

Abstract
Organisms that perform the de novo biosynthesis of cobalamin (vitamin B12) do so via unique pathways depending on the presence of oxygen in the environment. The anaerobic biosynthesis pathway of 5,6-dimethylbenzimidazole, the so-called “lower ligand” to the cobalt center, has been recently identified. This process begins with the conversion of 5-aminoimidazole ribotide (AIR) to 5-hydroxybenzimidazole (HBI) by the radical S-adenosyl-L-methionine (SAM) enzyme BzaF, also known as HBI synthase. In this work we report the characterization of a radical intermediate in the reaction of BzaF using electron paramagnetic resonance (EPR) spectroscopy. Using various isotopologues of AIR, we extracted hyperfine parameters for a number of nuclei, allowing us to propose plausible chemical compositions and structures for this intermediate. Specifically, we find that an aminoimidazole radical is formed in close proximity to a fragment of the ribose ring. These findings induce the revision of past proposed mechanisms and illustrate the ability of radical SAM enzymes to tightly control the radical chemistry that they engender.
Graphical Abstract
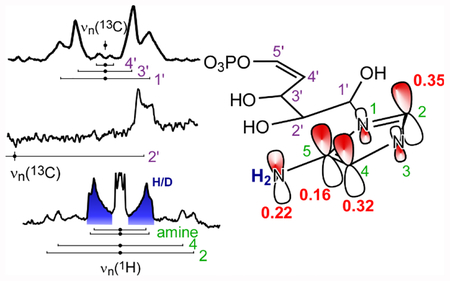
Introduction.
Vitamin B12, or cobalamin, is a biological cofactor1 used in radical-initiated rearrangements,2–7 methyl transfers8–10 and dehalogenation reactions.11–16 Organisms in all domains of life have a requirement for B12, but animals, plants, and fungi are unable to synthesize the vitamin and instead must acquire it from bacteria and archaea.17 The B12 cofactor consists of a hexacoordinate cobalt ion equatorially ligated by the four nitrogens of the corrin macrocycle (Figure 1, right).18 A number of ligands can bind in the so-called upper axial position designated R in Figure 1. For methyl-transfer reactions, this ligand is a CH3 group derived from either methyltetrahydrofolate or S-adenosyl-L-methionine (SAM). For the radical rearrangement reactions, the “upper” ligand is an adenosine sourced from ATP. In the isolated cobalamin cofactor, the “lower” axial position is often occupied by the nitrogen of a 5,6-dimethylbenzimidazole (DMB, Figure 1, shown in red) that is tethered to the corrin ring periphery by an intramolecular loop. In some B12-dependent enzymes, the DMB ligand is retained when the cofactor binds to the protein. In others, DMB dissociates from the cobalt center and is bound to a distinct pocket, anchoring the cofactor to the enzyme. In these cases, DMB is replaced by either a histidine side chain,19 a water molecule,20 or nothing in which case the cobalt is five-coordinate.21
Figure 1.

AIR is converted to hydroxymethylpyrimidine phosphate (HMPP) by ThiC (left side) and to hydroxybenzimidazole (HBI) by BzaF (right side), which are, respectfully, precursors of thiamine (vitamin B1) and cobalamin (R = CN is vitamin B12).
The biosynthesis of the complete corrinoid—including DMB—occurs via distinct pathways depending on the presence or absence of oxygen (Table S1).22,23 In the aerobic pathway, the enzyme BluB catalyzes the O2-dependent cleavage of reduced flavin mononucleotide to form DMB and erythrose-4-phosphate.23 Under anaerobic conditions, 5-aminoimidazole ribotide (AIR, Figure 1, center) is converted to DMB through reactions catalyzed by a set of recently discovered enzymes.24,25 In Desulfuromonas acetoxidans, these enzymes include BzaF, a radical SAM enzyme, which carries out the dramatic rearrangement of AIR to form 5-hydroxybenzimidazole (HBI, Figure 1, right).24 This HBI synthase reaction is initiated by a single electron transfer from the [Fe4S4]+ cluster in BzaF to a bound SAM molecule.26 The resultant homolytic cleavage of the C(5’-adenosine)—S(methionine) bond generates a fleeting 5’-deoxyadenosyl (5’-dAdo)27,28 radical that abstracts the pro-S hydrogen atom from 5’-C of AIR.24 Isotope tracing has shown that four of the five ribose carbons form the benzyl portion of HBI. The 1’ carbon (where the ’ denotes a ribose carbon) is lost as formate. The 2’ and 5’ carbons form bonds to the 4 and 5 carbons of the imidazole. HBI synthase (BzaF) also bears 44% protein sequence identity (66% protein sequence similarity) to ThiC from D. acetotoxidans (see SI section II), another radical SAM enzyme that converts AIR to hydroxymethylpyrimidine phosphate (HMPP), a precursor towards vitamin B1 (thiamine) biosynthesis (Figure 1, left). That two radical SAM enzymes operate on the same substrate molecule, AIR, yet deliver dramatically different products, presents a unique opportunity to examine the role radical SAM enzymes play in orchestrating the substrate radical rearrangement to yield the desired product after the initial H-atom abstraction.
In this paper we report an electron paramagnetic resonance (EPR) spectroscopic characterization of a radical intermediate trapped during the HBI synthase-catalyzed rearrangement of AIR. Using site-specific isotope labeling strategies, we measured numerous hyperfine interactions that map out the spin density distribution and help us to construct a geometric model of the intermediate. These findings provide new constraints on possible mechanisms for this complex rearrangement.
Experimental Section.
Aminoimidazole ribotide synthesis.
Synthesis of AIR and its isotopologues are described in references 24, 29, and 30.29,30 Synthesis and characterization of 2,3-2H2-AIR is included in the supplementary information section III. Also included are the NMR data (Figures S1 and S2). A list of the various AIR isotopologues in this study is provided in Table S2.
Protein Expression and Purification.
The nucleotide sequence of the Desulfuromonas acetoxidans ThiC homolog, BzaF (HBI synthase),24 was synthesized and cloned into a THT vector (pET 28 vector that has a TEV protease cleavage site for removing the His tag). HBI synthase was co-expressed in the presence of a plasmid encoding the suf operon for enhanced in vivo [Fe4S4] formation in E. coli BL21 (DE3).31–34 An overnight 15 mL culture was grown in LB medium in the presence of kanamycin (40 mg/L) and chloramphenicol (25 mg/L). This was then added to a 1.8 L minimal medium (M9 minimal salts 1X, 27 mL of 50% glucose, 7 mL of 1 M MgSO4, 200 μL of 1 M CaCl2, 72 mg kanamycin, and 45 mg chloramphenicol were added to 1.8 L water). The cultures were incubated at 37°C with shaking (180 rpm) until the OD600 reached 0.6 to 0.65. The cultures were then incubated at 4°C without shaking for 3 hrs. Then 50 mg of ferrous ammonium sulfate and 50 mg of cysteine were added. This was followed by induction of the culture with 70 μM IPTG. The culture was then incubated at 15°C with shaking (50 rpm) for 18 to 20 hours. The cultures were then incubated at 4°C for 3 hours without shaking. The cells were then harvested and stored in liquid nitrogen overnight before enzyme purification. For enzyme purification, the cell pellets were thawed at room temperature in an anaerobic chamber and suspended in lysis buffer (100 mM Tris-HCl, pH 7.5) in the presence of 2 mM DTT, lysozyme (0.2 mg/mL), and benzonase (100 units). This mixture was then cooled in an ice-bath for 2 hrs. The suspension of cells was then sonicated and centrifuged to give the cell-free extract. The enzyme was purified using standard Ni-NTA chromatography. The column was first incubated with the lysis buffer. The cell-free extract was passed over the column, which was then washed with 8 to 9 column volumes of wash buffer (100 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 2 mM DTT, pH 7.5). The enzyme was eluted using 100 mM Tris-HCl, 300 mM NaCl, 250 mM imidazole, 2 mM DTT, pH 7.5 and desalted using an Econo-Pac 10 DG desalting column (commercially available from Bio-Rad). The purified enzyme was stored in liquid nitrogen.
Deuterated buffer was prepared by first lyophilizing 20 mL of a 1 M K3PO4 solution, and then redissolving the residue into 20 mL of 2H2O. This was repeated twice to give a 1 M potassium phosphate stock solution that then was used to make deuterated buffer containing 100 mM potassium phosphate, 25% natural abundance glycerol, 2 mM DTT (pH 7.5). For experiments in deuterated buffer, HBI synthase was buffer exchanged twice using Bio-Rad centrifugal spin columns.
HPLC conditions for HBI synthase reaction time course experiments.
HPLC-1260 series Agilent equipped with a LC-18 column (SUPELCOSIL LC-18-T column (15 cm × 3 mm × 3 μm)) was used. The following solvents were used in the gradient: A. water, B. 5 mM ammonium formate, C. methanol. The following gradient was used: 0 min, 100% B; 25 min 7% A, 70% B 23% C; 27 min, 25% A, 75% C; 29 min 25% A, 75% C; 30 min, 100% B.
Sample Preparation.
All procedures were carried out in an N2-atmosphere glove box. Samples were prepared in 1.5 mL Eppendorf tubes. An excess of dithionite was used, 3 to 4 mM in the final solution. Final protein concentration was 700 μM. The final concentration of SAM and AIR were 1.6 mM and 1.1 mM, respectively. The protein was first allowed to incubate with dithionite for 2 minutes followed by the addition of SAM and another 2 min incubation time. AIR was added to initiate the reaction. Samples were quickly transferred to quartz EPR tubes (clear fused quartz 2.0 × 2.4 mm, 10 cm length, Vitrocom) using a 9 1/4” disposable glass pipette. The sample was freeze-quenched in liquid nitrogen 4 min after the addition of AIR. All reactant solutions were prepared in 100 mM potassium phosphate buffer, pH 7.5. All samples were stored in liquid nitrogen.
EPR Experimental.
Continuous-wave (CW) experiments were carried out using a Bruker E500 EPR spectrometer equipped with an Oxford Instruments ESR900 continuous-flow liquid helium cryostat. A Bruker super high QE (SHQE) resonator was used with a cavity resonance frequency of approximately 9.4 GHz. Spectra were collected under slow passage conditions using a non-saturating 200 μW of microwave power with a sample temperature of 20 K. Modulation frequency was 100 kHz with a 2 G amplitude (10 G = 1 mT). Data points were collected in 0.5 G increments using 60 ms time constant resulting in a 24 s sweep time.
Pulse EPR experiments were carried out on a Bruker E580 pulse EPR spectrometer with X (≈9–10 GHz) and Q band (≈33–35 GHz) capabilities. The E580 is equipped with an Oxford Instruments CF935 continuous-flow liquid helium cryostat. A laboratory-built Q band ENDOR probe was used with an approximate resonance frequency of 34.1 GHz.35 For RF pulse amplification, an ENI A1000 (0.3–35 MHz) or ENI LPI10 (10–86 MHz) amplifier provided up to 1 kW peak pulse power. Pulse data were collected at temperature of 15 K with a shot repetition time of 5.1 ms. Standard Davies36 and Mims37,38 ENDOR pulse sequences were used. Davies ENDOR was collected utilizing a microwave πinv pulse length of 64 ns and detection pulse lengths being 12 ns for π/2 and 24 ns for π pulses. The microwave π/2 pulse lengths were 12 ns for Mims ENDOR. For nuclear excitation, the RF π pulse lengths were 15 μs for 1H and 20 μs for 2H and 13C. The τ value was 300 ns for Davies ENDOR.
EPR Theory.
The results of an EPR experiment can be interpreted using the phenomenological spin Hamiltonian.
| (1) |
The terms in order are the electron Zeeman interaction, electron-nuclear hyperfine interaction, and nuclear Zeeman interaction. βe is the Bohr magneton, B the static magnetic field, g the electron g-value, S the electron spin, h is the Planck constant, Ai the hyperfine interaction for the i th nucleus, Ii the nuclear spin for the i th nucleus, βn the nuclear magneton, and gn the nuclear g-value for the i th nucleus. Simulations of the ENDOR spectra were carried out using the EasySpin toolbox for MATLAB (The Mathworks Inc.).39
The electron-nuclear hyperfine tensor A consists of isotropic (aiso) and anisotropic (T) components. The isotropic component arises from the Fermi contact interaction and is proportional to the spin population at the nucleus. The anisotropy T of the hyperfine interaction arises from a non-local through-space dipolar interaction (Tnon-loc) of the nuclear magnetic dipole with the unpaired electron magnetic dipole and a local component (Tloc) from asymmetric spin distribution in the p and d orbitals centered on the magnetic nucleus. The simplest description of Tnon-loc for an S = 1/2, I = 1/2 system is the point dipole-approximation:
| (2) |
where μ0 is the magnetic vacuum permeability, r is the distance between the two magnetic point dipoles, and θ is the angle between the vector that connects the two point-dipoles and the applied magnetic field. When nuclear and electron spins are well separated (r > 2.5 Å), this dipolar interaction is axial, T = [−Tnon-loc −Tnon-loc +2Tnon-loc] MHz. When the spin density is distributed over several atoms, one must sum the Tnon-loc over all i atoms with each interaction weighted by ρi, the spin density on the i th atom.
If the magnetic nucleus is near (r < 2.5 Å) to an electron spin-carrying center where the spin is found in a 2pπ orbital, Tnon-loc is not an axial tensor, and the following approximation is used:
| (3) |
Here d is the distance from that nearby nucleus of the spin-center to the probabilistic center of the lobes of its 2pπ orbital—typically taken to be about 0.68 Å.40,41 ρ is the unpaired spin density in the 2pπ orbital.
A proton bound to such a 2pπ spin-center exhibits a 1H hyperfine coupling proportional to the spin population of that 2pz-orbital via theory developed by McConnell et al.42,43
| (4) |
Where an is the measured isotropic hyperfine coupling for a proton on carbon n, Qn is empirically derived to be 63 MHz for a carbon atom and −70 MHz for a 14N atom, and ρn is the unpaired spin density on atom n. It is important to note that a small distribution of Qn values has been used for carbon depending on the chemical bonding environment, and an even larger distribution of values have been used for 14N.
Density Functional Theory.
Density functional theory (DFT) calculations were carried out using Orca 4.0.44 A variety of molecular models based on an imidazole or aminoimidazole based were generated that had a variety of protonation states and charges (summarized in Figure S8). Models were built using Avogadro 1.1.145 and pre-optimized using the universal force field for molecular mechanics.46 Further optimization was done with Orca utilizing the BP86 functional and cc-pVTZ basis set with appropriate atom polarization functions, and the resolution of identity approximation.47–51 EPR calculations were done using the B3LYP functional and EPR-III basis set.52–54 Examples of input files for geometry optimization and EPR property calculations are included in the SI.
Results.
CW EPR of a Radical Intermediate.
Dithionite (DT)-treated BzaF gives rise to an EPR spectrum (Figure S3, cf. first and second traces) assigned to the reduced [Fe4S4]+ that binds and cleaves SAM. This [Fe4S4]+ signal is not dramatically affected by the addition of SAM (Figure S3, fourth trace). However, when a solution is prepared containing BzaF, DT, SAM, and AIR and frozen approximately four minutes after mixing, the [Fe4S4]+ signal is replaced with a new organic radical signal (Figure 2). The organic radical signal decays at a rate that is coincident with formation of 5-hydroxybenzimidazole (HBI) formation (Figure S4). To establish the chemical identity of this organic radical intermediate we performed CW EPR spectroscopy on reaction mixtures made with various isotopologues of the AIR substrate (Figure 2A–D). When the reaction with natural abundance (NA) AIR is run in buffer that is 75% enriched in 2H2O, the spectral breadth narrows significantly (Figure 2B), indicating that there are one or more exchangeable protons coupled to the unpaired electron spin. The magnitude of a hyperfine interaction is proportional to the gyromagnetic ratio (γ) of the magnetic nucleus, thus the narrowing observed upon deuteration is caused by the approximately 6.5 fold smaller γ of 2H (γ = 6.54 MHz/T) compared to 1H (γ = 42.58 MHz/T). The remaining hyperfine splittings (more easily seen in the numerical derivative, green trace, Figure 2B) result from non-exchangeable 1H (e.g. C—H) and/or 14N (I = 1) couplings with the three nitrogens of the aminoimidazole moiety. The most dramatic narrowing of the spectral shape comes from selective deuteration of the 2 and 4 positions on the imidazole of AIR (Figure 2C). Labeling the ribose with 13C at the 1’,2’,3’,4’, and 5’ positions results in broadening of the CW EPR (Figure 2D) spectrum and obscures the 1H/14N hyperfine structure in the numerical derivative spectrum (green traces in Figure 2). Selectively labeling only the 2’ carbon accounts for a large portion of the 13C-induced broadening (Figure S5). Thus, from the continuous-wave EPR spectra of the radical intermediate generated with various AIR isotopologues, we observe that the electron spin interacts strongly with nuclei from the imidazole fragment and 2’-C of the ribose.
Figure 2.

X band CW EPR spectra of the HBI synthase reaction intermediate prepared using various isotopologues of AIR. Traces are scaled such that the double integral of each spectrum is set to 1. Green traces correspond to the derivative of the CW spectrum and accentuates hyperfine-induced structure. A narrow radical signal centered at 335 mT was removed from the traces of the numerical derivatives for clarity. A, natural abundance AIR in H2O buffer. B, natural abundance AIR in 75% 2H2O buffer. C, 2,4-2H2-AIR in H2O buffer. D, 1’,2’,3’,4’,5’−13C5-AIR in H2O buffer.
Proton and Deuterium ENDOR.
To quantitatively assess these hyperfine interactions, we performed electron-nuclear double-resonance (ENDOR) spectroscopic studies. The Q-band ENDOR spectrum of the radical intermediate generated with NA AIR in H2O buffer contains five hyperfine-split doublets centered at the proton Larmor frequency (≈51.1 MHz at 1.2 T, Figure 3A, doublets s, t, u, v, and w; Table S3). The two most strongly coupled protons (doublet s with peaks at ≈30 and 74 MHz and doublet t with peaks at ≈33 and 71 MHz) are assigned to protons bound to carbons at the 2 and 4 positions of the imidazole as these features disappear when these carbons are deuterated (Figure 3B). The corresponding deuterium ENDOR spectrum can be compared directly to the proton ENDOR spectrum by multiplying the excitation-frequency axis by the ratio of 1H/2H gyromagnetic ratios (Figure 3D, blue). The deuterium ENDOR spectrum allows for easier simulation as there is no overlap by other proton resonances. No quadrupolar splitting of the 2H ENDOR peaks due to the I = 1 deuteron nucleus is resolved. Notably these 1H hyperfine coupling constants (aiso(1H) = 38.2 MHz, and 33.1 MHz) are very similar to those found for protons in equivalent positions of the imidazole radical (aiso(1H) = 37 MHz and 30 MHz)55 and histidine imidazole radical (aiso = 33.9 and 29.8 MHz);56 suggesting that the BzaF radical intermediate is also an imidazole π-radical.
Figure 3.

Left, 1H and frequency-scaled 2H Davies ENDOR. A, NA-AIR H2O. B, 2,4-2H2-AIR H2O. C, NA-AIR 2H2O. D, blue trace is the frequency scaled 2H ENDOR of 2,4-2H2-AIR H2O. Instrument settings: microwave frequency = 34.1 GHz, πinv = 64 ns, π/2 = 12 ns, τ = 300 ns, πRF = 15 μs, T = 15 K. Center, 13C Davies ENDOR. E, 2’−13C-AIR. F, 1’,2’,3’,4’,5’−13C5-AIR. G, NA-AIR. Right 13C Mims ENDOR. H, 1’,2’,3’,4’,5’−13C5-AIR. I, 1’−13C-AIR. Spectral simulations are shown in red.
The next two most strongly coupled protons (Figure 3, doublet u with peaks at ≈42 and 61 MHz, and doublet v with peaks at ≈43.5 and 59.5 MHz) are solvent-exchangeable (cf. Figure 3 traces A and C, difference spectrum is presented in Figure S6). The parallel turning points in the ENDOR spectrum for these protons are not obvious, leading to a range of possible values for the hyperfine interaction. We achieve acceptable simulations using aiso = 11–13 MHz. Since these protons have a fairly large coupling, they must be located on or near the imidazole π-radical—either bound to the two ring nitrogens, to the nitrogen of the amine, to the two oxygens of the diol of the ribose, or to a nearby amino acid.
One last prominent doublet in the 1H ENDOR spectrum (labeled ‘w’ in Figure 3, traces A-C) is unaffected by preparing the radical in 2H2O (cf. Figure 3 traces A and C); therefore the corresponding proton must be covalently bound. Since all of the non-exchangeable protons on the aminoimidazole moiety are accounted for, this proton is either of proteinaceous origin or resides on the ribose moiety. Notably in the ENDOR spectra of the radical intermediate prepared using 5’,5’−2H2-AIR and 4’−2H-AIR, this doublet is not affected (Figure S7). AIR isotopologues deuterated at the other ribose carbons were unavailable.
13C ENDOR.
The proximity of the ribose fragment to the putative imidazole radical was tested using AIR labeled with 13C at various positions on the ribose. The 13C ENDOR spectra of the radical intermediate prepared using 1’,2’,3’,4’,5’−13C5-AIR have features corresponding to one strongly coupled carbon (Figure 3 trace F, peak at ≈24 MHz) and three weakly-coupled carbons (Figure 3 traces H and I; peaks from ≈12–14 MHz, doublets labeled x, y, and z). The strongly coupled carbon signal appears in samples prepared from both 1’,2’,3’,4’,5’−13C5-AIR and 2’−13C-AIR isotopologues, but not the natural abundance sample (1% 13C), verifying that it arises from a carbon nucleus and not a proton, nitrogen (99.6% 14N, I = 1), or phosphorus (100% 31P, I = 1/2). The isotropic hyperfine interaction at 2’-C is large (|aiso(13C)| = 20.9 MHz) compared to the anisotropic contribution (aaniso = [−1.3 −0.7 2.1] MHz) suggesting that the 2’ carbon is not part of a π-radical (π-radicals typically exhibit a ratio of aiso:Tloc ≈ 1.5), but rather in close proximity to a spin center leading to σ-radical character (with aiso:Tloc > 8).57,58 This is an important distinction as in the final HBI product, the 2’-C has formed a bond with the 5-C of imidazole and participates in the bicyclic π-system, meaning that the radical intermediate we observe has not yet incorporated the ribose moiety into the conjugated ring.
The following hyperfine tensors were determined for the remaining three weakly coupled 13C ENDOR doublets (Figure 3, traces H-I, peaks from ≈12–14 MHz): for doublet x, |A(13C)| = [1.91 2.58 0.09] MHz; for doublet y, A(13C) = [1.25 1.35 0.80] MHz; and for doublet z, A(13C) = [−0.2 −0.2 +0.4] MHz. The assignment of negative values to a1 and a2 for doublet z is based on the point-dipole approximation (Equation 2). The other tensors are reported as absolute values since these experiments are not reliably sensitive to the sign of the hyperfine tensors. Running the reaction with 1’−13C-AIR reveals that doublet x arises from the 1’-C (Figure 3, trace I; peaks at ≈12 and 14 MHz), indicating that the formate byproduct has not yet been formed (Figure 1). We do not have specifically 13C-labeled AIR at the 3’, 4’, or 5’ positions that would unambiguously assign doublets y and z to specific carbons on the ribose moiety. However, as stated earlier, using either the 5’,5’−2H2 nor 4’−2H-AIR substrates did not alter the 1H ENDOR spectrum or introduce new deuterium resonances, implying that their bonding partners, the 5’ and 4’ carbons, are also not strongly interacting with the unpaired electron spin.
Density Functional Theory of Imidazole-Like Radicals
To aid in our analysis of the experimentally-determined HFI described above, we explored a set of DFT-derived imidazole radical models. The predicted HFI for the most relevant of these models (Figure S8) are summarized in Tables 1–3 (Tables S4–S8 contain predicted HFI and spin populations for these models and others whose parameters matched those from experiment much less well). We performed benchmark calculations for NH3 and CH3 radicals as well as neutral and cation imidazole π-radicals (Tables 1 and S6).55,59 DFT predicts, almost quantitatively, the experimental 1H HFI for protons bound to carbon in these small molecule models. Interestingly, the predicted 1H HFI for nitrogen-bound protons is much less accurate (underestimated by ≈25%). Thus we expect calculated values for the amino group protons of a putative aminoimidazole radical to be significantly less accurate. Given these observations, in the Discussion section that follows, we arrive at our preferred model of the observed paramagnetic intermediate by relying heavily on the predicted HFI for protons bound to carbon and look more qualitatively at HFI for protons bound to nitrogen.
Table 1.
Experimental and DFT-Predicted 1H HFI for Imidazole-Type Radicals
| molecule | model | aiso 2-C 1H (MHz) | aiso 4-C 1H (MHz) | aiso 5-C 1H (MHz) |
|---|---|---|---|---|
| imidazole+ (exp)55 | 37.0 | 30.0 | 30.5 | |
| imidazole | A | −40.3 | −29.5 | −29.5 |
| imidazole+ | B | −37.4 | −28.4 | −35.6 |
| BzaF radical (this work) | −38.2 | −33.1 | n.a. | |
| 5-amino-Im | C | −29.5 | −24.3 | n.a. |
| 5-amino-Im+ 1-NH | D | −34.1 | −30.1 | n.a. |
| 1-N-methyl-5-amino-Im | G | −34.4 | −30.3 | n.a. |
| 1-N-glyceraldehyde-5-amino-Im | J | −32.0 | −25.5 | n.a. |
| 1-N-methanol-5-amino-Im | L | −33.0 | −29.6 | n.a. |
Table 3.
Experimental and DFT-Predicted 1’−13C HFI for 1-N Alkyl Imidazole Radicals
| molecule | model | ai 13C (MHz) |
|---|---|---|
| BzaF radical | [0.09, 1.91, 2.58] | |
| 1-N-methyl-Im | F | [−0.08, −0.40, −1.47] |
| 1-N-methyl-5-amino-Im | G | [0.51, 1.09, 1.85] |
| 1-N-formyl-5-ammonium-Im | P | [0.46, 1.30, 1.85] |
| 1-N-methanol-5-amino-Im | L | [0.74, 0.34, 1.54] |
| 1-N-acetyl-5-amino-Im (ϕ = 0°) | U | [0.40, 1.32, 1.63] |
| 1-N-(β-hydroxylpropan-α-al)-5-amino-Im (ϕ = 0°) | N | [0.85, 1.64, 2.61] |
| 1-N-glycolaldehyde-5-amino-Im (ϕ = 0°) | M | [0.83, 1.62, 2.56] |
Discussion.
Geometric and Electronic Structure of the Radical Intermediate
The two strongly coupled protons bound to carbons 2 and 4 of the imidazole ring are key in identifying this paramagnetic intermediate of the HBI synthase reaction as an imidazole-centered π-radical. For a π-radical, the isotropic hyperfine coupling measured for the in-plane protons arises from a spin polarization mechanism that is proportional to the spin in the 2pz orbital of the carbon to which the proton is bound (via the McConnell relation, Equation 4).42 Notably, however, the McConnell factor, QHCH, can vary from 56–84 MHz depending on the charge of the molecule and bonding environment for the carbon. Further, the protonation state of the imidazole nitrogens and the presence of other functional groups on the ring modulates these pz orbital populations. Therefore using DFT, we computed the HFI for several imidazole-like radicals and used these results to put forward some structural possibilities for the BzaF radical intermediate (Figure 4).
Figure 4.

Proposed structures for the radical intermediate.
With these considerations in mind, we first judged the quality of the various protonated and functionalized imidazole π-radical DFT models by how well they predicted the experimentally-determined 1H HFI for protons bound to the 2- and 4-carbons. The models we considered are illustrated in Figure S8. The predicted 1H HFI are given in Table 1 for selected structures. We found the most accurate predictions for the HFI of protons bound to 2- and 4-C to be for a neutral imidazole radical (model A), an imidazole cation radical (model B, protonated at one of the nitrogens), 5-amino imidazole protonated at 1-N (model D), 1-N-methyl imidazole (model F), 1-N-methyl-5-amino imidazole (model G), 1-N-methyl-5-amonium imidazole cation (model H), and 1-N-methanol-5-amino imidazole (model L). We note that 1-N-methyl-5-amino imidazole (model G) is predicted to have essentially identical spin distribution as the 5-amino imidazole protonated at 1-N (model D), and its corresponding methyl 13C HFI interaction is small, A = [0.51 1.09 1.85] MHz (aiso = 1.15 MHz, aaniso = [−0.64 −0.06 0.70] MHz) (Table 3). This is consistent with the measured aiso for 1’-C (1.5 MHz) of the BzaF radical, which is slightly larger and has a slightly more rhombic aaniso = [−0.38 −1.05 1.44] MHz. Alternatively, if a proton were bound to the imidazole nitrogen (1-N) as in model F, the predicted 1H HFI = [−1.21 3.36 4.02] MHz. This coupling would give rise to features in the 1H ENDOR spectrum that should be sensitive to exchange into deuterated solvent. Though, no features corresponding to a modestly coupled 1H are lost in the spectrum of the BzaF radical made in 2H2O (cf. Figure 3, traces A and C, and Figure S6). Therefore, we favor a structure for the BzaF radical that maintains 1’-C bound to 1-N (Figure 4).
From the above discussion and DFT models we can establish that the two strongly-coupled, solvent-exchangeable protons (doublet u and v, Figure 3, trace C) are unlikely to be coordinated to the imidazole nitrogens. In all cases we tested, except for the one in which both methyl and amino groups are bound to 5-C (model S), there is a node in the spin distribution at both nitrogens, leading to too small 1H hyperfine coupling for any protons bound at these positions. The largest predicted aiso for a proton bound to an imidazole nitrogen is ≈ 7 MHz for model E with the proton bound to 3-N. All the 5-aminoimidazole models (C through E and G through U) are predicted to have Mulliken spin population in the amino nitrogen pz orbital between 0.18 and 0.29, which would correspond to isotropic hyperfine couplings between 17 and 32 MHz for the amino protons (Table 2 and Table S5). The aaniso for these protons results from the dipolar interaction of the magnetic nucleus with the unpaired spin housed in the two lobes of the nitrogen pz orbital (Equation 3) plus dipolar interactions with the other spin bearing centers the π-radical (carbons 2, 4, and 5). The ENDOR spectral features u and v are simulated to be very axial whereas in-plane protons bound to a spin carrying nitrogen should give more rhombic HFI. For example, using a spin population of ρ = 0.2 for the nitrogen pz orbital and Equation 3, we would predict 1H aaniso = [−8.76 −0.56 9.32] MHz (using d = 0.68 Å and rN—H = 1.01 Å), with corresponding aiso = −13.4 to −15.0 MHz using QHNH = −67 to −75 MHz. The precise fate of the 5-amino group is not known except for that it can scramble with the 2-N of the imidazole, and that the eliminated nitrogen is perhaps released as ammonia.24 If the BzaF radical represented a species partway along this protonation pathway, the 1H HFI of the corresponding amino protons is predicted to become more axial as they move out of the nodal plane of the π-system. Up to this point, the BzaF radical intermediate is best described as an imidazole π-radical that likely retains 1’-C bound at 1-N and an amino group bound to 5-C that is participating in a hydrogen bonding interaction. In what follows we will construct a model of the positions of the other ribose carbons relative to this 1-N-methyl-5-amino imidazole radical. Only those models that most closely predict the spectroscopic observables are presented in the main text. Other, chemically possible, but unlikely based on spectroscopic grounds are presented in the SI.
Table 2.
Experimental and DFT-Predicted 1H HFI for Amino-Type Radicals
| molecule | model | aiso 1H (MHz) |
|---|---|---|
| NH2 (exp)60 | 76.5 | |
| NH2 (DFT) | −61.3 | |
| BzaF radical | ≈ −12 | |
| 5-amino-Im | C | −17.4 |
| 1-N-methyl-5-amino-Im | G | −20.0 |
| 1-N-glycoladehyde-5-amino-Im | M | −27.4 |
| 1-N-(β-hydroxylpropan-α-al)-5-amino-Im | N | −27.1 |
Recall that the measured HFI for 2’−13C is [19.6 20.3 23.0] MHz. This largely isotropic hyperfine coupling is perhaps indicative of a covalent interaction with the imidazole radical. However, computational model S, which illustrates a possible intermediate in which the 2’-C has formed a bond to the 5 carbon as in the final product, yields 1H and 13C HFI values that are inconsistent with what is measured due to the breaking of ring conjugation. Therefore, we prefer a description that has the 2’-C in close enough proximity to 5-C to interact with the magnetic orbital, but without formation of a bond. This is a similar mechanism to that causing the transannular hyperfine interaction seen in Mo(V) complexes59 and hyperfine induced by magnetic nuclei being in Van der Waals contact with a paramagnetic species.61 The anisotropic hyperfine is the sum of local (Tloc) and non-local (inter-atom dipolar, Tnon-loc) contributions. The non-local term consists of the sum of all the through-space dipolar interactions of the 2’-C with the four main spin-bearing centers on the imidazole fragment—Nam, 5-C, 4-C, and 2-C—which are predicted by the McConnell relation and deduction to possess approximately 0.18(+/−0.08), 0.20(−/+0.08), 0.29, and 0.33 spins in their respective pz orbitals for a model resembling G, P, or L. If we assume that the observed hyperfine anisotropy is wholly due to non-local contributions, we can calculate the closest possible positions of the 2’-C about the imidazole radical (Figure S9). These calculations use an eight point-dipole approximation in which each of the four spin-carrying pz orbitals are approximated as two spin dipoles situated at ±0.68 Å away from the parent nucleus with half of the corresponding spin population.40,41,43,62 In the final product of the HBI synthase reaction, we know that the 2’-C forms a bond with the 5-C. A subset of the points are between 2.0 and 3.5 Å of 5-C (Figure S9B and S9C)—geometric solutions that are consistent with all measured hyperfine interactions and illustrate how the radical intermediate could be structurally constrained by the BzaF enzyme to be pre-organized for breaking the 1’-C—2’-C bond and forming subsequent bonds. Some of these positions for the 2’-C are also within a normal C—C bond length (1.525±0.05 Å) of the 1’-C bound to 1-N (Figure S9C) leaving open the possibility that the1’-C—2’-C bond remains intact even though 1’-C is ultimately lost as formate (Figure 1).24 A few examples of this possibility are shown in Figure 4 structure A, C, and D. As noted above the McConnell QCH value used to estimate spin density on each carbon has some variance based on the angle of the other bonds to the carbon-hydrogen bond in question. This variance in QNH is even larger for estimating the spin density on the nitrogen. These small variations lead to the locations of 2’-C being an estimate of possibilities.
There are two remaining 13C hyperfine tensors that are unassigned to a specific atom in the AIR substrate. The first tensor is simulated as having hyperfine components of [1.25 1.35 0.80] MHz. The non-zero aiso of 1.1 MHz suggests that this hyperfine coupling tensor belongs to a carbon that is in close proximity or covalently bound to a minor spin carrying center for example 2’-C (ρ = 0.01). Based on this assumption we assign this tensor to 3’-C. The second unassigned 13C hyperfine interaction is simulated with components of [−0.2 −0.2 +0.4] MHz. That this hyperfine interaction is axial and lacks an isotropic contribution suggests it arises from a through-space point dipole–dipole interaction. Using Equation 2, we can estimate a distance of 4.63 Å separating this 13C nucleus from the geometrical centroid of the spin distributed over the aminoimidazole. This distance is reasonable for an atom at least three bonds away from the aminoimidazole and is thus consistent with the coupling arising from either the 4’ or 5’ carbon. Since the proton region of the ENDOR spectrum is not affected by deuteration at 5’-C or 4’-C and site specific 13C labeling was not available at these positions, we cannot unambiguously assign this hyperfine interaction to either carbon.
The aggregate of these observations allows us to propose a series of structures for the HBI synthase radical intermediate (Figure 4). The non-exchangeable 1H HFI establish that the radical character is mostly born by the π-system of the imidazole ring, a feature common to each of the models. The 13C hyperfine coupling of 1’-C requires its being bound to 1-N and the anisotropy of this interaction is best predicted using hydroxylpropanal or glycolaldehyde functional groups at this position, akin to Radicals a and c (DFT models N and M in Table 3). The strong, relatively isotropic 13C HFI observed for 2’-C points to its close proximity a lobe of the spin-carrying orbital of the imidazole radical and the 2’−13C hyperfine anisotropy confirms this close association as shown in Figure S9. The two remaining 13C HFI we measured could not be definitively assigned to a particular carbon center (they belong to either the 3’-, 4’-, or 5’-C). Interestingly, these two tensors have nearly identical 13C hyperfine anisotropies meaning their corresponding carbon nuclei are the same effective distance (r ≈ 4.6 Å) from the imidazole radical. The models presented in Figure 4 are flexible enough (owing to there being many single bonds and the potential for 1’-C—2’-C bond cleavage) that the ribose carbons could adopt positions that satisfy the above structural constraints imposed by the various 13C couplings.
To discriminate between the proposed radical structures in Figure 4, we have to rely on mechanistic arguments. A reaction mechanism has been previously described for HBI synthase that was consistent with extensive labeling studies;24 however, the radical character was presumed to stay on the ribose moiety throughout. Given that the current EPR results unequivocally show that an imidazole radical intermediate is formed, this mechanistic proposal must be updated and can be readily modified to incorporate the intermediacy of Radical a, but not Radicals b, c or d shown in Figure 4. In our revised mechanistic proposal (Figure 5), hydrogen atom abstraction from 5’-C of AIR 1 gives the substrate radical 4. This then undergoes a beta scission reaction to generate 5. Electron transfer from the electron rich aminoimidazole to the enol phosphate radical cation of 5 gives 6, the proposed radical observed in the EPR experiment. Radical facilitated cleavage of the aminal of 6 gives 7. Addition to the resulting aminoimidazole radical to 5’-C of the enol phosphate of 7’ gives 8. Loss of phosphate, by a beta scission, gives the radical cation 9 which then tautomerizes to 10. Hydrogen atom abstraction (possibly from deoxyadenosine 3) gives 11. Imine hydrolysis to 12, cyclization to 13 and sequential loss of ammonia and formic acid give 15. Cyclization to 16, loss of water and two tautomerizations give 19. Loss of water from 19, followed by two tautomerization reactions complete the formation of HBI 22. The fragmentation that occurs going from 6 to 7 could be triggered by proton donation to 3-N of the imidazole, a step that could be slow, allowing for build-up of the observed intermediate.
Figure 5.

Mechanistic proposal for the formation of HBI via a pathway that goes through a radical intermediate corresponding to Structure a (Figure 4).
Conclusions.
The dizzying array of chemistries catalyzed by radical SAM enzymes is well illustrated by the pair of radical SAM enzymes BzaF and ThiC that each employ the adenosyl radical to abstract the pro-S atom from the 5’ carbon of the AIR substrate, but yield the dramatically different products 5-hydroxybenzimidazole (HBI) and hydroxymethylpyrimidine phosphate (HMPP), respectively.30,63,64 These two proteins are largely homologous with 44% sequence identity and 66% sequence similarity in Desulfuromonas acetoxidans. This implies that the precise identities of amino acids in the substrate-binding region are crucial in dictating which pathway the initial substrate radical proceeds down. In a first step toward unraveling this predestination, we have used freeze quench EPR techniques in combination with DFT to trap and characterize a radical intermediate in the BzaF-catalyzed biosynthesis of HBI. The measured 1H HFI point to its identity as an aminoimidazole π–radical. 13C–labeling of the ribose moiety reveals that the 2’-C is in Van der Waals contact with the π–radical, and that the 1’-C is likely still bound to the 1-N of the aminoimidazole nitrogen. Another carbon, likely the 5’-C, sits approximately 4.63 Å from the center of the aminoimidazole spin density. No 31P hyperfine interaction was detected, but that does not preclude the phosphate group from still being bound to the ribose fragment if it is too distant from the aminoimidazole to be detected. In the crystal structure of ThiC with AIR (PDB: 4S27), the 5’-C is 4.0 Å from 2-C, 5.2 Å from 5-C, and 6.0 Å from 4-C of the aminoimidazole moiety. If BzaF binds AIR similarly to ThiC, then these distances and the distribution of the spin density are consistent with the estimate from the hyperfine interaction.
One important difference of the ThiC mechanism from that for BzaF is that one equivalent of SAM appears to be able to perform two H-atom abstractions from AIR in ThiC.30 The first H-atom abstraction in ThiC is identical to BzaF where the pro-S hydrogen of the 5’ carbon is targeted. Both enzymes direct AIR to lose the 2’ hydroxyl group and to lose the 1’ carbon as a formate. We can speculate that both enzymes guide this portion of the reaction in a similar manner to encourage these eliminations. In the crystal structure of ThiC, the phosphate group is observed to be the primary anchor for the substrate;65 the rest of the substrate participates in relatively few hydrogen bonds. One can speculate that the active sites of both enzymes do not bind the ribose or aminoimidazole moieties too restrictively in order to accommodate the large movements needed to form their respective final products. A homology model of HBI synthase generated using the ThiC crystal structure notes only three major differences in the hydrogen bonding network to AIR:24 the 3’-O contact Asn228 in ThiC is replaced by Ser66 in BzaF; Thr320 that hydrogen bonds to Glu422 which in turns bonds to 2’-O is replaced with Ala161; Asp383 which possibly hydrogen bonds with 3-N is replaced with Asn227. Somehow, these modest differences yield a dramatic effect on the product outcome.
Supplementary Material
Acknowledgements.
Funding provided by the National Institutes of Health: National Institute of General Medical Sciences (R01GM104543) to R.D.B. and the National Institute of Diabetes and Digestive and Kidney Diseases (DK44083) to T.P.B. This research was also supported by the Robert A. Welch Foundation (A-0034 to T.P.B.). The authors would like to thank Daniel L. M. Suess for useful discussions.
Footnotes
Supporting information contains additional EPR spectra, synthesis of 2,4-2H2-AIR, DFT models, and select amino acid sequences.
References.
- (1).Chemistry and Biochemistry of B12; Banerjee R, Ed.; Wiley: New York, 1999. [Google Scholar]
- (2).Kolhouse JF; Utley C; Allen RH Isolation and Characterization of Methylmalonyl-CoA Mutase from Human Placenta. J. Biol. Chem 1980, 255 (7), 2708–2712. [PubMed] [Google Scholar]
- (3).Toraya T Radical Catalysis in Coenzyme B12 -Dependent Isomerization (Eliminating) Reactions. Chem. Rev 2003, 103 (6), 2095–2128. [DOI] [PubMed] [Google Scholar]
- (4).Barker HA; Weissbach H; Smyth RD A COENZYME CONTAINING PSEUDOVITAMIN B12. Proc. Natl. Acad. Sci 1958, 44 (11), 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ludwig and ML; Matthews RG STRUCTURE-BASED PERSPECTIVES ON B12 -DEPENDENT ENZYMES. Annu. Rev. Biochem 1997, 66 (1), 269–313. [DOI] [PubMed] [Google Scholar]
- (6).Stubbe JA Mechanism of B12-Dependent Ribonucleotide Reductase. Mol. Cell. Biochem 1983, 50 (1), 25–45. [DOI] [PubMed] [Google Scholar]
- (7).Gerfen GJ; Licht S; Willems J-P; Hoffman BM; Stubbe J Electron Paramagnetic Resonance Investigations of a Kinetically Competent Intermediate Formed in Ribonucleotide Reduction: Evidence for a Thiyl Radical-Cob(II)Alamin Interaction. J. Am. Chem. Soc 1996, 118 (35), 8192–8197. [Google Scholar]
- (8).Banerjee RV; Matthews RG Cobalamin-Dependent Methionine Synthase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 1990, 4 (5), 1450–1459. [DOI] [PubMed] [Google Scholar]
- (9).Chen LH; Liu ML; Hwang HY; Chen LS; Korenberg J; Shane B Human Methionine Synthase. CDNA Cloning, Gene Localization, and Expression. J. Biol. Chem 1997, 272 (6), 3628–3634. [PubMed] [Google Scholar]
- (10).Zhang Q; van der Donk WA; Liu W Radical-Mediated Enzymatic Methylation: A Tale of Two SAMS. Acc. Chem. Res 2012, 45 (4), 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Parthasarathy A; Stich TA; Lohner ST; Lesnefsky A; Britt RD; Spormann AM Biochemical and EPR-Spectroscopic Investigation into Heterologously Expressed Vinyl Chloride Reductive Dehalogenase (VcrA) from Dehalococcoides Mccartyi Strain VS. J. Am. Chem. Soc 2015, 137 (10), 3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sjuts H; Fisher K; Dunstan MS; Rigby SE; Leys D Heterologous Expression, Purification and Cofactor Reconstitution of the Reductive Dehalogenase PceA from Dehalobacter Restrictus. Protein Expr. Purif 2012, 85 (2), 224–229. [DOI] [PubMed] [Google Scholar]
- (13).Schmitz RPH; Wolf J; Habel A; Neumann A; Ploss K; Svatos A; Boland W; Diekert G Evidence for a Radical Mechanism of the Dechlorination of Chlorinated Propenes Mediated by the Tetrachloroethene Reductive Dehalogenase of Sulfurospirillum Multivorans. Environ. Sci. Technol 2007, 41 (21), 7370–7375. [DOI] [PubMed] [Google Scholar]
- (14).van de Pas B; Gerritse J; de Vos W; Schraa G; Stams A Two Distinct Enzyme Systems Are Responsible for Tetrachloroethene and Chlorophenol Reductive Dehalogenation in Desulfitobacterium Strain PCE1. Arch. Microbiol 2001, 176 (3), 165–169. [DOI] [PubMed] [Google Scholar]
- (15).van de Pas BA; Smidt H; Hagen WR; van der Oost J; Schraa G; Stams AJM; de Vos WM Purification and Molecular Characterization Ofortho-Chlorophenol Reductive Dehalogenase, a Key Enzyme of Halorespiration in Desulfitobacterium Dehalogenans. J. Biol. Chem 1999, 274 (29), 20287–20292. [DOI] [PubMed] [Google Scholar]
- (16).Magnuson JK; Romine MF; Burris DR; Kingsley MT Trichloroethene Reductive Dehalogenase from Dehalococcoides Ethenogenes: Sequence of TceA and Substrate Range Characterization. Appl. Environ. Microbiol 2000, 66 (12), 5141–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Roth J; Lawrence J; Bobik T COBALAMIN (COENZYME B12): Synthesis and Biological Significance. Annu. Rev. Microbiol 1996, 50 (1), 137–181. [DOI] [PubMed] [Google Scholar]
- (18).Brink C; Hodgkin DC; Lindsey J; Pickworth J; Robertson JH; White JG Structure of Vitamin B12: X-Ray Crystallographic Evidence on the Structure of Vitamin B12. Nature 1954, 174 (4443), 1169–1171. [DOI] [PubMed] [Google Scholar]
- (19).Drennan CL; Huang S; Drummond JT; Matthews RG; Lidwig ML How a Protein Binds B12: A 3.0 A X-Ray Structure of B12-Binding Domains of Methionine Synthase. Science 1994, 266 (5191), 1669–1674. [DOI] [PubMed] [Google Scholar]
- (20).Svetlitchnaia T; Svetlitchnyi V; Meyer O; Dobbek H Structural Insights into Methyltransfer Reactions of a Corrinoid Iron-Sulfur Protein Involved in Acetyl-CoA Synthesis. Proc. Natl. Acad. Sci 2006, 103 (39), 14331–14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).St. Maurice M; Mera P; Park K; Brunold TC; Escalante-Semerena JC; Rayment I Structural Characterization of a Human-Type Corrinoid Adenosyltransferase Confirms That Coenzyme B12 Is Synthesized through a Four-Coordinate Intermediate† ‡. Biochemistry 2008, 47 (21), 5755–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Moore SJ; Warren MJ The Anaerobic Biosynthesis of Vitamin B12. Biochem. Soc. Trans 2012, 40 (3), 581–586. [DOI] [PubMed] [Google Scholar]
- (23).Taga ME; Larsen NA; Howard-Jones AR; Walsh CT; Walker GC BluB Cannibalizes Flavin to Form the Lower Ligand of Vitamin B12. Nature 2007, 446 (7134), 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mehta AP; Abdelwahed SH; Fenwick MK; Hazra AB; Taga ME; Zhang Y; Ealick SE; Begley TP Anaerobic 5-Hydroxybenzimidazole Formation from Aminoimidazole Ribotide: An Unanticipated Intersection of Thiamin and Vitamin B12 Biosynthesis. J. Am. Chem. Soc 2015, 137 (33), 1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hazra AB; Han AW; Mehta AP; Mok KC; Osadchiy V; Begley TP; Taga ME Anaerobic Biosynthesis of the Lower Ligand of Vitamin B12. Proc. Natl. Acad. Sci 2015, 112 (34), 10792–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen D; Walsby C; Hoffman BM; Frey PA Coordination and Mechanism of Reversible Cleavage of S-Adenosylmethionine by the [4Fe-4S] Center in Lysine 2,3-Aminomutase. J. Am. Chem. Soc 2003, 125 (39), 11788–11789. [DOI] [PubMed] [Google Scholar]
- (27).Magnusson OT; Reed GH; Frey PA Spectroscopic Evidence for the Participation of an Allylic Analogue of the 5’-Deoxyadenosyl Radical in the Reaction of Lysine 2,3-Aminomutase. J. Am. Chem. Soc 1999, 121 (41), 9764–9765. [Google Scholar]
- (28).Magnusson OT; Reed GH; Frey PA Characterization of an Allylic Analogue of the 5’-Deoxyadenosyl Radical: An Intermediate in the Reaction of Lysine 2,3-Aminomutase†. Biochemistry 2001, 40 (26), 7773–7782. [DOI] [PubMed] [Google Scholar]
- (29).Tate S; Kubo Y; Ono A; Kainosho M Stereospecific Measurements of the Vicinal 1H-31P Coupling Constants for the Diastereotopic C5’ Methylene Protons in a DNA Dodecamer with a 13C/2H Doubly Labeled Residue. Conformational Analysis of the Torsion Angle .Beta. J. Am. Chem. Soc 1995, 117 (27), 7277–7278. [Google Scholar]
- (30).Chatterjee A; Hazra AB; Abdelwahed S; Hilmey DG; Begley TPA “Radical Dance” in Thiamin Biosynthesis: Mechanistic Analysis of the Bacterial Hydroxymethylpyrimidine Phosphate Synthase. Angew. Chem. Int. Ed 2010, 49 (46), 8653–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lanz ND; Grove TL; Gogonea CB; Lee K-H; Krebs C; Booker SJ RlmN and AtsB as Models for the Overproduction and Characterization of Radical SAM Proteins In Methods in Enzymology; Elsevier, 2012; Vol. 516, pp 125–152. [DOI] [PubMed] [Google Scholar]
- (32).Bhandari DM; Fedoseyenko D; Begley TP Mechanistic Studies on Tryptophan Lyase (NosL): Identification of Cyanide as a Reaction Product. J. Am. Chem. Soc 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Joshi S; Mahanta N; Fedoseyenko D; Williams H; Begley TP Aminofutalosine Synthase: Evidence for Captodative and Aryl Radical Intermediates Using β-Scission and SRN 1 Trapping Reactions. J. Am. Chem. Soc 2017, 139 (32), 10952–10955. [DOI] [PubMed] [Google Scholar]
- (34).Wang Y; Schnell B; Baumann S; Müller R; Begley TP Biosynthesis of Branched Alkoxy Groups: Iterative Methyl Group Alkylation by a Cobalamin-Dependent Radical SAM Enzyme. J. Am. Chem. Soc 2017, 139 (5), 1742–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sienkiewicz A; Smith BG; Veselov A; Scholes CP Tunable Q-Band Resonator for Low Temperature Electron Paramagnetic Resonance/Electron Nuclear Double Resonance Measurements. Rev. Sci. Instrum 1996, 67 (6), 2134. [Google Scholar]
- (36).Davies ER A New Pulse Endor Technique. Phys. Lett. A 1974, 47 (1), 1–2. [Google Scholar]
- (37).Mims WB Pulsed Endor Experiments. Proc. R. Soc. Math. Phys. Eng. Sci 1965, 283 (1395), 452–457. [Google Scholar]
- (38).Mims WB Some Applications of Pulse Techniques in EPR. Appl. Magn. Reson 1991, 2 (4), 595–609. [Google Scholar]
- (39).Stoll S; Schweiger A EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson 2006, 178 (1), 42–55. [DOI] [PubMed] [Google Scholar]
- (40).Stoll S; NejatyJahromy Y; Woodward JJ; Ozarowski A; Marletta MA; Britt RD Nitric Oxide Synthase Stabilizes the Tetrahydrobiopterin Cofactor Radical by Controlling Its Protonation State. J. Am. Chem. Soc 2010, 132 (33), 11812–11823. [DOI] [PubMed] [Google Scholar]
- (41).Gordy W Theory and Applications of Electron Spin Resonance; Techniques of chemistry ; v. 15; Wiley: New York, 1980. [Google Scholar]
- (42).McConnell HM; Chesnut DB Theory of Isotropic Hyperfine Interactions in π-Electron Radicals. J. Chem. Phys 1958, 28 (1), 107. [Google Scholar]
- (43).McConnell HM; Strathdee J Theory of Anisotropic Hyperfine Interactions in π -Electron Radicals. Mol. Phys 1959, 2 (2), 129–138. [Google Scholar]
- (44).Neese F The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2 (1), 73–78. [Google Scholar]
- (45).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4 (1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Rappe AK; Casewit CJ; Colwell KS; Goddard WA; Skiff WM UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc 1992, 114 (25), 10024–10035. [Google Scholar]
- (47).Vosko SH; Wilk L; Nusair M Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys 1980, 58 (8), 1200–1211. [Google Scholar]
- (48).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38 (6), 3098–3100. [DOI] [PubMed] [Google Scholar]
- (49).Perdew JP Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33 (12), 8822–8824. [DOI] [PubMed] [Google Scholar]
- (50).Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7 (18), 3297. [DOI] [PubMed] [Google Scholar]
- (51).Neese F; Wennmohs F; Hansen A; Becker U Efficient, Approximate and Parallel Hartree–Fock and Hybrid DFT Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree–Fock Exchange. Chem. Phys 2009, 356 (1–3), 98–109. [Google Scholar]
- (52).Rega N; Cossi M; Barone V Development and Validation of Reliable Quantum Mechanical Approaches for the Study of Free Radicals in Solution. J. Chem. Phys 1996, 105 (24), 11060. [Google Scholar]
- (53).Becke AD A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys 1993, 98 (2), 1372. [Google Scholar]
- (54).Becke AD Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98 (7), 5648. [Google Scholar]
- (55).Kar L; Bernhard WA The Imidazole π Cation and Barbital π Anion Trapped in a Cocrystalline Complex X-Irradiated at 12 K: An ESR–ENDOR Study. J. Chem. Phys 1980, 73 (8), 3625. [Google Scholar]
- (56).Ngo FQ Free Radical Formation in X-Irradiated Histidine HCl. J. Chem. Phys 1974, 60 (9), 3373. [Google Scholar]
- (57).Carrington A; McLachlan AD Introduction to Magnetic Resonance: With Applications to Chemistry and Chemical Physics; A Science paperback; Chapman and Hall ; Wiley: London : New York, 1979. [Google Scholar]
- (58).Weil JA; Bolton JR Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, 2nd ed.; Wiley-Interscience: Hoboken, N.J, 2007. [Google Scholar]
- (59).Stiefel EI; Newton W; Pariyadath, N. EPR Ligand Superhyperfine Interactions in Mononuclear Mo(V) Complexes: Implications for Mo Enzymes. J. Common Met 1977, 54 (2), 513–525. [Google Scholar]
- (60).Vansant EF; Lunsford JH Electron Paramagnetic Resonance Study of Copper(II)-Ammonia Complexes in Y-Type Zeolites. J. Phys. Chem 1972, 76 (20), 2860–2865. [Google Scholar]
- (61).Horitani M; Byer AS; Shisler KA; Chandra T; Broderick JB; Hoffman BM Why Nature Uses Radical SAM Enzymes so Widely: ENDOR Studies of Lysine 2,3-Aminomutase Show the 5’-DAdo• ‘Free Radical’ Is Never Free. J. Am. Chem. Soc 2015, 150429153813005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Stull JA; Stich TA; Service RJ; Debus RJ; Mandal SK; Armstrong WH; Britt RD 13 C ENDOR Reveals That the D1 Polypeptide C-Terminus Is Directly Bound to Mn in the Photosystem II Oxygen Evolving Complex. J. Am. Chem. Soc 2010, 132 (2), 446–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Martinez-Gomez NC; Poyner RR; Mansoorabadi SO; Reed GH; Downs DM Reaction of AdoMet with ThiC Generates a Backbone Free Radical†. Biochemistry 2009, 48 (2), 217–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Chatterjee A; Li Y; Zhang Y; Grove TL; Lee M; Krebs C; Booker SJ; Begley TP; Ealick SE Reconstitution of ThiC in Thiamine Pyrimidine Biosynthesis Expands the Radical SAM Superfamily. Nat. Chem. Biol 2008, 4 (12), 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Fenwick MK; Mehta AP; Zhang Y; Abdelwahed SH; Begley TP; Ealick SE Non-Canonical Active Site Architecture of the Radical SAM Thiamin Pyrimidine Synthase. Nat. Commun 2015, 6, 6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.