB and WML volume together [30]. Our analyses revealed similar but stronger mediation effect of vascular pathologies on the relationship between SNPs and cognition. The major strength of the present study is that we assessed the effects of PRS, aggregating multiple possible risk alleles for AD across the whole genome, within or beyond the APOE-linkage region, weighted by their estimated effect sizes. Moreover, we considered both cerebral small-vessel and systemic large-vessel imaging markers, which have been previously associated with dementia or poor cognition, as potential mediators.
Several limitations in the present study should be noted. In the population-based sample, in which most subjects were cognitively normal or mildly impaired, mean scores of cognitive tests reflect both lifelong cognitive variability and recent pathological changes, and the former may overwhelm the latter. However, with our relatively large sample size, we were able to detect small signals and parse these signals into what appear to be meaningful mediation relationships. Nonetheless, the sample may have only been large enough to detect ^POF-related signals, even if other causal SNPs are present. In any event, in the setting of small signals, another major limitation is the possible violation of the no-unmeasured-confounding assumption necessary for causal mediation analyses. However, our sensitivity analyses suggest that given the expected direction of unmeasured confounding, our estimated indirect effects may underestimate the true mediated effects. In addition, the ADPRS, including only common genetic variants, cannot account for all the genetic effects on cognitive performance and AD dementia. Although our SNP-based PRS were strongly associated with vascular and cognitive phenotypes, and PRS for AD dementia has been reported to be capable of capturing nearly all common genetic risk for AD [50], there are still causal genomic variants (e.g., rare variants) that are not well-tagged by GWAS SNPs. However, the genetic effects not captured by SNP-based risk scores can also be seen as a type of unmeasured mediator-outcome confounding. Therefore, the sensitivity analyses mentioned above may help minimize these concerns. Finally, although our use of causal mediation analysis appears to imply mechanistic causality, we note that our design is ultimately correlational. In future research, an experimental-causal-chain approach may help to develop a more fundamental understanding of causal mechanisms.
This is the first study, to our knowledge, that combined polygenic profiling and causal mediation methods to identify the causal relationship between two genetically correlated phenotypes and their shared genetic factors. Our findings support the hypothesis of a genetic overlap, mostly due to APOE, between AD dementia and vascular pathologies, especially SVD. Our results also showed that in older individuals, CMB, WML, and CAC may causally affect cognitive function and partially mediate the polygenic effects of AD-related genes on cognition, underscoring the potential role of vascular factors in cognitive decline, and suggesting vascular pathologies as a target for future mechanistic research in this area.
Supplementary Material
Research in Context
Systematic review:
A large literature, updated with a recent PubMed search, includes extensive epidemiological and neuropathological evidence suggesting shared mechanisms between vascular pathologies and Alzheimer’s disease (AD) dementia. However, few studies have examined their polygenic overlap, and no published research has focused on whether vascular pathologies mediate the relationship between AD-associated genes and late-life cognition.
Interpretation:
Our findings support the hypothesis of a genetic overlap, mostly due to APOE, between AD dementia and vascular pathologies. The cumulative effect of AD-related genes on late-life cognition was partially but significantly mediated by cerebral microbleeds, white matter lesions, and coronary calcification, underscoring the potential role of vascular factors in cognitive decline.
Future directions:
These results should be confirmed in larger samples. Research is also needed on the relationship of specific genes and pathways with different domains of cognitive function. In the meantime, these findings suggest vascular pathologies as a target for future mechanistic research on AD.
ACKNOWLEDGEMENTS
The authors thank all the participants of the AGES-Reykjavik Study and the Icelandic Heart Association clinic staff for their invaluable contributions.
Study funding: The AGES-Reykjavik Study was funded by the NIH [contract N01-AG-12100]; the Intramural Research Program of the National Institute on Aging and the National Eye Institute [ZIAEY000401], NIH; and the Icelandic Heart Association and the Icelandic Parliament. None of the funding organizations or sponsors were involved in study design; in the collection, analysis, or interpretation of data; in writing of the report; or in the decision to submit the manuscript for publication.
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136:2697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schneider JA, Bennett DA. Where vascular meets neurodegenerative disease. Stroke. 2010;41:S144–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70:1208–14. [DOI] [PubMed] [Google Scholar]
- [4].Goos JD, Kester MI, Barkhof F, Klein M, Blankenstein MA, Scheltens P, et al. Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke. 2009;40:3455–60. [DOI] [PubMed] [Google Scholar]
- [5].Qiu C, Cotch MF, Sigurdsson S, Jonsson PV, Jonsdottir MK, Sveinbjrnsdottir S, et al. Cerebral microbleeds, retinopathy, and dementia: the AGES-Reykjavik Study. Neurology. 2010;75:2221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011;134:335–44. [DOI] [PubMed] [Google Scholar]
- [8].Inaba M, White L, Bell C, Chen R, Petrovitch H, Launer L, et al. White matter lesions on brain magnetic resonance imaging scan and 5-year cognitive decline: the Honolulu-Asia aging study. J Am Geriatr Soc. 2011;59:1484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Prins ND, van Dijk EJ, den Heijer T, Vermeer SE, Koudstaal PJ, Oudkerk M, et al. Cerebral white matter lesions and the risk of dementia. Arch Neurol. 2004;61:1531–4. [DOI] [PubMed] [Google Scholar]
- [10].Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ. 2010;341:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].de Jong FJ, Schrijvers EM, Ikram MK, Koudstaal PJ, de Jong PT, Hofman A, et al. Retinal vascular caliber and risk of dementia: the Rotterdam study. Neurology. 2011;76:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ikram MK, De Jong FJ, Van Dijk EJ, Prins ND, Hofman A, Breteler MM, et al. Retinal vessel diameters and cerebral small vessel disease: the Rotterdam Scan Study. Brain. 2006;129:182–8. [DOI] [PubMed] [Google Scholar]
- [13].Ikram MK, de Jong FJ, Vernooij MW, Hofman A, Niessen WJ, van der Lugt A, et al. Retinal vascular calibers associate differentially with cerebral gray matter and white matter atrophy. Alzheimer Dis Assoc Disord. 2013;27:351–5. [DOI] [PubMed] [Google Scholar]
- [14].de la Torre JC. Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev. 2010;9:218–25. [DOI] [PubMed] [Google Scholar]
- [15].Sabayan B, van Buchem MA, Sigurdsson S, Zhang Q, Meirelles O, Harris TB, et al. Cardiac and Carotid Markers Link With Accelerated Brain Atrophy: The AGES-Reykjavik Study (Age, Gene/Environment Susceptibility-Reykjavik). Arterioscler Thromb Vasc Biol. 2016;36:2246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].van Oijen M, de Jong FJ, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Atherosclerosis and risk for dementia. Ann Neurol. 2007;61:403–10. [DOI] [PubMed] [Google Scholar]
- [17].Wendell CR, Waldstein SR, Ferrucci L, O’Brien RJ, Strait JB, Zonderman AB. Carotid atherosclerosis and prospective risk of dementia. Stroke. 2012;43:3319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bos D, Vernooij MW, Elias-Smale SE, Verhaaren BF, Vrooman HA, Hofman A, et al. Atherosclerotic calcification relates to cognitive function and to brain changes on magnetic resonance imaging. Alzheimers Dement. 2012;8:S104–11. [DOI] [PubMed] [Google Scholar]
- [19].Reis JP, Launer LJ, Terry JG, Loria CM, Zeki Al Hazzouri A, Sidney S, et al. Subclinical atherosclerotic calcification and cognitive functioning in middle-aged adults: the CARDIA study. Atherosclerosis. 2013;231:72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vidal JS, Sigurdsson S, Jonsdottir MK, Eiriksdottir G, Thorgeirsson G, Kjartansson O, et al. Coronary artery calcium, brain function and structure: the AGES-Reykjavik Study. Stroke. 2010;41:891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Michaelson DM. APOE epsilon4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014;10:861–8. [DOI] [PubMed] [Google Scholar]
- [22].Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta analysis. Neurology. 2013;81:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–11. [DOI] [PubMed] [Google Scholar]
- [24].Khan TA, Shah T, Prieto D, Zhang W, Price J, Fowkes GR, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. Int J Epidemiol. 2013;42:475–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sveinbjornsdottir S, Sigurdsson S, Aspelund T, Kjartansson O, Eiriksdottir G, Valtysdottir B, et al. Cerebral microbleeds in the population based AGES-Reykjavik study: prevalence and location. J Neurol Neurosurg Psychiatry. 2008;79:1002–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012;124:305–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Desikan RS, Schork AJ, Wang Y, Thompson WK, Dehghan A, Ridker PM, et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation. 2015;131:2061–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Traylor M, Adib-Samii P, Harold D, Alzheimer’s Disease Neuroimaging I, International Stroke Genetics Consortium UKYLSDNAr, Dichgans M, et al. Shared genetic contribution to Ischaemic Stroke and Alzheimer’s Disease. Ann Neurol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu G, Yao L, Liu J, Jiang Y, Ma G, Genetic, et al. Cardiovascular disease contributes to Alzheimer’s disease: evidence from large-scale genome-wide association studies. Neurobiol Aging. 2014;35:786–92. [DOI] [PubMed] [Google Scholar]
- [30].Sajeev G Mediation Analysis in Understanding Mechanism of Alzheimer’s Disease Risk. Boston: Harvard T.H. Chan School of Public Health; 2015. [Google Scholar]
- [31].Harris TB, Launer LJ, Eiriksdottir G, Kjartansson O, Jonsson PV, Sigurdsson G, et al. Age, Gene/Environment Susceptibility-Reykjavik Study: multidisciplinary applied phenomics. Am J Epidemiol. 2007;165:1076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].VanderWeele TJ. Mediation Analysis: A Practitioner’s Guide. Annu Rev Public Health. 2016;37:17–32. [DOI] [PubMed] [Google Scholar]
- [34].Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3:143–55. [DOI] [PubMed] [Google Scholar]
- [35].Emsley R, Liu H. PARAMED: Stata module to perform causal mediation analysis using parametric regression models. Boston College Department of Economics; 2013. [Google Scholar]
- [36].Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, et al. Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology. 2012;78:326–33. [DOI] [PubMed] [Google Scholar]
- [37].Staals J, Booth T, Morris Z, Bastin ME, Gow AJ, Corley J, et al. Total MRI load of cerebral small vessel disease and cognitive ability in older people. Neurobiol Aging. 2015;36:2806–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yates PA, Villemagne VL, Ellis KA, Desmond PM, Masters CL, Rowe CC. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol. 2014;4:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smith EE, Nandigam KR, Chen YW, Jeng J, Salat D, Halpin A, et al. MRI markers of small vessel disease in lobar and deep hemispheric intracerebral hemorrhage. Stroke. 2010;41:1933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm (Vienna). 2002;109:813–36. [DOI] [PubMed] [Google Scholar]
- [41].Esiri M, Chance S, Joachim C, Warden D, Smallwood A, Sloan C, et al. Cerebral amyloid angiopathy, subcortical white matter disease and dementia: literature review and study in OPTIMA. Brain Pathol. 2015;25:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–34. [DOI] [PubMed] [Google Scholar]
- [43].de Leeuw FE, de Groot JC, Oudkerk M, Witteman JC, Hofman A, van Gijn J, et al. Hypertension and cerebral white matter lesions in a prospective cohort study. Brain. 2002;125:765–72. [DOI] [PubMed] [Google Scholar]
- [44].Liu W, Zhang Y, Yu CM, Ji QW, Cai M, Zhao YX, et al. Current understanding of coronary artery calcification. Journal of geriatric cardiology : JGC. 2015;12:668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, Morris AW, et al. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016;131:709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72 Pt A:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wisdom NM, Callahan JL, Hawkins KA. The effects of apolipoprotein E on nonimpaired cognitive functioning: a meta-analysis. Neurobiol Aging. 2011;32:63–74. [DOI] [PubMed] [Google Scholar]
- [49].Mormino EC, Sperling RA, Holmes AJ, Buckner RL, De Jager PL, Smoller JW, et al. Polygenic risk of Alzheimer disease is associated with early- and late-life processes. Neurology. 2016;87:481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Escott-Price V, Shoai M, Pither R, Williams J, Hardy J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging. 2017;49:214 e7–e11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.