

Abstract
Metabolic bone diseases are global public health concerns and are primarily caused by uncontrolled osteoclast (OC) formation and activation. During OC differentiation, intracellular reactive oxygen species (ROS) stimulated by receptor activator of nuclear factor kappa-B ligand (RANKL) can serve as the signaling molecules to promote osteoclastic genes expression. Nuclear factor erythroid-2 related factor 2 (NRF2), a master mediator of cellular antioxidant response, also plays a critical role in OC differentiation through the regulation of redox homeostasis. In this study, we investigated the effects of three NRF2 inducers on osteoclastogenesis, including Bardoxolone methyl (CDDO-Me), Sulforaphane (SFN), and tert-butylhydroquinone (tBHQ). By treating RAW cells with three compounds, we found that NRF2 was activated and its downstream antioxidant genes were upregulated, and the RANKL-induced intracellular ROS production and osteoclastogenesis were impaired. Additionally, the expression of nuclear factor of activated T cells c1 (NFATC1), C-FOS and tumor necrosis factor alpha (TNFα) were inhibited after acute exposures (6 hours) to the three compounds. Furthermore, suppressed expression of osteoclast differentiation associated genes, tartrate-resistant acid phosphatase (TRAP), cathepsin K (CTSK), matrix metalloproteinase-9 (MMP-9) and dendritic cell specific transmembrane protein (DC-STAMP) were observed after prolonged exposures (5 days) to the compounds. Taken together, these results suggest that CDDO-Me, SFN and tBHQ attenuate RANKL-induced osteoclastogenesis via activation of NRF2-mediated antioxidant response. Among these compounds, relatively low concentrations of CDDO-Me showed stronger active and inhibitory effects on antioxidant response and osteoclastogenesis, respectively.
Keywords: CDDO-Me, Sulforaphane, tert-butylhydroquinone, NRF2, ROS, Osteoclast differentiation
1. Introduction
Metabolic bone diseases such as osteoporosis, periodontitis and Paget’s disease of bone are global public health concerns and are primarily caused by uncontrolled osteoclast formation and activation [1–3]. Osteoclasts (OC) are large and multinucleated cells that can resorb bones and differentiate from mononuclear progenitors of monocyte/macrophage lineage, which is strictly regulated by receptor activator of nuclear factor kappa-B ligand (RANKL). During osteoclastogenesis, RANKL is associated with its receptor RANK, forming to the RANKL/RANK axis which plays a central role in physiological bone remodeling via activation of various signaling pathways [4]. Recent studies have demonstrated that intracellular reactive oxygen species (ROS), which are generated by RANKL, can act as signaling molecules to promote OC differentiation and resorption, implicating a critical role for ROS and their mediators in bone homeostasis [5–9]. Thus, controlling the intracellular ROS signaling may provide a promising therapeutic strategy for osteoclast-associated bone diseases. Low/moderate levels of ROS can mediate normal biochemical pathways; conversely, elevated levels of ROS may lead to oxidative stress and even intracellular damage [10]. In physiological and pathological conditions, intracellular redox state is primarily regulated by Nuclear factor erythroid-2 related factor 2 (NRF2). After translocating to the nucleus, NRF2 can form heterodimers with other bZIP proteins, then bind to cis-acting element(s) called antioxidant response elements (ARE) and lead to activation of antioxidant and detoxification genes, such as NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase1 (HO-1), glutamate cysteine ligase catalytic subunit (GCLC) and ligase modifier subunit (GCLM) [10]. Studies with NRF2 global knockout mice showed that NRF2 deficiency was associated with increased intracellular ROS, activation of the RANKL-induced pathways and expression of osteoclastic genes through the downregulation of antioxidant response [7]. Additionally, in vitro studies utilizing NRF2 inducers such as curcumin [7], quercetin [11], dimethyl fumarate [4] and Carnosic acid [12] demonstrated inhibitory effects of these compounds on RANKL-induced OC differentiation, suggesting a critical regulatory role of NRF2 on osteoclastogenesis.
Bardoxolone methyl (CDDO-Me), a newly identified activator of NRF2 pathway, is a triterpenoid analog of oleanolic acid that exhibits promising anti-cancer and anti-inflammatory activities [13]. It was described to affect cellular differentiation in rat neuronal cells [14] and human leukemia cells [15]. However, it has not been reported whether CDDO-Me affects the differentiation of mouse osteoclasts. In this study, we sought to determine and compare the effects of CDDO-Me and two well-known NRF2 inducers: Sulforaphane (SFN) and tert-butylhydroquinone (tBHQ), on the differentiation of mouse RAW 264.7 cells. All three compounds attenuated the RANKL-induced osteoclastogenesis through activating the NRF2 pathway; CDDO-Me, specifically, elicited its effects on antioxidant activation and osteoclastogenesis inhibition at much lower concentrations than those required for SFN and tBHQ.
2. Materials and Methods
2.1. Cell Culture
RAW cells were cultured and expanded in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin at 37 ⁰C in a humidified 5% CO2 atmosphere. For primary osteoclast precursors culture, 8–12 weeks old male C57BL/6 mice were euthanatized by CO2, and tibias and femurs were collected. Then the bone marrow cells were flushed out into α-MEM medium containing 10% FBS and penicillin/streptomycin. After one day, the non-adherent cells were removed and reseeded into plates with 30 ng/mL M-CSF for growing osteoclast precursors. All animal procedures were approved by the Institutional Animal Care and Use Committees at the University of North Carolina, and performed in accordance with the Care and Use of Laboratory Animals of the National Institutes of Health.
2.2. Cell Viability Assay
Cell viability was detected by the RealTime-Glo™ MT Cell Viability Assay (Promega) according to the user manual. The cell viability was monitored using the Cytation 5 imaging reader (BioTek) which measures the luminescence in response to various concentrations of compounds.
2.3. Osteoclast differentiation and TRAP staining.
RAW cells and primary osteoclast precursors were seeded into 12-well plates. After being treated with the compounds in the presence of RANKL (10 ng/mL, R&D systems) and M-CSF (30 ng/mL, only applied to primary cells, R&D systems) for 5 days, the cells were fixed with 10% paraformaldehyde for 10 minutes, and stained with TRAP staining solution for 10–30 minutes. The stained plates were scanned and the images were taken using Eclipse Ti microscope (Nikon).
2.4. RT-qPCR
RAW cells were treated with the compounds for the indicated time points. Total mRNA was extracted using RNAzol (Molecular Research Center) and reverse-transcribed with the iScript™ cDNA Synthesis Kit (Bio-Rad). The PCR reactions were performed on a StepOnePlus Real-time PCR system (Applied Biosystems) using the iTaq™ Universal SYBR Green Supermix (Bio-Rad). The primers for RT-qPCR are listed in Supplementary Table S1. Threshold cycles of primer probes were normalized to a housekeeping gene β-actin, and the relative values were calculated based on Comparative-Ct method (2−∆∆Ct method).
2.5. Western Blot
RAW cells were treated with the compounds for 6 hours, and then the whole protein were extracted. For immunoblot analysis, the Criterion Vertical Electrophoresis Cell and Trans-Blot Turbo Transfer System (Bio-Rad) were used. The primary antibodies used in the study were against NRF2 (Cell Signaling Technology), HMOX1 and β-ACTIN (Santa Cruz Biotechnology).
2.6. Measurement of intracellular ROS
The qualitative analysis of intracellular ROS in compounds-treated RAW cells were determined by fluorescence microscopy using CellROX Green (Thermo Fisher) and Nucblue stain reagents (Invitrogen). The quantitative analysis of intracellular ROS was conducted using 2′,7′-Dichlorofluorescin diacetate (DCF, Sigma-Aldrich). After pretreated with CDDO-Me, SFN and tBHQ for 6 hours, RAW cells were incubated with 10 µM DCF for 15 minutes at 37°C. Then, the cells were stimulated with RANKL (100 ng/mL) for 5 min and homogenized using 0.01 M Tris-HCl with 0.5% Triton X-100 (pH 7.4). The fluorescence of the lysate was measured using plate reader (BioTek) at the following settings: 485 nm excitation and 535 nm emission.
2.7. Statistical Analyses
All statistical analyses were performed using Graphpad Prism 5 (GraphPad Software, San Diego, CA). Significance was determined as p<0.05. The comparisons with a specific control were assessed using one-way analysis of variance (ANOVA) followed by the Bonferroni t-test. Data are expressed as mean ± SEM.
3. Results
3.1. CDDO-Me, SFN and tBHQ attenuated the RANKL-induced differentiation in RAW cells and primary osteoclast precursors.
To determine the effects of the compounds on osteoclastogenesis, RAW cells and osteoclast precursors were treated with CDDO-Me, SFN, and tBHQ at non-cytotoxic concentrations (Supplementary Fig. S1) in the presence of RANKL. As shown in Fig. 1, the formation of TRAP-positive multinucleated osteoclasts was observed in the control groups, but was dramatically suppressed in compounds-treated groups in a concentration-dependent manner. Interestingly, by comparison, CDDO-Me showed significant inhibitory effects on osteoclastogenesis at a relatively low concentration of 0.01 µM. To the best of our knowledge, this is the first report demonstrating the inhibitory effect of CDDO-Me on osteoclastogenesis.
Fig. 1.
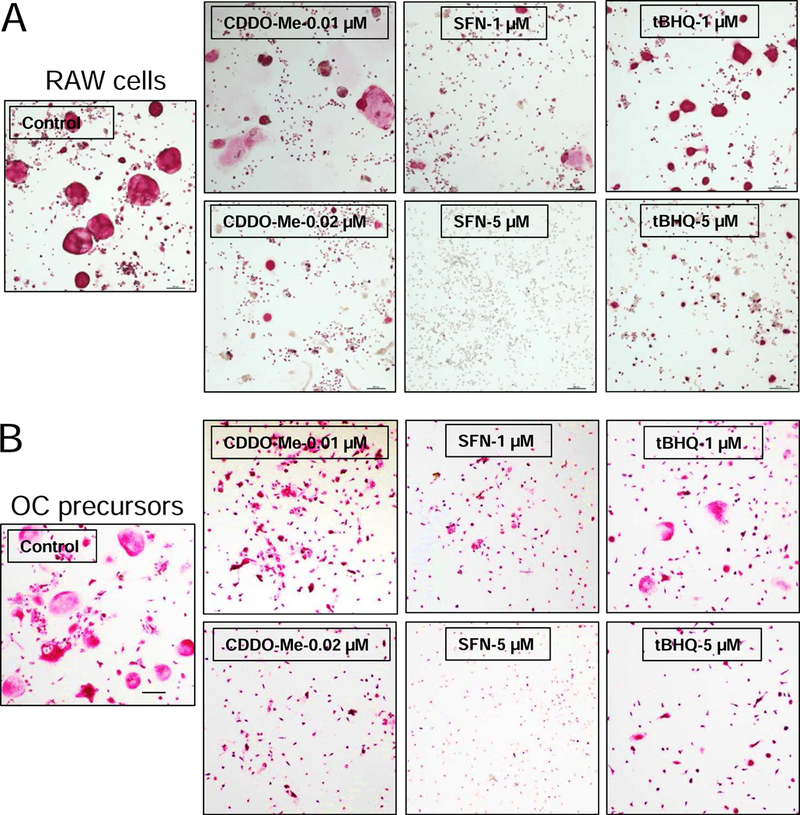
CDDO-Me, SFN and tBHQ attenuate the RANKL-induced differentiation in RAW cells and primary osteoclast precursors. (A) RAW cells and (B) primary osteoclast precursors were treated with various concentrations of CDDO-Me, SFN, and tBHQ in the presence of RANKL (10 ng/ml) and M-CSF (30 ng/mL, only applied to primary cells) for 5 days. The cells were then fixed with 10% paraformaldehyde, and stained with TRAP solution. Images were taken using microscope. Representative images are shown in panel. (bar = 200 µm). Control (only treated with RANKL).
3.2. CDDO-Me, SFN and tBHQ activated NRF2 pathway and upregulated the expression of NRF2-mediated antioxidant genes.
Accumulated evidence shows that NRF2 plays negative regulatory roles in osteoclastogenesis through abolishing RANKL-induced intracellular ROS [4, 7, 12, 16, 17]. To investigate whether CDDO-Me, SFN, and tBHQ activate the NRF2 pathway in RAW cells, the accumulation of NRF2 protein and its downstream targets were assessed. As shown in Fig. 2A, all there compounds activated NRF2 and HO1 in a concentration-dependent fashion in RAW cells. Consistent with these findings, significant upregulation of NRF2-mediated genes, including HO1, NQO1, GCLC, GCLM, Sulfiredoxin (SRX) and Glutathione reductase (GSR), were observed in compound-treated cells (Fig. 2B and C, and Supplementary Fig. S2). Specifically, in the time-response studies shown in Fig. 2B, although the quantitative profile of gene expression varied with these compounds, all the genes tended to reach peak expression between 4 and 6 hours after exposure onset. Overall, these results indicate that the NRF2-mediated antioxidant response in RAW cells can be activated by CDDO-Me, SFN, and tBHQ in generally time- and concentration-dependent manners. Furthermore, CDDO-Me shows stronger capability to activate NRF2 pathway than SFN and tBHQ.
Fig. 2.
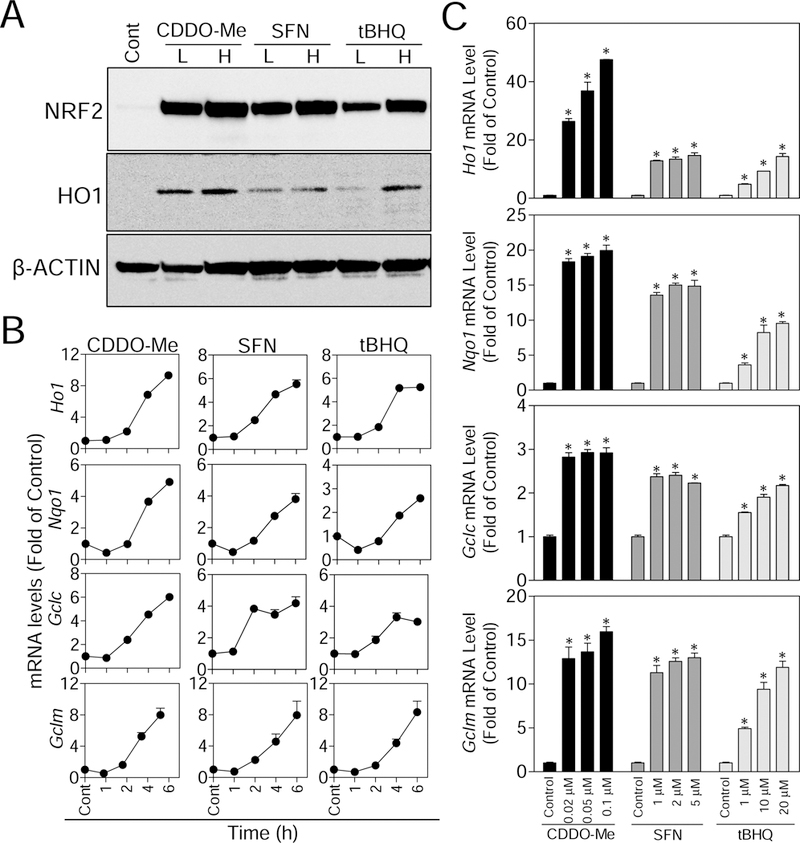
CDDO-Me, SFN and tBHQ promote the NRF2 protein accumulation and stimulate the expression of NRF2-mediated antioxidant genes in time- and concentration-dependent manners. (A) RAW cells were treated with low (L) and high (H) concentrations of CDDO-Me (0.01 and 0.02 µM), SFN (1 and 5 µM), and tBHQ (1 and 5 µM) for 6 hours, then western blotting were performed. (B) In the time-response study, RAW cells were treated with CDDO (0.01 µM), SFN (5 µM) and tBHQ (10 µM) for the indicated time points (1, 2, 4 and 6 hours). Total mRNA was extracted, and NRF2-mediated antioxidant genes (HO1, NQO1, GCLC and GCLM) were measured by RT-qPCR. (C) In the concentration-response study, RAW cells were treated with various concentrations of CDDO-Me, SFN and tBHQ for 6 hours. Then total mRNA was extracted, and NRF2-mediated antioxidant genes (HO1, NQO1, GCLC and GCLM) were measured by RT-qPCR. All results were normalized to β-Actin. Cont, Control (group without any treatments); n = 3. *p < 0.05 vs. Cont.
3.3. CDDO-Me, SFN and tBHQ suppressed the intracellular ROS generation induced by RANKL and the expression of osteoclastic and inflammatory genes.
Intracellular ROS levels were determined to investigate whether CDDO-Me, SFN and tBHQ exposure affect RANKL-stimulated ROS signaling in RAW cells. Consistent with the premise that ROS are signaling molecules regulating osteoclastogenesis, as shown in Fig. 3A and B, RANKL significantly increased the intracellular ROS levels in control cells when compared with the vehicle. While in the compounds-treated cells, intracellular ROS levels were significantly attenuated in a concentration-dependent manner (Fig. 3B), suggesting that upregulated antioxidants (Fig. 2C and D) may impair the production of ROS that are stimulated by RANKL. The abolished ROS molecules is likely responsible, at least partly, for the attenuated differentiation observed in compound-treated cells (Fig. 1).
Fig. 3.
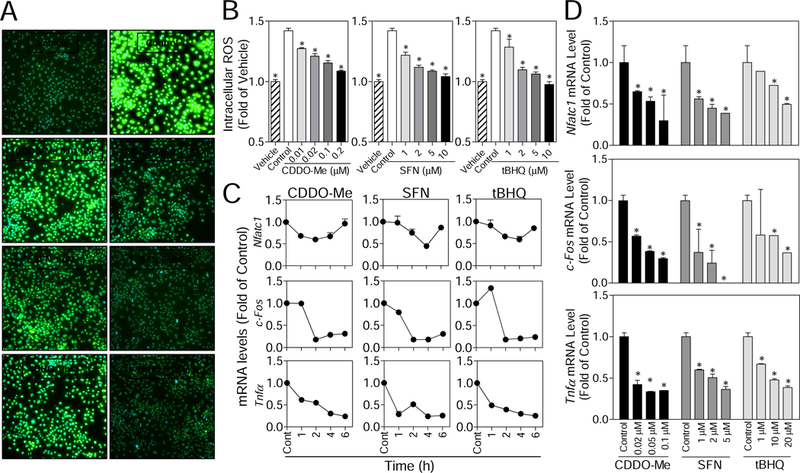
CDDO-Me, SFN and tBHQ suppress the generation of RANKL-induced intracellular ROS and the expression of osteoclastic and inflammatory genes in RAW cells. (A) Merged images (100X) for CellROX Green fluorescence and DAPI in compounds-treated RAW cells. (B) Following the pretreatment of the compounds, the intracellular ROS levels induced by RANKL (100 ng/mL) were determined using DCF in RAW cells. Vehicle (without treatment); Control (only treated with RANKL); CDDO-Me, SFN and tBHQ (pretreated with indicated compounds and then RANKL). (C) RAW cells were treated with CDDO-Me (0.01 µM), SFN (5 µM) and tBHQ (10 µM) for the indicated time points (1, 2, 4 and 6 hours), and the osteoclastic genes (NFATC1, C-FOS and TNFα) were measured by RT-qPCR. (D) RAW cells were treated with various concentrations of CDDO-Me, SFN and tBHQ for 6 hours, and the osteoclastic genes (NFATC1, C-FOS and TNFα) were measured by RT-qPCR. All results were normalized to β-Actin. Cont, Control (normal culture medium); n = 3. *p < 0.05 vs. Cont.
NFATC1 and C-FOS are expressed abundantly in bone tissues, and are considered to be the master regulators in osteoclast differentiation and function. Additionally, pro-inflammatory factor, such as tumor necrosis factor alpha (TNFα), showed to stimulate osteoclastogenesis via a RANKL/RANK-independent mechanism [18]. To further explore whether the osteoclastic and inflammatory genes were affected by three compounds, RT-qPCR was performed to detect gene expression of NFATC1, C-FOS, TNFα, interleukin 1β (IL1β), and interleukin 6 (IL6). As shown in Fig. 3C, the mRNA levels of NFATC1 were decreased by all compounds between 1 and 4 hours of treatment, though the levels interestingly recovered after 6 hours of treatment. The expressions of C-FOS and TNFα were significantly suppressed after 1- or 2-hours of exposure to all compounds. Fig. 3D shows that mRNA levels of NFATC1, C-FOS and TNFα were all decreased by these compounds in a concentration-dependent manner. The compounds have not shown the effects on the expression of IL6 and IL1β (Supplementary Fig. S3). Overall, the significant inhibitory effects of CDDO-Me, SFN, and tBHQ on the expressions of NFATC1, C-FOS, and TNFα have been observed within the first 6 hours of treatment, suggesting that the inhibition of these compounds on RAW cell differentiation may occur at the initial stage of osteoclastogenesis.
3.4. Effects of CDDO-Me, SFN and tBHQ on the expression of OC differentiation associated genes.
To further determine the effects of CDDO-Me, SFN and tBHQ on the expressions of OC differentiation associated genes, we treated RAW cells with compounds in the presence of RANKL (10 ng/ml) for 5 days. As shown in Fig. 4A, the overall expressions of NFATC1 were inhibited by all compounds, while the tBHQ-treated group showed less inhibitory effect than CDDO-Me- and SFN-treated groups. Interestingly, the C-FOS were significantly inhibited by CDDO-Me and tBHQ, but not by SFN. In addition, we measured the gene expressions of terminal stage and cell-cell fusion markers in OC differentiation, including TRAP, cathepsin K (CTSK), matrix metalloproteinase-9 (MMP9), and dendritic cell specific transmembrane protein (DC-STAMP). We found that all the genes have been significantly suppressed by the compounds. Specifically, CDDO-Me (0.01 µM) and SFN (5 µM) showed stronger inhibitory effects on expression of TRAP, CTSK, MMP9 and DC-STAMP than tBHQ (5 µM). In conclusion, these results are consistent with the attenuated phenotypes of OC differentiation (Fig. 1A) including less TRAP-positive multinucleated and smaller size osteoclasts in compounds-treated groups.
Fig. 4.
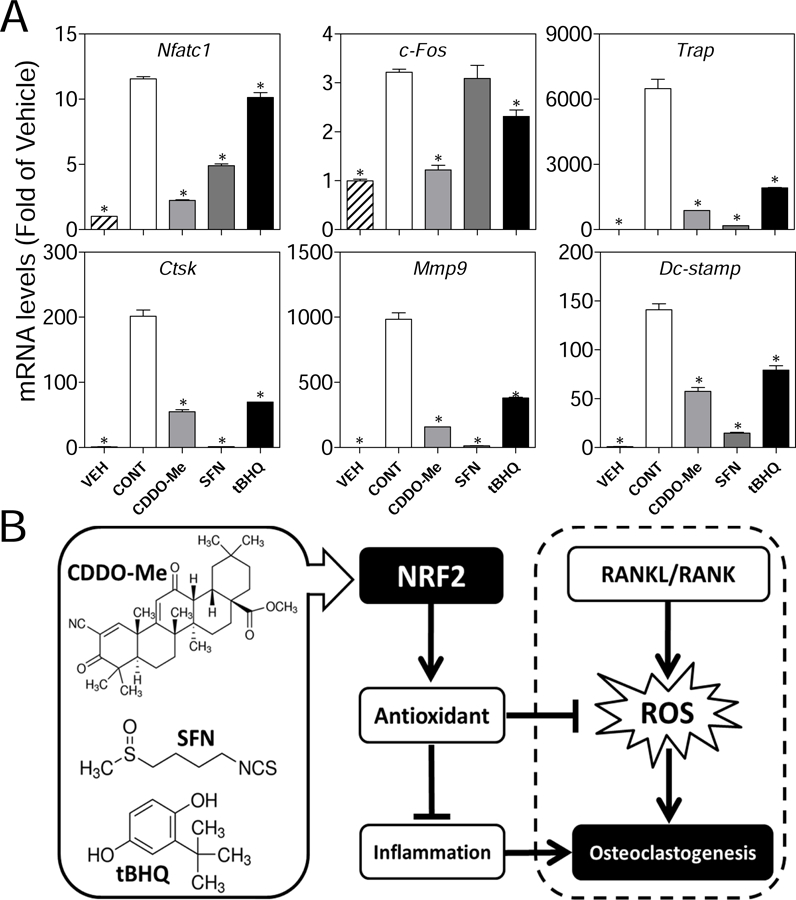
The effects of CDDO-Me, SFN and tBHQ on gene expression of OC differentiation associated markers and schematic presentation of the proposed mechanism. (A) RAW cells were treated with RANKL and non-cytotoxic concentrations of CDDO-Me (0.01 µM), SFN (5 µM) and tBHQ (5 µM) for 5 days. Then total mRNA was extracted and the OC differentiation associated genes (NFATC1, C-FOS, TRAP, MMP9, CTSK and DC-STAMP) were determined by RT-qPCR. All results were normalized to β-Actin. Vehicle (group without treatments); CONT, Control (group treated with RANKL); n = 3. *p < 0.05 vs. Cont.. (B) CDDO-Me, SFN, and tBHQ activate NRF2, which then upregulates the expression of antioxidant enzymes. The elevated antioxidants block RANKL-induced intracellular ROS signaling and suppress pro-inflammatory cytokines which modulate osteoclast differentiation and function, ultimately leading to attenuated osteoclastogenesis.
4. Discussion
Bone is a dynamic tissue, which is constantly remodeled by osteoclasts that resorb bone and by osteoblasts that form new bone. An uncontrolled osteoclast formation causes abnormal bone homeostasis, leading to diseases such as osteoporosis, periodontitis and Paget’s disease of bone [1–3, 19]. Thus, drug/compound that can effectively inhibit osteoclast differentiation and formation will provide a promising strategy for the prevention and treatment of bone resorbing diseases. In this study, we aimed to elucidate the inhibitory effects of three NRF2 inducers CDDO-Me, SFN, and tBHQ on OC differentiation in mouse macrophage RAW 264.7 cells. The concurrence of these compounds-induced NRF2 activation and antioxidant response, attenuated RANKL-induced ROS production and OC differentiation suggests that the inhibitory effects of these compounds on osteoclastogenesis may result, at least partially, from the activation of NRF2 and upregulation of its downstream antioxidant enzymes, that block the intracellular ROS signaling triggered by RANKL (Fig. 4B). To our knowledge, this is the first report showing that CDDO-Me inhibits osteoclastogenesis in RAW cells.
ROS such as superoxide and hydrogen peroxide are by-products of mitochondrial respiration and are traditionally considered deleterious to multiple organ systems. An excessive amount of ROS may result in oxidative stress and cause damage to lipids, proteins, and DNA, implicating in various disorders such as cancer, neurodegeneration and aging [20]. Despite the deleterious effects of ROS, growing evidence suggests that they can also act as secondary messengers in the receptor-mediated signaling cascades, such as insulin-stimulated glucose uptake (ISGU) [10] and RANKL-induced osteoclastogenesis. During osteoclastogenesis, RANKL binds to its receptor RANK and generates intracellular ROS through the activation of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), Ras-related C3 botulinum toxin substrate 1 (RAC1), and NADPH oxidase 1 (NOX1). The induced ROS messengers then trigger Calcium (Ca2+) oscillations and bring about the auto-amplification of NFATC1 [8, 9, 21]. Additionally, RANKL can induce BTB and CNC homology 1 (Bach1), a competitor of NRF2, and attenuate NRF2-mediated antioxidant response, leading to an augmentation of intracellular ROS signaling and osteoclastogenesis [16]. Based on these studies, we propose that the NRF2-mediated cellular adaptive response plays dual roles in RANKL-induced osteoclastogenesis: 1) it protects cells against the oxidative damage; and 2) it blocks RANKL-stimulated ROS signaling, and thus, attenuates osteoclastogenesis.
Previous studies have demonstrated that CDDO-Me increases the NRF2 or NRF2-mediated antioxidant genes in human lung, white blood and breast epithelial cells [22–24]. Here, in RAW cells, we observed that CDDO-Me exposure led to accumulation of NRF2, upregulation of antioxidants (Fig. 2B and C), and attenuation of RANKL-triggered ROS (Fig. 3A and B). These results are consistent with the previous report that both CDDO-Me and CDDO-imidazolide (a CDDO analogue) activated the NRF2 pathway, induced NQO1 and HO1, and reduced ROS in RAW cells [25]. Consistent with the inhibited intracellular ROS, we also observed decreased expressions of NFATC1 and C-FOS after a short-term exposure to CDDO-Me (6 hours), suggesting that CDDO-Me may play an inhibitory role at the initial stages of OC differentiation. In addition to RANKL and M-CSF, pro-inflammatory cytokines such as TNFα, IL1β, and IL6, have been demonstrated to either directly or indirectly play regulatory roles in osteoclastogenesis and inflammatory bone destruction [18, 26]. Therefore, to investigate other inhibitory mechanisms by which CDDO-Me might affect osteoclastogenesis, the anti-inflammatory effects of CDDO-Me in RAW cell were also examined. We found that CDDO-Me decreased the basal gene expression of TNFα (Fig. 3C and D), but not IL1β and IL6 (Fig. S3). Considering the inhibitory effect of CDDO-Me on NFκB signaling by blocking IκBα kinase β (IKKβ) activity in cells, it is possible that CDDO-Me might have attenuated osteoclastogenesis partially through suppressing NFκB-mediated TNFα (Fig. 4B), however, the underlying mechanism needs further investigation.
Consistent with the previous observations, the observed antioxidative effects of SFN and tBHQ in our study are not surprising, since both compounds are widely used antioxidants and NRF2 activators, and have been demonstrated to interact with the NRF2/ARE pathway in many cell types [17]. It has been previously reported that SFN inhibits osteoclastogenesis in vitro through regulating NRF2/HO1 signaling and suppressing the expression of cell-cell fusion markers DC-STAMP and OC-STAMP [27]. Other studies showed that tBHQ had inhibitory effects on OC differentiation and osteoclastic resorption via augmentation of HO1, attenuation of high mobility group box 1 (HMGB1) release and NFATC1 [28]. In our study, though less potent, we found that both SFN and tBHQ exhibited similar activities as CDDO-Me. However, lower concentrations of CDDO-Me showed stronger activities in the induction of HO1 and NQO1 (Fig. 2A and B) and the inhibition of RANKL-induced ROS (Fig. 3A and B) than SFN and tBHQ, which is consistent with previous studies reporting that gene inhibition of HO1 by RNA interference augments OC differentiation and, conversely, overexpression of HO1 inhibits osteoclastogenesis [29]. Furthermore, these results suggest that the inhibitory effects of these compounds on osteoclastic genes may be associated with their abilities to upregulate HO1 and NQO1. Interestingly, 5 days-exposure of SFN (1 µM) showed stronger inhibitory effects on TRAP, CTSK, MMP9, and DC-STAMP than that of CDDO-Me and tBHQ, and it is consistent with the observed inhibition on TRAP staining in both cell types (Fig. 1A and B), indicating a different mechanism or other factors such as compound stability and metabolism may be involved in the prolonged exposure (5 days) of the compounds. However, the underlying mechanism needs further investigation.
In summary, we propose a possible mechanism by which CDDO-Me, SFN, and tBHQ attenuate RANKL-induced osteoclastogenesis in RAW cells (Fig. 4B), providing the foundational information for the development of therapeutic strategy for osteoclast-associated bone diseases.
Supplementary Material
Highlights.
We first investigated the effects and mechanisms of CDDO-Me in osteoclast differentiation
CDDO-Me, SFN and tBHQ attenuate the RANKL-induced osteoclast differentiation in RAW cell
CDDO-Me, SFN and tBHQ activate NRF2-mediated antioxidant response and suppress RANKL-induced intracellular ROS production in concentration-dependent manners.
CDDO-Me elicits its effects on antioxidant activation and osteoclastogenesis inhibition at much lower concentrations than those required for SFN and tBHQ.
Acknowledgments
This work was supported by the National Institutes of Health grant [NIH/NIDCR R01DE022816, 2014].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Compston JE, McClung MR, Leslie WD, Osteoporosis, Lancet, 393 (2019) 364–376. [DOI] [PubMed] [Google Scholar]
- [2].Silva IAL, Conceicao N, Gagnon E, et al. , Effect of genetic variants of OPTN in the pathophysiology of Paget’s disease of bone, Biochim Biophys Acta, 1864 (2018) 143–151. [DOI] [PubMed] [Google Scholar]
- [3].Lee DE, Kim JH, Choi SH, et al. , Periodontitis mainly increases osteoclast formation via enhancing the differentiation of quiescent osteoclast precursors into osteoclasts, J Periodontal Res, 50 (2015) 256–264. [DOI] [PubMed] [Google Scholar]
- [4].Yamaguchi Y, Kanzaki H, Katsumata Y, et al. , Dimethyl fumarate inhibits osteoclasts via attenuation of reactive oxygen species signalling by augmented antioxidation, J Cell Mol Med, 22 (2018) 1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim HS, Nam ST, Mun SH, et al. , DJ-1 controls bone homeostasis through the regulation of osteoclast differentiation, Nat Commun, 8 (2017) 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kanzaki H, Shinohara F, Kajiya M, et al. , The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling, The Journal of biological chemistry, 288 (2013) 23009–23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hyeon S, Lee H, Yang Y, et al. , Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation, Free radical biology & medicine, 65 (2013) 789–799. [DOI] [PubMed] [Google Scholar]
- [8].Kim MS, Yang YM, Son A, et al. , RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis, The Journal of biological chemistry, 285 (2010) 6913–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee NK, Choi YG, Baik JY, et al. , A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation, Blood, 106 (2005) 852–859. [DOI] [PubMed] [Google Scholar]
- [10].Xue P, Hou Y, Zhang Q, et al. , Prolonged inorganic arsenite exposure suppresses insulin-stimulated AKT S473 phosphorylation and glucose uptake in 3T3-L1 adipocytes: involvement of the adaptive antioxidant response, Biochemical and biophysical research communications, 407 (2011) 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wattel A, Kamel S, Prouillet C, et al. , Flavonoid quercetin decreases osteoclastic differentiation induced by RANKL via a mechanism involving NF kappa B and AP-1, J Cell Biochem, 92 (2004) 285–295. [DOI] [PubMed] [Google Scholar]
- [12].Thummuri D, Naidu VGM, Chaudhari P, Carnosic acid attenuates RANKL-induced oxidative stress and osteoclastogenesis via induction of Nrf2 and suppression of NF-kappaB and MAPK signalling, J Mol Med (Berl), 95 (2017) 1065–1076. [DOI] [PubMed] [Google Scholar]
- [13].Wang YY, Yang YX, Zhe H, et al. , Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties, Drug Des Devel Ther, 8 (2014) 2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tran TA, McCoy MK, Sporn MB, et al. , The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection, J Neuroinflammation, 5 (2008) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Konopleva M, Tsao T, Ruvolo P, et al. , Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia, Blood, 99 (2002) 326–335. [DOI] [PubMed] [Google Scholar]
- [16].Kanzaki H, Shinohara F, Itohiya K, et al. , RANKL induces Bach1 nuclear import and attenuates Nrf2-mediated antioxidant enzymes, thereby augmenting intracellular reactive oxygen species signaling and osteoclastogenesis in mice, FASEB J, 31 (2017) 781–792. [DOI] [PubMed] [Google Scholar]
- [17].Kanzaki H, Shinohara F, Itohiya-Kasuya K, et al. , Nrf2 activation attenuates both orthodontic tooth movement and relapse, J Dent Res, 94 (2015) 787–794. [DOI] [PubMed] [Google Scholar]
- [18].Zhang YH, Heulsmann A, Tondravi MM, et al. , Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways, The Journal of biological chemistry, 276 (2001) 563–568. [DOI] [PubMed] [Google Scholar]
- [19].Ishii T, Ruiz-Torruella M, Ikeda A, et al. , OC-STAMP promotes osteoclast fusion for pathogenic bone resorption in periodontitis via up-regulation of permissive fusogen CD9, FASEB J, 32 (2018) 4016–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moreira PI, Smith MA, Zhu X, et al. , Oxidative stress and neurodegeneration, Annals of the New York Academy of Sciences, 1043 (2005) 545–552. [DOI] [PubMed] [Google Scholar]
- [21].Kajiya H, Calcium signaling in osteoclast differentiation and bone resorption, Adv Exp Med Biol, 740 (2012) 917–932. [DOI] [PubMed] [Google Scholar]
- [22].El-Ashmawy M, Delgado O, Cardentey A, et al. , CDDO-Me protects normal lung and breast epithelial cells but not cancer cells from radiation, PLoS One, 9 (2014) e115600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Noel S, Zheng L, Navas-Acien A, et al. , The effect of ex vivo CDDO-Me activation on nuclear factor erythroid 2-related factor 2 pathway in white blood cells from patients with septic shock, Shock, 42 (2014) 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thimmulappa RK, Fuchs RJ, Malhotra D, et al. , Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils, Antioxidants & redox signaling, 9 (2007) 1963–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].To C, Ringelberg CS, Royce DB, et al. , Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis, Carcinogenesis, 36 (2015) 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Michou L, Chamoux E, Couture J, et al. , Gene expression profile in osteoclasts from patients with Paget’s disease of bone, Bone, 46 (2010) 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takagi T, Inoue H, Takahashi N, et al. , Sulforaphane inhibits osteoclast differentiation by suppressing the cell-cell fusion molecules DC-STAMP and OC-STAMP, Biochemical and biophysical research communications, 483 (2017) 718–724. [DOI] [PubMed] [Google Scholar]
- [28].Yamaguchi Y, Sakai E, Sakamoto H, et al. , Inhibitory effects of tert-butylhydroquinone on osteoclast differentiation via up-regulation of heme oxygenase-1 and down-regulation of HMGB1 release and NFATc1 expression, Journal of applied toxicology : JAT, 34 (2014) 49–56. [DOI] [PubMed] [Google Scholar]
- [29].Sakai E, Shimada-Sugawara M, Nishishita K, et al. , Suppression of RANKL-dependent heme oxygenase-1 is required for high mobility group box 1 release and osteoclastogenesis, J Cell Biochem, 113 (2012) 486–498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.