

Abstract
A mechanically strong composite hydrogel was produced based on the interpenetrating network (IPN) between gelatin and silk fibroin. When two layers of IPN was created, the resulting hydrogel exhibited much improved mechanical properties. This hydrogel is biodegradable, non-cytotoxic and allows cell adhesion and proliferation on the surface.
Graphical Abstract
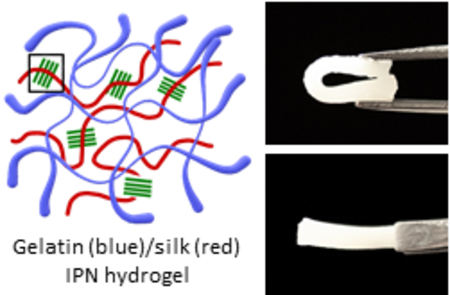
A mechanically tough and biofunctional hydrogel was created by the multi-interpenetrating network between gelatin and silk fibroin.
Hydrogel is a crosslinked network of hydrophilic polymers. Due to their high water contents, hydrogels mimic the natural extracellular matrix (ECM) and can serve as scaffolds for a variety of applications in tissue engineering and regenerative medicine1–6. However, the high water content also makes it challenging to produce mechanically robust hydrogels. Applications in regenerating load-bearing tissues, such as bone and cartilage require the materials that can withstand high stresses7, 8. In bone tissue engineering, for instance, stem cells (e.g. mesenchymal stem cells) should be seeded on stiff substrates for proper osteogenic differentiation9. Even for soft tissues such as skin and cornea, the biomaterials should withstand mechanical stresses10, 11. Most hydrogels fail to meet the mechanical requirement of such applications.
Several strategies have been developed to improve the mechanical properties of hydrogels. The incorporation of nanomaterials such as inorganic nanoparticles12, 13, graphene oxides14, and nanoclays15–17 in the hydrogel has been demonstrated to increase the stiffness of the hydrogel. However, these materials suffer from high cost of production18, cytotoxicity19, 20 and slow (or non-)degradation21.
Alternatively, forming interpenetrating network (IPN) between two independently crosslinked polymers results in much stronger hydrogels compared to the hydrogels made of individual polymers22–26. However, most of the IPN hydrogels in the literature, utilized synthetic polymers such as polyacrylamide or poly(ethylene glycol) as a component of IPN hydrogels, which is less desirable than the natural polymers. Even when natural polymers were used to make IPN hydrogels, chemical modifications were added to the polymers, such as methacrylate groups, for photo-crosslinking27. This complicates the synthesis of hydrogels and increases the cost.
Here, we introduce an IPN hydrogel made of two widely-used natural polymers – gelatin and silk fibroin (Figure 1). In order to enhance the mechanical properties, two layers of IPNs were formed in the hydrogel successively. Gelatin was chosen due to its ready-availability and its excellent bioactivity, which allows cell adhesion and proliferation without addition of external cell adhesive ligands28, 29. Silk fibroin was chosen because of its excellent mechanical properties and proven biocompatibility30, 31. Previously reported gelatin-silk IPN hydrogels were either purely physically crosslinked32, resulting in sub-optimal mechanical strength of the hydrogel, or involved chemical modifications to the proteins27 which complicates the process and increases the cost. The method described in this communication is simple, cost-effective, and the resulting hydrogels are mechanically robust and biofunctional.
Figure 1.
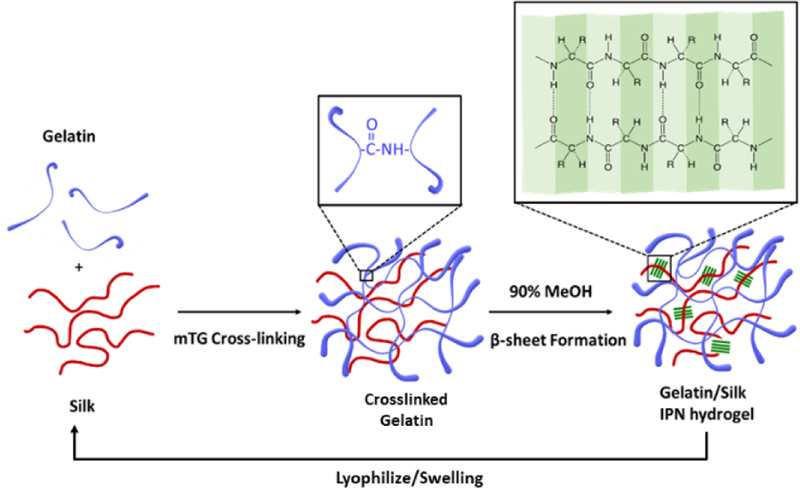
Schematic of the synthesis of gelatin-silk fibroin IPN hydrogel. Gelatin is crosslinked covalently by the action of mTG, and silk is crosslinked physically by the formation of β-sheet structures. This process of IPN formation is repeated on the same hydrogel.
To make an IPN hydrogel, an aqueous mixture of gelatin and silk fibroin was first treated by microbial transglutaminase (mTG), which creates covalent crosslinks between glutamine and lysine within gelatin33. This results in a somewhat weak hydrogel. In addition to the covalent crosslinks within gelatin, silk fibroin is crosslinked physically by the induction of beta-sheets by immersing the hydrogel in 90% ethanol. The resulting IPN hydrogel is swelled in water and lyophilized, and the second layer of IPN is formed by swelling the lyophilized IPN in the gelatin-silk fibroin mixture and repeating the dual crosslinking procedure. (The detailed methods are provided in the ESI.) For convenience of our further discussions, the gelatin-silk fibroin hydrogel with only one IPN will be called “1-IPN” and the hydrogel with two layers of IPN will be called “2-IPN”.
Rheology was used as indirect evidence for the formation of covalent crosslinks within gelatin by the action of mTG (Figure 2a). G’ and G’’ of the mixture of gelatin and silk fibroin in the presence of mTG continued to rise with time, with G’ reaching ~ 10,000 Pa after 1 hour. On the other hand, G’ and G’’ remained two orders of magnitude smaller without mTG (< 100 Pa), despite a constant increase in G’ and G’’. This increase in moduli without mTG is most likely due to the β-sheet formation of silk fibroin at an elevated temperature (37 °C)34. The substantial difference in shear moduli by mTG is evidence for the covalent bond formation within gelatin by mTG. Both G’ and G’’ increased with angular frequencies, which is typical of hydrogels that are crosslinked by both covalent and physical crosslinks (Figure S1a)35. Although the hydrogel was not treated with ethanol during the rheology, β-sheet may have formed in silk at an elevated temperature, and gelatin itself naturally forms physical crosslinks by hydrogen bonding. In the temperature sweep, G’ and G’’ of mTG-crosslinked gelatin-silk fibroin hydrogel decreased with temperature (Figure S1b). This is because the physical crosslinks within gelatin dissociated as the thermal energy increased. However, G’ and G’’ reached a plateau instead of continuing to decrease, which proves the existence of permanent covalent crosslinks. The shear moduli of gelatin-silk fibroin mixture without mTG increased with temperature then decreased at high temperatures. The initial increase is likely due to the formation of beta sheets as temperature increased, but at high temperatures, the physical crosslinks of gelatin dissociates, resulting in a decrease in shear moduli. Based on the substantial increase in G’ and the melting resistance of the hydrogel at elevated temperatures, we conclude that the addition of mTG resulted in covalent crosslinks within the gelatin polymers.
Figure 2.
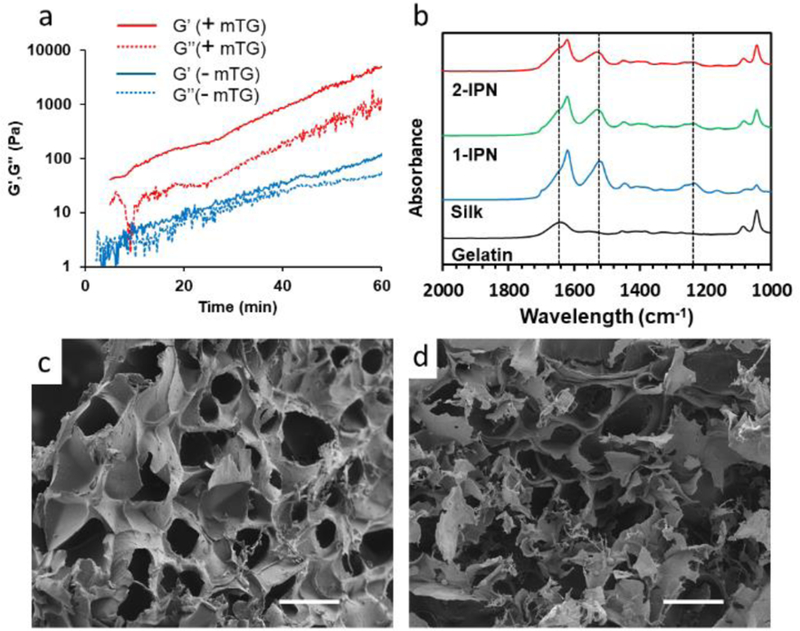
(a) Shear moduli of an aqueous mixture of gelatin and silk fibroin as a function of time. (b) FT-IR of hydrogels. The three vertical dotted lines are (from left to right) amide I, amide II and amide II peaks. (c,d) SEM images of (c) 1-IPN and 2-IPN hydrogels. Scale bar = 100 µm.
The formation of β-sheet structure of silk fibroin within the IPN hydrogels was confirmed by FT-IR (Figure 2b, Figure S2). Gelatin-only hydrogel that was crosslinked by mTG exhibited a random coil conformation (amide I at 1640 cm−1). Silk fibroin hydrogel that was crosslinked by β-sheet showed a shift of amide I peaks to 1620 cm−1. Additionally, new peaks at 1530 cm−1 (amide II) and 1260 cm−1 (amide III) appeared corresponding to the β-sheets. Both 1-IPN and 2-IPN hydrogels showed similar peaks that corresponded to β-sheets due to the presence of physical crosslinks within silk fibroin. There was no significant difference between 1-IPN and 2-IPN because the molecular compositions were similar.
Taken together, the results from rheology and FT-IR spectra prove that the IPN hydrogels had both covalent crosslinks (within gelatin) and physical crosslinks (within silk fibroin).
Scanning electron microscope (SEM) images of the IPN hydrogels show the presence of interconnected micropores, which is a characteristic of lyophilized hydrogels (Figure 2c,d). We did not observe significant morphological differences between 1-IPN and 2-IPN.
The pore size ranged between 40–60 µm. There was no distinct difference between 1-IPN and 2-IPN hydrogels. The IPN hydrogels had similar structures as silk only hydrogel (Figure S3). This microporous structure provides the space for cellular growth within the hydrogels.
The IPN hydrogels were made of natural protein polymers – gelatin and silk fibroin- and can naturally degrade by various proteolytic enzymes and phagocytic cells if implanted in an in vivo environment36, 37. One important mechanism of degradation is through the actions of matrix metalloproteinases (MMPs). Biodegradation of the IPN hydrogels was measured by incubating them in collagenase type II solution (0.15 % w/v) at 37 °C by measuring the weight of the residual gels (Figure S4). Collagenase II is a bacterial analogue of MMPs. As expected, gelatin degraded most rapidly because gelatin possesses the amino sequences that are recognized by collagenases38. There was no significant decrease in the amount of residual gel for silk hydrogel. This result is consistent with the fact that silk fibroin is minimally sensitive to collagenases36. 1-IPN and 2-IPN degraded at similar rates as silk hydrogel although it contained 50% gelatin by weight. Slower degradation of IPNs than the gelatin-only hydrogel is important because the gelatin hydrogels can degrade much more quickly than desired39.
The IPN hydrogels were highly elastic and mechanically tough. As can be seen in Figure 3a, b, the hydrogel restored its original shape without any damage even after significant bending. Figure 3c,d (and Figure S5 for more complete compressive modulus information) is the summary of the mechanical tests in the compression mode. Disc shaped hydrogels were compressed at a constant velocity (5 mm/min) as the force applied to the hydrogel was measured. Stress-strain curves clearly show that IPN hydrogels are much stiffer than silk only or gelatin only hydrogels (Figure 3c). The gelatin hydrogel was the softest with the compressive modulus 5.87 kPa at 10% strain, 22.6 kPa at 30% strain and 101 kPa at 50% strain (Figure 3d). Compressive modulus of 1-IPN hydrogel was much higher with its compressive modulus being 848 kPa at 50% strain. An addition of another layer of IPN to 1-IPN further increased its stiffness to 1.56 GPa at 50% strain. The compressive moduli of 2-IPN hydrogels are comparable to or greater than other silk-based hydrogels reported in the literature27, 31, 40, proving the benefit of forming multiple IPNs within a hydrogel. The compressive moduli of 2-IPN is comparable to that of soft tissues such as cartilage and skin (~0.1–1 MPa)41, and some hard tissues such as cancellous bone (1–10 MPa)42.
Figure 3.
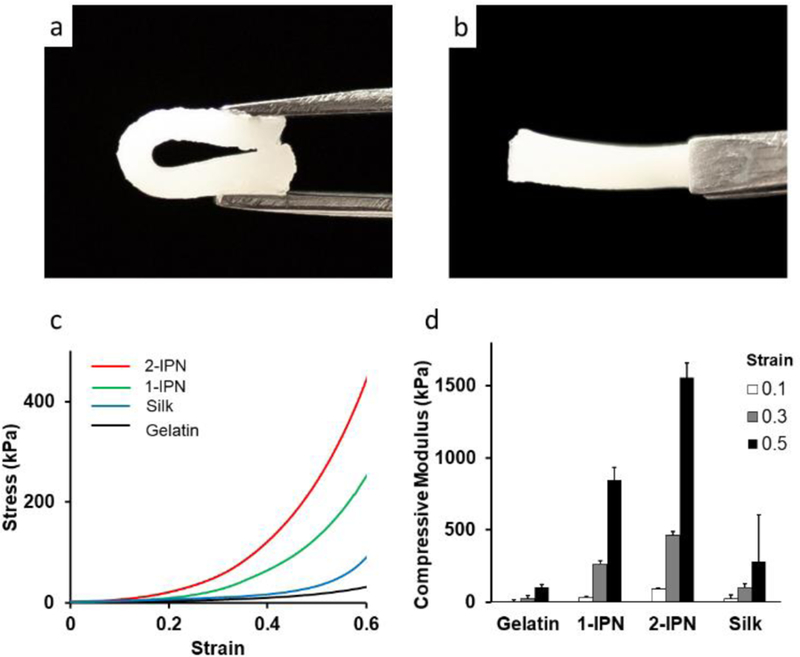
(a,b) Demonstration of the elasticity of the IPN hydrogel. 2-IPN was completely bent, restored its original shape when released. (c) Strain-stress curve from the compressive mechanical tests. (d) Compressive modulus at 0.1, 0.3 and 0.5 strain.
Biocompatibility of the IPN hydrogels was tested by culturing human dermal fibroblasts (hDFs) on the hydrogels in vitro (Figure 4a). At all time points (1, 3, 7 days post seeding), hDF proliferation was the highest on gelatin hydrogels, followed by 2-IPN, 1-IPN and silk hydrogels. Only the silk hydrogel group showed statistically significant differences compared to the gelatin hydrogel group. In general, silk hydrogels are considered as biocompatible, but silk fibroin does not possess inherent cell adhesive ligands. This proliferation assay clearly shows the advantage of incorporating gelatin in the hydrogel since gelatin provides the natural cell adhesive ligands, such as RGD and allows cell adhesion and proliferation. Some silk fibroin-only hydrogels were reported to have comparable mechanical properties as the IPN hydrogels reported here. However, the clear advantage of the gelatin-silk fibroin IPN hydrogel is the inclusion of gelatin, which significantly enhanced its biological activity, manifested by superior cell adhesion and proliferation.
Figure 4.
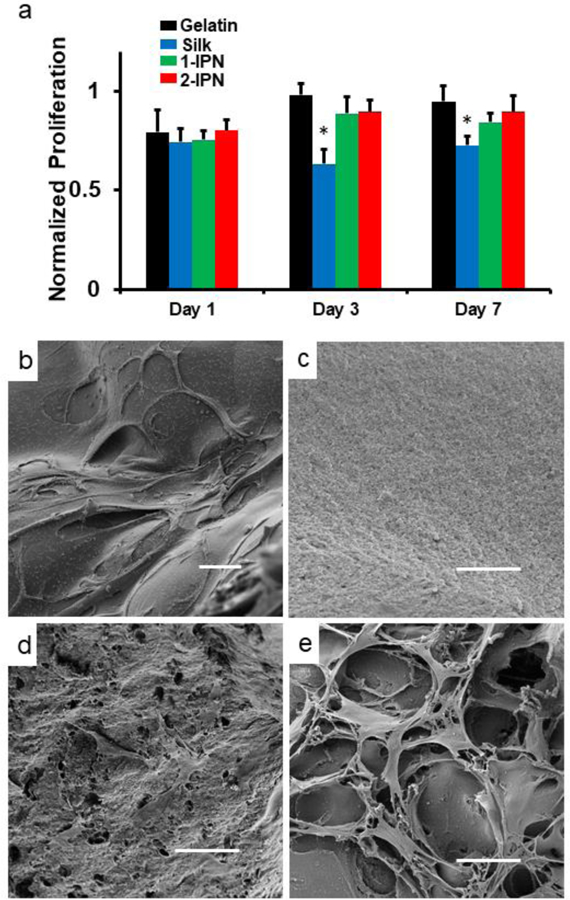
(a) Proliferation of hDFs on hydrogels. The results were normalized to the cells on tissue culture polystyrene (TCPS). * denote p < 0.05 compared to the gelatin group (n = 4). (b-e) SEM images on day 3. (b) Gelatin hydrogel (c) Silk hydrogel (d) 1-IPN (e) 2-IPN. Scale bar = 50 µm. Larger versions of SEM images can be found in ESI.
Due to the optical opacity of the IPN hydrogels, scanning electron microscope (SEM) was used for visualization of hDFs instead of optical microscopy. The frequency of cells in the SEM images was largely consistent with the proliferation assay. On silk hydrogels, very few cells were found (Figure 4b), whereas on both 1-IPN and 2-IPN, a large number of cells could be identified on day 3 (Figure 4c, d).
Conclusions
The gelatin-silk fibroin IPN hydrogels displayed much improved mechanical properties compared to gelatin or silk hydrogels. Addition of another layer of IPN further enhanced the mechanical properties. These biodegradable hydrogels were made of naturally-occurring proteins which were used without any chemical modifications. They were non-cytotoxic and allowed cell adhesion and proliferation of hDFs in vitro. These hydrogels will be useful biomaterials in tissue engineering and regenerative medicine.
Supplementary Material
Acknowledgement
This work was supported in part by an NIH COBRE Center of Integrated Biomedical and Bioengineering Research (CIBBR, P20 GM113131) through an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Tan H, Chu CR, Payne KA and Marra KG, Biomaterials, 2009, 30, 2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rafat M, Li F, Fagerholm P, Lagali NS, Watsky MA, Munger R, Matsuura T and Griffith M, Biomaterials, 2008, 29, 3960–3972. [DOI] [PubMed] [Google Scholar]
- 3.Qi C, Xu L, Deng Y, Wang G, Wang Z and Wang L, Biomaterials science, 2018, 6, 2859–2870. [DOI] [PubMed] [Google Scholar]
- 4.Xavier JR, Thakur T, Desai P, Jaiswal MK, Sears N, Cosgriff-Hernandez E, Kaunas R and Gaharwar AK, ACS nano, 2015, 9, 3109–3118. [DOI] [PubMed] [Google Scholar]
- 5.Carvalho CR, Wrobel S, Meyer C, Brandenberger C, Cengiz IF, Lopez-Cebral R, Silva-Correia J, Ronchi G, Reis RL, Grothe C, Oliveira JM and Haastert-Talini K, Biomaterials science, 2018, 6, 1059–1075. [DOI] [PubMed] [Google Scholar]
- 6.Choi WI, Hwang Y, Sahu A, Min K, Sung D, Tae G and Chang JH, Biomaterials science, 2018, 6, 2627–2638. [DOI] [PubMed] [Google Scholar]
- 7.Liu M, Zeng X, Ma C, Yi H, Ali Z, Mou X, Li S, Deng Y and He N, Bone research, 2017, 5, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuah YJ, Peck Y, Lau JE, Hee HT and Wang DA, Biomaterials science, 2017, 5, 613–631. [DOI] [PubMed] [Google Scholar]
- 9.Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S and Engler AJ, Nature materials, 2014, 13, 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma L, Gao C, Mao Z, Zhou J, Shen J, Hu X and Han C, Biomaterials, 2003, 24, 4833–4841. [DOI] [PubMed] [Google Scholar]
- 11.Long K, Liu Y, Li W, Wang L, Liu S, Wang Y, Wang Z and Ren L, Journal of biomedical materials research. Part A, 2015, 103, 1159–1168. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Barrett DG, Messersmith PB and Holten-Andersen N, ACS nano, 2016, 10, 1317–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H-J, Jiang H and Haraguchi K, Macromolecules, 2018, 51, 529–539. [Google Scholar]
- 14.Cha C, Shin SR, Gao X, Annabi N, Dokmeci MR, Tang XS and Khademhosseini A, Small (Weinheim an der Bergstrasse, Germany), 2014, 10, 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haraguchi K and Takehisa T, Advanced materials, 2002, 14, 1120–1124. [Google Scholar]
- 16.Gaharwar AK, Rivera CP, Wu CJ and Schmidt G, Acta biomaterialia, 2011, 7, 4139–4148. [DOI] [PubMed] [Google Scholar]
- 17.Sheikhi A, Afewerki S, Oklu R, Gaharwar AK and Khademhosseini A, Biomaterials science, 2018, 6, 2073–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranjan P, Agrawal S, Sinha A, Rao TR, Balakrishnan J and Thakur AD, Scientific reports, 2018, 8, 12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao KH, Lin YS, Macosko CW and Haynes CL, ACS applied materials & interfaces, 2011, 3, 2607–2615. [DOI] [PubMed] [Google Scholar]
- 20.Wagner A, White AP, Stueckle TA, Banerjee D, Sierros KA, Rojanasakul Y, Agarwal S, Gupta RK and Dinu CZ, ACS applied materials & interfaces, 2017, 9, 32323–32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomas H, Alves CS and Rodrigues J, Nanomedicine : nanotechnology, biology, and medicine, 2018, 14, 2407–2420. [DOI] [PubMed] [Google Scholar]
- 22.Sun JY, Zhao X, Illeperuma WR, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ and Suo Z, Nature, 2012, 489, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniele MA, Adams AA, Naciri J, North SH and Ligler FS, Biomaterials, 2014, 35, 1845–1856. [DOI] [PubMed] [Google Scholar]
- 24.Branco da Cunha C, Klumpers DD, Li WA, Koshy ST, Weaver JC, Chaudhuri O, Granja PL and Mooney DJ, Biomaterials, 2014, 35, 8927–8936. [DOI] [PubMed] [Google Scholar]
- 25.Fares MM, Shirzaei Sani E, Portillo Lara R, Oliveira RB, Khademhosseini A and Annabi N, Biomaterials science, 2018, 6, 2938–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macdougall LJ, Perez-Madrigal MM, Shaw JE, Inam M, Hoyland JA, O’Reilly R, Richardson SM and Dove AP, Biomaterials science, 2018, 6, 2932–2937. [DOI] [PubMed] [Google Scholar]
- 27.Xiao W, He J, Nichol JW, Wang L, Hutson CB, Wang B, Du Y, Fan H and Khademhosseini A, Acta biomaterialia, 2011, 7, 2384–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidenko N, Schuster CF, Bax DV, Farndale RW, Hamaia S, Best SM and Cameron RE, Journal of materials science. Materials in medicine, 2016, 27, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou S, Lake R, Park S, Edwards S, Jones C and Jeong KJ, ACS Applied Bio Materials, 2018, 1, 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rockwood DN, Preda RC, Yucel T, Wang X, Lovett ML and Kaplan DL, Nature protocols, 2011, 6, 1612–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su D, Yao M, Liu J, Zhong Y, Chen X and Shao Z, ACS applied materials & interfaces, 2017, 9, 17489–17498. [DOI] [PubMed] [Google Scholar]
- 32.Gil ES, Frankowski DJ, Spontak RJ and Hudson SM, Biomacromolecules, 2005, 6, 3079–3087. [DOI] [PubMed] [Google Scholar]
- 33.Yung CW, Wu LQ, Tullman JA, Payne GF, Bentley WE and Barbari TA, Journal of biomedical materials research. Part A, 2007, 83, 1039–1046. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto A, Chen J, Collette AL, Kim UJ, Altman GH, Cebe P and Kaplan DL, The journal of physical chemistry. B, 2006, 110, 21630–21638. [DOI] [PubMed] [Google Scholar]
- 35.Jeong KJ and Panitch A, Biomacromolecules, 2009, 10, 1090–1099. [DOI] [PubMed] [Google Scholar]
- 36.Cao Y and Wang B, International journal of molecular sciences, 2009, 10, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tondera C, Hauser S, Kruger-Genge A, Jung F, Neffe AT, Lendlein A, Klopfleisch R, Steinbach J, Neuber C and Pietzsch J, Theranostics, 2016, 6, 2114–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao X, Lang Q, Yildirimer L, Lin ZY, Cui W, Annabi N, Ng KW, Dokmeci MR, Ghaemmaghami AM and Khademhosseini A, Advanced healthcare materials, 2016, 5, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hozumi T, Kageyama T, Ohta S, Fukuda J and Ito T, Biomacromolecules, 2018, 19, 288–297. [DOI] [PubMed] [Google Scholar]
- 40.Partlow BP, Hanna CW, Rnjak-Kovacina J, Moreau JE, Applegate MB, Burke KA, Marelli B, Mitropoulos AN, Omenetto FG and Kaplan DL, Advanced functional materials, 2014, 24, 4615–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffin M, Premakumar Y, Seifalian A, Butler PE and Szarko M, Journal of visualized experiments : JoVE, 2016, DOI: 10.3791/54872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gil ES, Kluge JA, Rockwood DN, Rajkhowa R, Wang L, Wang X and Kaplan DL, Journal of biomedical materials research. Part A, 2011, 99, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.